Introduction
Hepatocellular carcinoma (HCC) is one of the most
common malignancies with a poor prognosis and high recurrence rate.
Although the detection rate of early HCC has increased in recent
years and hepatectomy procedures have improved, the mortality rate
remains high. Due to hepatoma cell resistance to chemotherapy and
radiotherapy, the overall survival of HCC remains unsatisfactory
(1). The cancer stem cell (CSC)
hypothesis assumes that rare cells in tumors possess the ability of
self-renewal, unlimited proliferation and pluripotency, which are
the root of tumor recurrence and distant metastasis (2). Additionally, there is accumulating
evidence that CSCs have stronger resistance to traditional
therapies compared to cancer nonstem cells (3–9).
Some reports state that functional liver cancer stem cells (LCSCs)
were found in HCC cell lines (10–15), and CD133 has been used as a
surface maker to isolate LCSCs (12,16).
CD133 (also known as AC133 or prominin-1) is the
first identified gene in the prominin family of pentaspan membrane
proteins, which was originally classified as a marker of primitive
hematopoietic and neural stem cells (17). Subsequent reports showed that
CD133 was also expressed on some normal tissues (18–20). Furthermore, according to recent
reports on the expression and distribution of the antigens, CD133
was notably detected in many types of solid tumor cells,
particularly in their CSCs, such as brain tumors, renal tumors,
colon carcinomas and prostate carcinomas (21–24). As a key role in maintaining the
stemness properties of CSCs, CD133 possesses a feature that its
expression decreases with tumor cell differentiation, making it a
specific marker for isolating and identifying CSCs (25). There are reports that the
expression level of CD133 has a positive correlation with advanced
poorly differentiated HCC (26).
Furthermore, CD133+ HCC cells have stronger
proliferation capacity in vitro and tumorigenesis ability
in vivo (13). However,
knowledge on the functional role of CD133 in LCSCs remains
preliminary.
In the present study, we isolated CD133+
cells from the human HepG2 HCC cell line and identified them as
stem-like cells in HCC. Then we explored the functional role of
CD133 in the modulation of stemness properties and
chemoradiosensitivity in LCSCs by lentivirus-mediated CD133
silencing. Our findings strongly suggest that suppression of CD133
degrades the stemness properties and enhances the
chemoradiosensitivity of LCSCs, which may provide a new strategy
for future LCSC-targeted therapies and, potentially, for the
therapies of other CD133-expressing types of cancer.
Materials and methods
HCC cell lines and cell culture
The HCC cell line HepG2 was obtained from the Cell
Bank of Shanghai Institutes for Biological Sciences, the Chinese
Academy of Sciences (Shanghai, China). The Hep3B and SMMC-7721 HCC
cell lines were obtained from the Institute of Life Sciences of
Chongqing Medical University (Chongqing, China). The cell lines
were routinely cultured in RPMI-1640 medium (Gibco-BRL, USA)
containing 12% heat-inactivated fetal bovine serum (Gibco-BRL) and
maintained at 37°C in a humidified 5% CO2 incubator.
Magnetic sorting and culture of
HepG2-CD133+ cells
The cells for magnetic sorting were magnetically
labeled with CD133 MicroBeads (100 μl/10 million cells) and
separated on MACS MS column (were from Miltenyi Biotec). All
operations were in strict accordance with the manufacturer’s
instructions. The purity of sorted cells was evaluated by flow
cytometry and western blotting. Trypan blue staining was used for
assessing the viability of sorted cells and >90% of these cells
were acceptable for the following experiments.
The fresh isolated HepG2-CD133+ cells
were cultured before assay in a stem cell medium containing
serum-free DMEM/F12 (1:1) medium (Gibco-BRL), 20 ng/ml epidermal
growth factor (EGF), 20 ng/ml basic fibroblast growth factor
(bFGF), and 20 ng/ml leukemia inhibitor factor (LIF) (all were from
Miltenyi Biotec).
Flow cytometry analysis
The CD133 expression analyses were performed
according to the instructions. Briefly, the fresh sorted
CD133+ cells were incubated at 4°C for 30 min with
phycoerythrin (PE)-conjugated anti-human CD133/2 following
treatment with FcR Blocking Reagent kit (from Miltenyi Biotec).
Isotype-matched mouse IgG2b-PE antibodies served as controls. Cell
cycle and apoptosis analyses were performed using a FACSCalibur
apparatus (Becton-Dickinson, USA) through Annexin V and propidium
iodide (PI) dual staining.
Transfection of HepG2-CD133+
cells
The HIV lentiviral vector carrying interfering RNAs
against CD133 (shCD133) and non-silencing RNAs (shNC) was
commercially synthesized (Changsha Yingrun Biotechnologies, Inc.,
China). There were 3 groups: the shCD133-transfected group, the
shNC-transfected group and the blank control group (untreated
HepG2-CD133+ cells). Briefly, fresh sorted
HepG2-CD133+ cells were plated in 6-well plates in stem
cell medium. Twelve hours later, cells were transfected with
shCD133 or shNC, respectively, at a multiplicity of infection (MOI)
of 20. Polybrene (5 μg/ml) was supplemented to promote the
cell transfection efficiency. Twenty-four hours post-transfection,
the medium was changed to fresh culture medium.
Total-RNA isolation and RT-PCR
analysis
Total-RNA was isolated from cells using TRIzol
(Invitrogen, USA) according to the manufacturer’s instructions.
Total-RNA (2 μl) was used for reverse transcription. Random
hexamer primers GAPDH served as a control for RNA integrity. CD133
expression was analyzed by PCR. The primers were, CD133: forward,
5′-GAT TCA TAC TGG TGG CTG GGT GG-3′ and reverse, 5′-GCA GGT GAA
GAG TGC CGT AAG T-3′; GAPDH: forward, 5′-ACC ACA GTC CAT GCC ATC
AC-3′ and reverse, 5′-TCC ACC ACC CTG TTG CTG TA-3′. cDNA was used
in PCR reactions of 30 cycles (94°C 5 min, 94°C 20 sec, 55°C 25
sec, 72°C 30 sec). PCR products were subjected to electrophoresis
using 2% agarose gel and the results were analyzed by gel imaging
analysis system.
Western blot analysis
Cells were washed by PBS 3 times before being
harvested. Cell lysis, sample preparation, SDS-PAGE separation and
electro-transferring to PVDF membrane were performed with standard
methods. CD133/1 (W6B3C1) pure antibody (1:100; Miltenyi Biotec),
mouse anti-GAPDH mAb (1:1,000; KFP, USA), mouse anti-Bcl-2 mAb
(1:1,000; Santa Cruz Biotechnology, Inc., USA) and mouse anti-Bax
mAb (1:1,000; Santa Cruz Biotechnology, Inc.) were used.
Tumorsphere-forming and colony-forming
assays
For tumor-sphere formation assay, cells were seeded
in 6-well plates (Corning Inc., USA) in the form of single cell
suspensions (10,000 cells/well) and supported with serum-free stem
cell medium as mentioned. All plates were maintained at 37°C in a
humidified incubator and fed with 0.1 ml medium every 3 days.
Tumorspheres were observed by inverted microscopy (Olympus, Tokyo,
Japan) on Days 1, 3, 5 and 7. After culturing for 7 days,
serum-free stem cell medium was replaced by serum-based medium and
the morphological changes of tumor-spheres were observed by
inverted microscopy (Olympus).
Colony-forming assay was carried out 5 days after
lentivirus infection as previously described (27). Briefly, 1,000 cells were seeded in
6-well plates (Corning Inc.) and 14 days later the colonies were
stained with Giemsa solution and scored if they contained >50
cells under an inverted microscope (Olympus).
Cell proliferation assays
Cell proliferation was examined on Days 0, 1, 2, 3,
4 and 5 after inoculation by means of a cell proliferation assay
using Cell Counting kit-8 (CCK-8) (Beyotime Institute of
Biotechnology, China) according to the manufacturer’s instructions.
The optical density was measured using a Multiskan Spectrum (Thermo
Scientific, USA) at a wavelength of 450 nm.
Animal preparation and xenograft
tumorigenicity assay
All procedures involving animals were in accordance
with the institutional animal welfare guidelines of the Animal Care
and Use Committee. Female 6- to 8-week-old NOD/SCID mice were
purchased from the Animal Experiment Center of Chongqing Medical
University (Chongqing, China) and randomly divided into 3 groups (5
mice/group). To determine the tumorigenesis ability of sorted
CD133+ HCC cells in vivo, increasing number of
HepG2-CD133+ cells (100–10,000 cells/mouse) were
suspended in 200 μl serum-free DMEM/F2 and Matrigel
(Invitrogen) mixture (1:1), and injected subcutaneously into the
NOD/SCID mice. HepG2-CD133− cells were used as control.
The incidence of subcutaneous tumors was recorded. Five weeks
later, the grafts were separated, fixed in 10% formaldehyde
solution, and embedded in paraffin for histological analysis. FACS
was used to examine the expression of CD133 in the transplanted
cells. The tumorigenesis ability of CD133-downregulated
HepG2-CD133+ cells was tested based on the minimal
number of HepG2-CD133+ cells which could form tumors in
NOD/SCID mice.
Drug susceptibility testing
To assess the chemosensitivity of CD133 silencing
cells, shCD133-transfected HepG2-CD133+ cells were
seeded in 96-well plates (8,000 cells/well) with serum-free stem
cell medium 5 days after lentivirus infection. Cisplatin (5
μg/ml) and doxorubicin (5 μg/ml) were simultaneously
added into the plates. After 24 and 48 h, the cell growth
inhibition rate (GIR) was studied by standard CCK-8 assay (28).
Radiation treatment and clonogenic
assay
X-ray irradiation (IR) was delivered by an
electro-linear accelerator (Varian 23EX, USA) at a dose rate of 2
Gy/min, with a surface to surface distance (SSD) of 100 cm.
Clonogenic assay was carried out 5 days after lentivirus infection.
Briefly, increasing number of cells were exposed to corresponding
radiation doses (0, 2, 4, 6, 8 and 10 Gy). Fourteen days after
incubation, colonies (>50 cells/colony) were counted and
analyzed. Plating efficiency (PE) and survival fraction (SF) were
calculated as follows: PE = (colony number/number of inoculated
cells) ×100%; SF = colonies counted/(cells seeded × [PE/100])
(29).
In vivo study
The in vivo experiment was performed for
further validation. Briefly, 5 days after lentivirus infection,
2×105 shCD133-transfected HepG2-CD133+ cells
were injected subcutaneously into 6- to 8-week-old NOD/SCID mice (3
mice/group). Twenty-four hours later, daily i.p. injection of
cisplatin (1.5 mg/kg, for 7 days) or 4 Gy ionizing irradiation were
administered (29,30). HepG2-CD133+ cells
served as blank control. Tumor size was measured using vernier
calipers every week after injection for a total of 4 weeks. The
volume (mm3) was calculated by the formula:
(width2 × length)/2.
Statistical analysis
Statistically significant values were determined
using SPSS17.0 software. Data are presented as the means ± standard
deviation (SD) and evaluated with the Student’s t-test. P<0.05
was considered to indicate statistically significant
differences.
Results
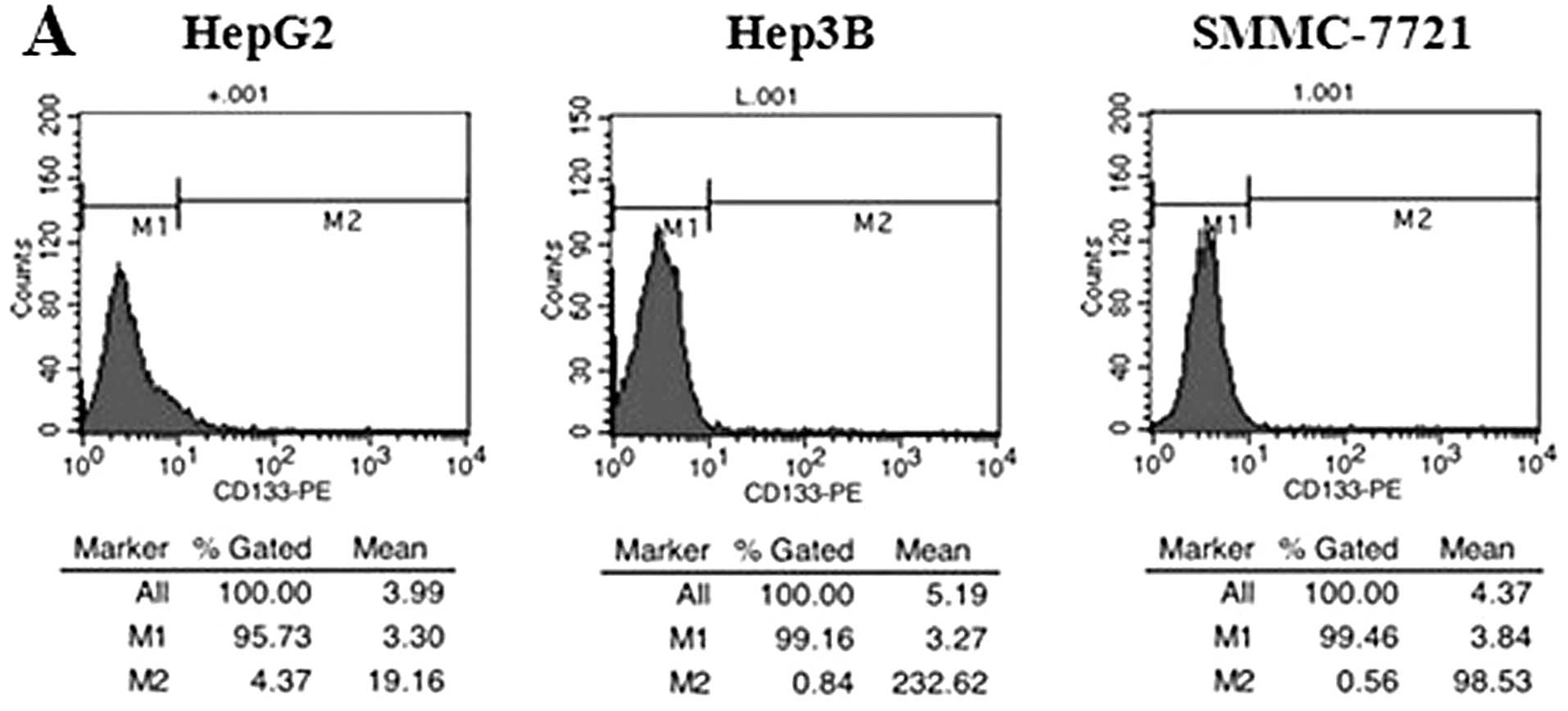
CD133 expression in HCC cell lines and
magnetic sorting of CD133+ cells from the HepG2 cell
line
Prior to sorting, CD133 expression was evaluated by
flow cytometry analysis to determine the frequency of
CD133+ cells in HCC cell lines. The results showed that
CD133+ cells made up 1.16–4.37% of unsorted HepG2 cells,
0.57–0.84% of unsorted Hep3B cells and 0.21–0.56% of unsorted
SMMC-7721 cells (Fig. 1A). Based
on these data and our experimental task needs, we chose the HepG2
cells for the current study. After sorting by MACS, we successfully
enriched a high purity of CD133+ (>91%) and
CD133− population from the HepG2 cell line (Fig. 1B). Then the expression level of
the CD133 protein in the isolated populations was validated by
western blotting. A significantly stronger expression of the CD133
protein was detected in the CD133+ population (Fig. 1C). Furthermore, we found a notable
cellular morphology change between CD133+ and
CD133− cells. The HepG2-CD133− cells grew
against the wall of the flask as distributed monolayers, whereas
the HepG2-CD133+ cells grew as aggregate cell clusters
(Fig. 1D).
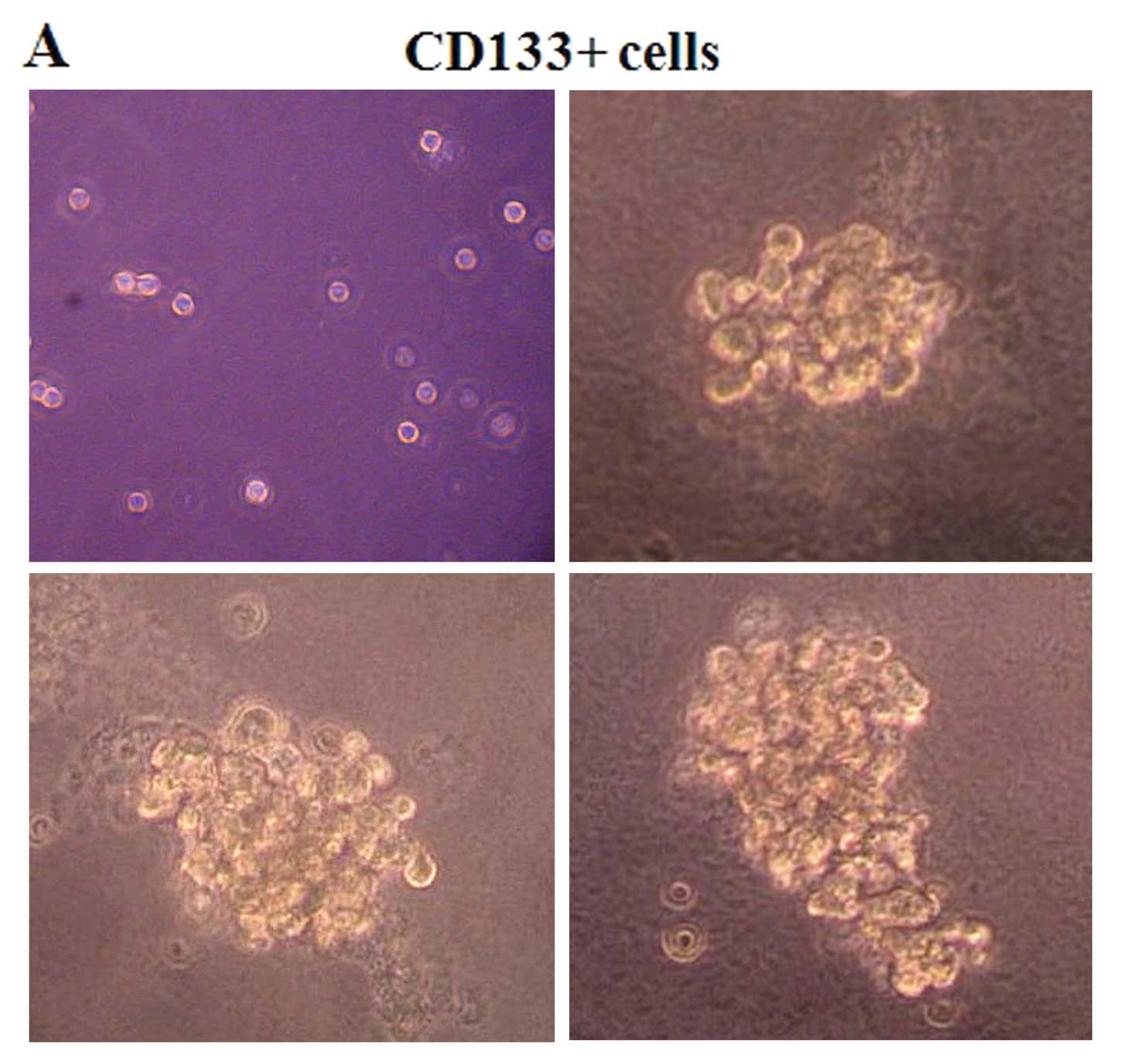
HepG2-CD133+ cells show higher
tumorsphere formation, colony-forming and proliferation
ability
To verify the stemness properties of
CD133+ HCC cells in vitro, tumorsphere formation,
colony-forming and cell proliferation assays were carried out. In
the sphere formation assay, we found that CD133+ and
CD133− cells grew in the form of suspended individual
cells on the first day. As time passed, CD133+ cells
grew in aggregate clusters and increased in size (Fig. 2A), whereas the CD133−
cells presented aberrant cell shapes after culturing in the
serum-free stem cell medium and failed to stay alive in it for more
than 1 week (Fig. 2B).
Forty-eight hours after replacing the serum-free stem cell medium
with serum-based medium, the non-adherent tumorspheres of cells
attached the bottom of the flask and grew into monolayers (Fig. 2C). Colony formation assay showed
that CD133+ cells possess higher colony-forming ability
than CD133− cells. CD133+ cells formed more
and larger colonies than their CD133− counterparts
(P<0.001) (Fig. 2D and E). In
addition, we pursued the proliferative activity of
CD133+ cells. The results showed that the proliferation
rates of CD133+ cells on Days 3, 4, 5 were significantly
higher than CD133− cells (P<0.01), and unsorted cells
showed higher proliferation rates than CD133− cells on
Day 5 (P<0.05) (Fig. 2F and
G).
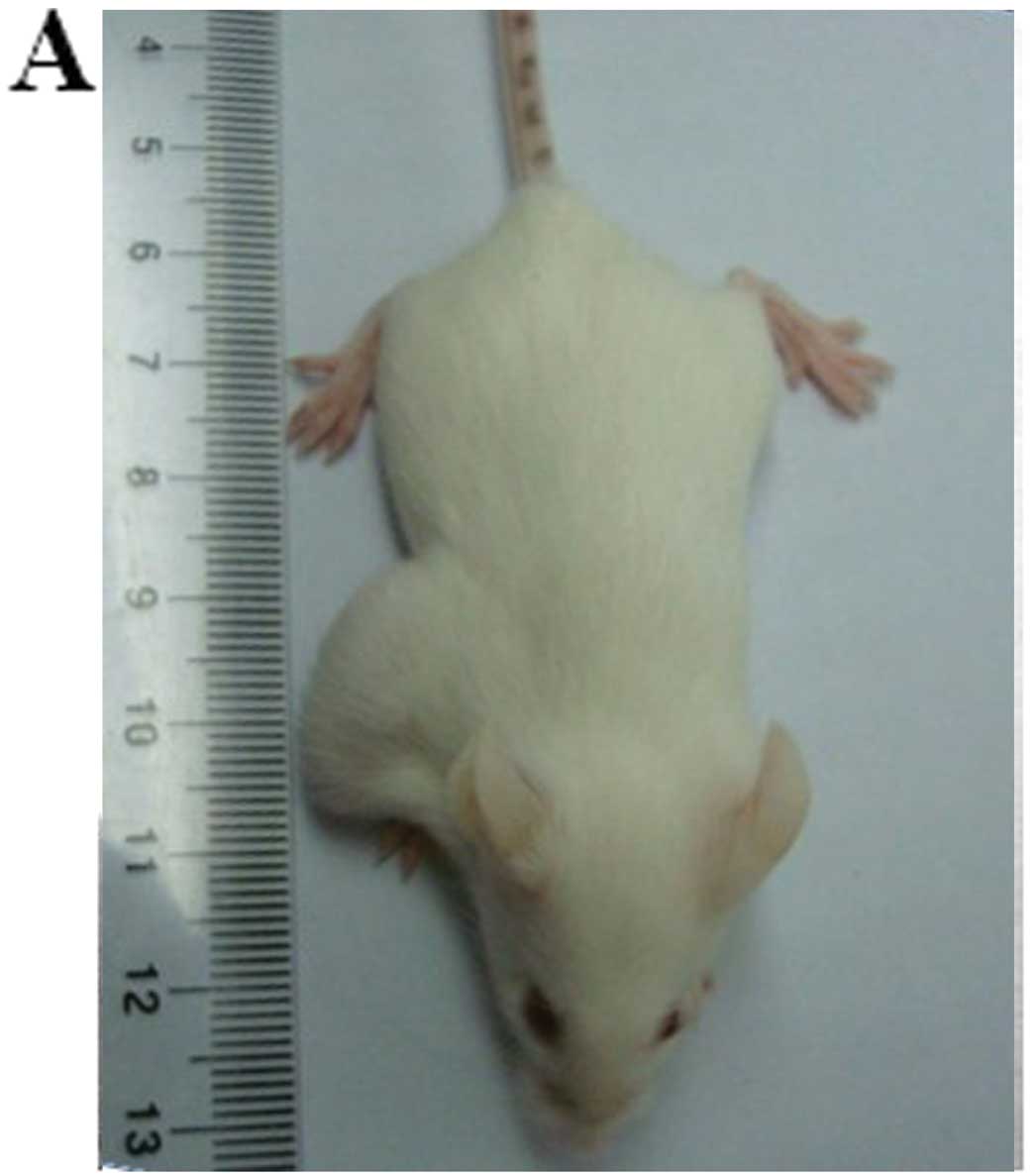
HepG2-CD133+ cells possess
higher capacity for tumorigenicity
To validate the capacity of initiating tumors of
CD133+ HCC cells in vivo, we compared the
abilities of CD133+ vs. CD133− HepG2 cells to
give rise to xenografts in NOD/SCID mice. A significant difference
in tumorigenicity was found between these 2 subpopulations
(Table I). As few as 1,000
CD133+ cells were sufficient to form subcutaneous
xenografts in 3 of 5 inoculated NOD/SCID mice 5 weeks after
inoculation, and 10,000 CD133+ cells formed grafts in 5
of 5 inoculated NOD/SCID mice. However, no tumors were observed in
inoculation of 10,000 CD133− cells (Fig. 3A). Hematoxylin and eosin (H&E)
staining analysis showed a highly cellular mass below the
CD133+ cell injection site (Fig. 3B). Furthermore, CD133 expression
was analyzed by flow cytometry to elucidate whether
CD133+ cells self-renew and generate CD133−
cells in vivo. The result revealed that the grafted cells
consisted of 4.30% CD133+ and 95.72% CD133−
cells (Fig. 3C), which resembled
the CD133 expression pattern of the original HepG2 cells.
 | Table I.Tumorigenicity study. |
Table I.
Tumorigenicity study.
| No. of cells
injected/mouse | CD133+
HCC cells | CD133−
HCC cells | shCD133-HCC
cells |
|---|
|
1×102 | 0/5a | 0/5 | - |
|
1×103 | 3/5 | 0/5 | 0/5 |
|
1×104 | 5/5 | 0/5 | 2/5 |
|
1×105 | - | - | 5/5 |
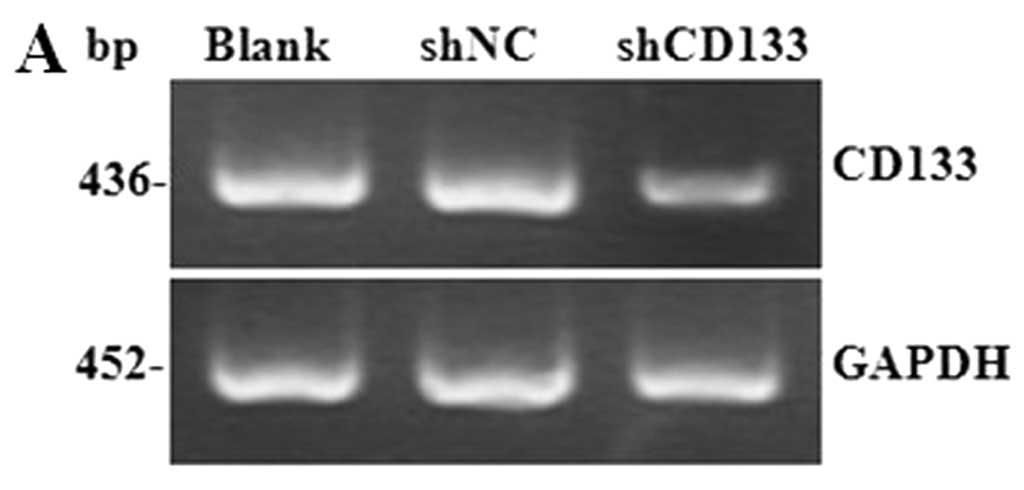
Infection of lentivirus containing shRNA
targeting CD133 in HepG2-CD133+ cells
To assess the role of CD133 in CD133+
LCSCs, expression of CD133 in HepG2-CD133+ cells was
downregulated by RNA interference (RNAi). GFP expression was
observed in shCD133-transfected cells 4 days after lentivirus
infection. RT-PCR results showed that the expression of CD133 mRNA
was decreased by 66.45% in CD133 knockdown cells, compared to the
blank control (P<0.01) (Fig.
4A), and western blotting results showed that the expression of
the CD133 protein was significantly decreased after 7 days of
lentivirus infection (P<0.01) (Fig. 4B). These results indicated that
CD133 was efficiently downregulated in HepG2-CD133+
cells by lentivirus-mediated shRNA.
Silencing of CD133 suppresses stemness
properties of HepG2-CD133+ cells
After 5 days of lentivirus infection, tumorsphere
formation, colony-forming and proliferation assays were performed
again to examine the biological changes in CD133-downregulated
LCSCs. Fig. 4C shows that
CD133+ cells transfected with shCD133 displayed a
significantly reduced proliferation rate, compared to the shNC and
the blank control group (P<0.01). Giemsa stained colonies
revealed that CD133 downregulation resulted in a dramatic decrease
in the number and size of colonies (P<0.001) (Fig. 4D and E). Furthermore, this
shCD133-induced CD133 downregulation was accompanied by a reduced
number and a smaller size of tumorspheres (Fig. 4F). Additionally, the tumorigenesis
ability of CD133 silenced cells was tested and the results showed
that 1,000 HepG2-CD133+ cells failed to form tumor
xenografts in NOD/SCID mice as they once did after CD133
downregulation. Also, 10,000 CD133-downregulated cells formed
tumors in 2 of 5 inoculated NOD/SCID mice. Only as many as
1×105 CD133 downregulated cells produced tumors in 5 of
5 NOD/SCID mice 5 weeks after inoculation (Table I).
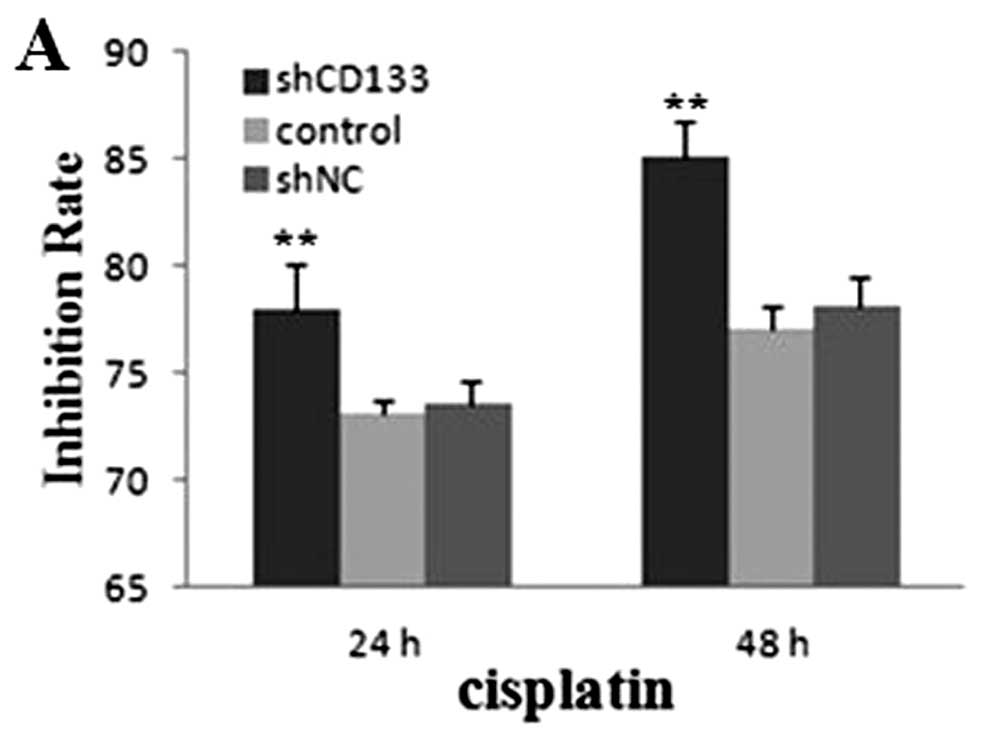
Suppression of CD133 enhances
chemoradiosensitivity of HepG2-CD133+ cells
To explore the potential role of CD133 in
chemoradiosensitivity in CD133+ LCSCs, experiments in
vitro and in vivo were employed. We assessed the
sensitivity of the cells to cisplatin and doxorubicin. Compared
with the shNC and the blank control group, both the cell GIR of
cisplatin and doxorubicin in the shCD133 group were higher
(P<0.01) (Fig. 5A and B). For
radiosensitivity testing in vitro, an apparent difference in
clone rate (GR) and survival fraction (SF) was observed after
radiation treatment (Table II).
The SF of the shCD133 group was significantly lower than the shNC
and the blank control group after 14 days of incubation (P<0.01)
(Fig. 5C). Furthermore, we
pursued the chemoradiosensitivity of CD133-downregulated LCSCs
in vivo. Our data showed that either cisplatin treatment or
4 Gy radiation treatment effectively restrained the growth speed of
tumors in the shCD133 group. Four weeks later, xenografts were
separated. Tumor volumes in the shCD133 group were significantly
smaller than in the blank control group (P<0.01) (Fig. 5D and E). These results support a
functional role of CD133 in LCSC chemoradioresistance.
Downregulation of CD133 significantly increased the sensitivity of
LCSCs to chemotherapy and radiotherapy.
| Table II.Clone rate and survival fraction
study. |
Table II.
Clone rate and survival fraction
study.
| | shCD133
| shNC
| Blank control
|
|---|
| No. of
cells/well | IR dose (Gy) | CR (%) | SF (%) | CR (%) | SF (%) | CR (%) | SF (%) |
|---|
| 200 | 0 | 18.00±2.78 | 100.00±15.47 | 35.50±3.77 | 101.43±10.79 | 35.00±4.92 | 100.00±14.07 |
| 400 | 2 | 7.50±1.40 | 41.67±7.73 | 31.00±2.61 | 88.57±7.46 | 31.58±2.65 | 90.24±7.57 |
| 600 | 4 | 2.67±0.88 | 14.81±4.90 | 23.83±5.00 | 68.10±14.29 | 23.38±3.78 | 66.83±10.80 |
| 800 | 6 | 1.13±0.25 | 6.25±1.39 | 14.25±1.19 | 40.71±3.41 | 15.75±1.35 | 45.00±3.86 |
| 1,000 | 8 | 0.16±0.06 | 0.93±0.32 | 7.30±0.46 | 20.86±1.31 | 8.07±1.00 | 23.05±2.86 |
| 1,200 | 10 | 0.06±0.05 | 0.15±0.28 | 1.25±0.22 | 3.75±0.63 | 1.50±0.44 | 4.29±1.26 |
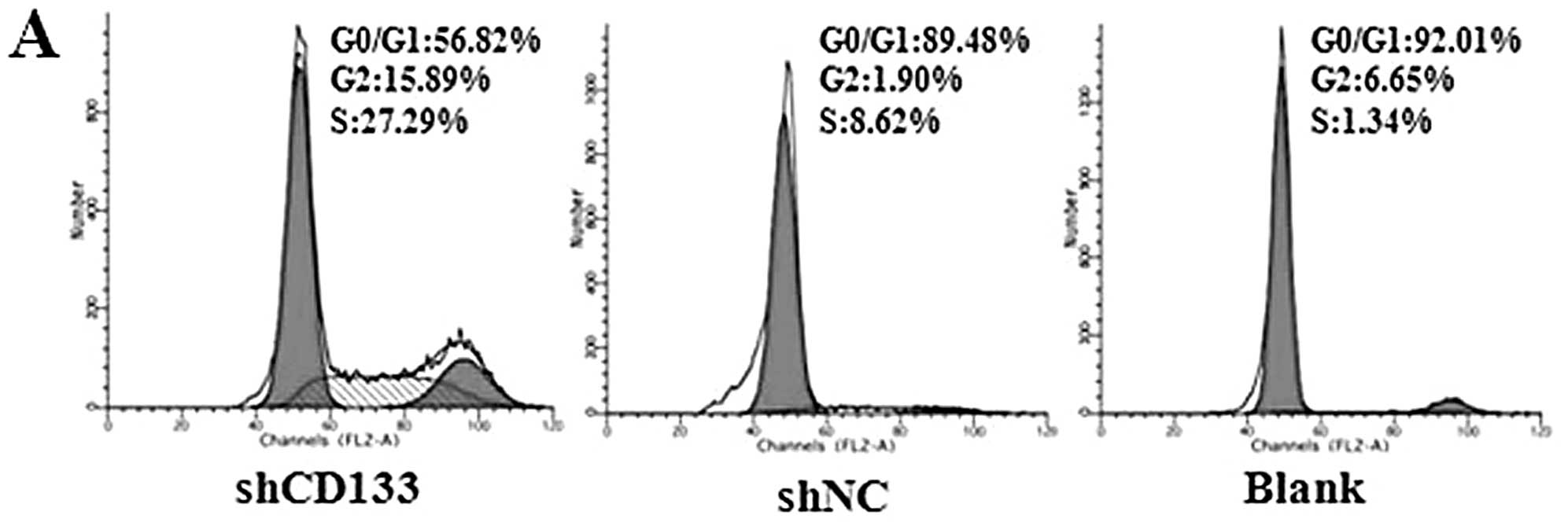
Effects of CD133 downregulation on cell
cycle and apoptosis
Effects of CD133 in the regulation of cell cycle and
apoptosis were assessed to explore the possible mechanism of
enhanced chemoradiosensitivity. The results of cell cycle analyses
showed that the fresh isolated CD133+ LCSCs were mostly
distributed in the static phase (G0/G1). Following downregulation
of CD133 by RNAi, cells in the G0/G1 phases of the cell cycle
decreased significantly, which revealed that silencing of CD133
initiated LCSC action to differentiate (Fig. 6A and B). We also measured the
apoptotic cells by flow cytometry analysis. It was observed that up
to 26.60±3.92% of the shCD133 group cells became apoptotic 5 days
after lentivirus infection, whereas the apoptosis rate was
3.71±0.47% in the shNC group and 2.77±0.94% in the blank control
group (P<0.05) (Fig. 6C and
D). We then examined the expression of several molecules
involved in cell survival regulation. Western blotting results
showed that downregulation of CD133 decreased the expression level
of Bcl-2 but increased the expression level of Bax (Fig. 6E).
Discussion
The cancer stem cell (CSC) hypothesis suggests that
only a small subset of cells within a tumor possess the ability to
self-renew and differentiate to multiple lineages, which were
defined as tumor-initiating cells (31). Studies have confirmed that CSCs
exist in both hematologic and solid tumors (32,33). CD133, a member of pentaspan
membrane proteins encoded by the PROM1 gene, represents a marker of
tumor-initiating cells in a number of human cancers (34), and has been used to isolate liver
cancer stem cells (LCSCs) from hepatocellular carcinoma in recent
years (28,35). Therefore, taking the CSC model as
a reference (6,36), LCSC-targeted therapy via this
marker may be an effective and curative strategy for eradicating
cancer. Here, we sorted CD133+ cells from HepG2 cells,
which demonstrated many stem-like properties. We downregulated
CD133 in HepG2 CD133+ cells by lentivirus-mediated RNAi
and analyzed the functional role of CD133 in the modulation of
stemness properties and chemoradiosensitivity in LCSCs.
Our analysis of CD133 expression in the HepG2, Hep3B
and SMMC-7721 cell lines showed that HepG2 cells possess higher
endogenic CD133 expression, compared to Hep3B and SMMC-7721 cells.
After sorting by MACS, high-purity HepG2-CD133+ cells
were obtained for subsequent experiments. In order to verify the
CSC-like properties of sorted CD133+ cells, the
proliferation, tumorsphere formation, colony-forming in
vitro and tumorigenicity in vivo abilities were compared
between HepG2-CD133+ and HepG2-CD133− cells.
The present study showed that HepG2-CD133+ cells possess
higher proliferation, tumorsphere-forming and colony-forming
abilities in vitro and tumorigenesis ability in vivo,
which revealed that HepG2-CD133+ cells have stem-like
features and could be treated as LCSCs. Suetsugu et al
(16) demonstrated
CD133+ HCC cells as cancer stem/progenitor cells, which
is consistent with our findings. Thus, CD133 could be regarded as a
potential target for stem-targeted therapy for HCC.
Next, we infected HepG2-CD133+ cells with
lentiviruses containing shRNA targeting CD133. After the silencing
of CD133, the cell proliferation, tumorsphere formation and colony
formation of LCSCs were significantly inhibited. Furthermore,
attenuated tumor formation in NOD/SCID mice was observed after
CD133-targeted RNAi, which supported the results of previous
studies that reported induced oncogenicity in metastatic melanoma
and glioblastoma cells after CD133 downregulation (37,38). However, CD133-silenced LCSCs could
still survive in serum-free stem cell medium with decreased
tumorsphere formation, compared to sorted CD133− cells
which failed to survive in serum-free stem cell medium. Excluding
the impacts of CD133 silencing efficiency and experimental errors,
one explanation might be that CD133 is not the only molecule
maintaining the stemness properties of CD133+ LCSCs.
Beyond CD133, the markers of LCSCs proposed at present include
CD90, CD44 and OV6 (39,40). Thus, we speculate that there are
inner links among these markers expressed in LCSCs and the
CD133+ cells are more likely to express other LCSC
markers than CD133− cells.
In addition, we assessed the chemoradiosensitivity
of CD133-downregulated LCSCs. Our present study in vitro and
in vivo showed that knockdown of CD133 could significantly
enhance the sensitivity of LCSCs to chemotherapy and radiotherapy.
Cell cycle and apoptosis were then analyzed by flow cytometry to
investigate the possible mechanism. Our present results showed that
G0/G1 phase LCSCs were dramatically decreased after CD133 silencing
by RNAi. Based on the stem cell theory, CSCs are almost exclusively
in the G0/G1 phases of the cell cycle (41), and dormant CSCs are more resistant
to chemotherapy and radiotherapy than differentiated cancer cells
(42). Initiating the LCSCs into
differentiation, which may be one of the causes of enhanced
sensitivity to traditional treatments in LCSCs. The results of
apoptosis analysis showed an increased apoptosis rate after
suppression of CD133, and western blotting showed an obviously
declined expression of anti-apoptotic protein Bcl-2 and increased
expression of apoptosis protein Bax after CD133 downregulation,
which may be another cause for enhancing LCSC
chemoradiosensitivity. Briefly, these results suggested that
enhanced chemoradio-sensitivity of LCSCs may be related to altered
cell cycle distribution and weakened anti-apoptosis ability.
However, the molecular mechanisms of CD133 in chemoradiosensitivity
remain insufficiently clear and it is necessary to develop further
studies on the relationship among LCSC markers.
In summary, downregulation of endogenous CD133
inhibits the stem-like properties and enhances
chemoradiotherapeutic response of LCSCs in vitro and in
vivo, supporting that CD133 acts as a crucial marker in
maintaining the LCSC performances. Stem-targeted therapy via CD133
silencing could be an effective way for the treatment of HCC and
potentially for other CD133-expressing cancer types.
Acknowledgements
This study was supported by Grant no.
81171365 from the National Natural Science Foundation of China. We
thank the Chongqing Cancer Institute (Chongqing, China) for
providing the electrolinear accelerator.
References
|
1.
|
Rich JN: Cancer stem cells in radiation
resistance. Cancer Res. 67:8980–8984. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Rosen JM and Jordan CT: The increasing
complexity of the cancer stem cell paradigm. Science.
324:1670–1673. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Bao S, Wu Q, McLendon RE, et al: Glioma
stem cells promote radioresistance by preferential activation of
the DNA damage response. Nature. 444:756–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Baumann M, Krause M and Hill R: Exploring
the role of cancer stem cells in radioresistance. Nat Rev Cancer.
8:545–554. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Eramo A, Ricci-Vitiani L, Zeuner A, et al:
Chemotherapy resistance of glioblastoma stem cells. Cell Death
Differ. 13:1238–1241. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Winquist RJ, Boucher DM, Wood M and Furey
BF: Targeting cancer stem cells for more effective therapies:
Taking out cancer’s locomotive engine. Biochem Pharmacol.
78:326–334. 2009.PubMed/NCBI
|
|
7.
|
Zhang Q, Shi S, Yen Y, et al: A
subpopulation of CD133+ cancer stem-like cells
characterized in human oral squamous cell carcinoma confer
resistance to chemotherapy. Cancer Lett. 289:151–160. 2010.
|
|
8.
|
Liu G, Yuan X, Zeng Z, et al: Analysis of
gene expression and chemoresistance of CD133+ cancer
stem cells in glioblastoma. Mol Cancer. 5:672006. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Jin Y, Bin ZQ, Qiang H, et al: ABCG2 is
related with the grade of glioma and resistance to mitoantone, a
chemotherapeutic drug for glioma. J Cancer Res Clin Oncol.
135:1369–1376. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Chiba T, Kamiya A, Yokosuka O, et al:
Cancer stem cells in hepatocellular carcinoma: Recent progress and
perspective. Cancer Lett. 286:145–153. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Chiba T, Kita K, Zheng Y, et al: Side
population purified from hepatocellular carcinoma cells harbors
cancer stem cell-like properties. Hepatology. 44:240–251. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Yin S, Li J, Hu C, et al: CD133 positive
hepatocellular carcinoma cells possess high capacity for
tumorigenicity. Int J Cancer. 120:1444–1450. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Ma S, Chan K, Hu L, et al: Identification
and characterization of tumorigenic liver cancer stem/progenitor
cells. Gastroenterology. 132:2542–2556. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Yang W, Yan HX, Chen L, et al:
Wnt/beta-catenin signaling contributes to activation of normal and
tumorigenic liver progenitor cells. Cancer Res. 68:4287–4295. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Yamashita T, Ji J, Budhu A, et al:
EpCAM-positive hepatocellular carcinoma cells are tumor-initiating
cells with stem/progenitor cell features. Gastroenterology.
136:1012–1024. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Suetsugu A, Nagaki M, Aoki H, et al:
Characterization of CD133+ hepatocellular carcinoma
cells as cancer stem/progenitor cells. Biochem Biophys Res Commun.
351:820–824. 2006.
|
|
17.
|
Yin AH, Miraglia S, Zanjani ED, et al:
AC133, a novel marker for human hematopoietic stem and progenitor
cells. Blood. 90:5002–5012. 1997.PubMed/NCBI
|
|
18.
|
Richardson GD, Robson CN, Lang SH, et al:
CD133, a novel marker for human prostatic epithelial stem cells. J
Cell Sci. 117:3539–3545. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Corbeil D, Roper K, Hellwig A, et al: The
human AC133 hematopoietic stem cell antigen is also expressed in
epithelial cells and targeted to plasma membrane protrusions. J
Biol Chem. 275:5512–5520. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Uchida N, Buck DW, He D, et al: Direct
isolation of human central nervous system stem cells. Pro Natl Acad
Sci USA. 97:14720–14725. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Singh SK, Clarke ID, Terasaki M, et al:
Identification of a cancer stem cell in human brain tumors. Cancer
Res. 63:5821–5828. 2003.PubMed/NCBI
|
|
22.
|
Bruno S, Bussolati B, Grange C, et al:
CD133+ renal progenitor cells contribute to tumor
angiogenesis. Am J Pathol. 169:2223–2235. 2006.
|
|
23.
|
O’Brien CA, Pollett A, Gallinger S, et al:
A human colon cancer cell capable of initiating tumour growth in
immunodeficient mice. Nature. 445:106–110. 2007.PubMed/NCBI
|
|
24.
|
Collins AT, Berry PA, Hyde C, et al:
Prospective indentification of tumorigenic prostate cancer stem
cells. Cancer Res. 65:10946–10951. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Song W, Li H, Tao K, et al: Expression and
clinical significance of the stem cell marker CD133 in
hepatocellular carcinoma. Int J Clin Pract. 62:1212–1218. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Peichev M, Naiyer AJ, Pereira D, et al:
Expression of VEGFR-2 and AC133 by circulating human CD34(+) cells
identifies a population of functional endothelial precursors.
Blood. 95:952–958. 2000.
|
|
27.
|
Xu Y, Wang Z, Wang J, et al:
Lentivirus-mediated knockdown of cyclin Y (CCNY) inhibits glioma
cell proliferation. Oncol Res. 18:359–364. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Tomuleasa C, Soritau O, Rus-Ciuca D, et
al: Isolation and characterization of hepatic cancer cells with
stem-like properties from hepatocellular carcinoma. J
Gastrointestin Liver Dis. 19:61–67. 2010.PubMed/NCBI
|
|
29.
|
Chen YW, Chen KH, Huang PI, et al:
Cucurbitacin I suppressed stem-like property and enhanced
radiation-induced apoptosis in head and neck squamous
carcinoma-derived CD44+ALDH1+ cells. Mol
Cancer Ther. 9:2879–2892. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Shafee N, Smith CR, Wei S, et al: Cancer
stem cells contribute to cisplatin resistance in Brca1/p53-mediated
mouse mammary tumors. Cancer Res. 68:3243–3250. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Reya T, Morrison SJ, Clarke MF, et al:
Stem cell, cancer, cancer stem cells. Nature. 414:105–111. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Bonnet D and Dick JE: Human acute myeloid
leukemia is organized as a hierarchy that originates from a
primitive hematopoietic cell. Nat Med. 3:730–737. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Clark MF, Dick JE, Dirks PB, et al: Cancer
stem cells - perspectives on current status and future directions:
AACR workshop on cancer stem cells. Cancer Res. 66:9339–9344. 2006.
View Article : Google Scholar
|
|
34.
|
Mizrak D, Brittan M and Alison MR: CD133:
molecule of the moment. J Pathol. 214:3–9. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Zhang J, Luo N, Luo Y, et al: MicroRNA-150
inhibits human CD133-positive liver cancer stem cells through
negative regulation of the transcription factor c-MyB. Int J Oncol.
40:747–756. 2012.PubMed/NCBI
|
|
36.
|
Korkaya H and Wicha MS: Selective
targeting of cancer stem cells: a new concept in cancer
therapeutics. BioDrugs. 21:299–310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Rappa G, Fodstad O and Lorico A: The stem
cell-associated antigen CD133 (promini-1) is a molecular
therapeutic target for metastatic melanoma. Stem Cells.
26:3008–3017. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Wang CH, Chiou SH, Chou CP, et al:
Photothermolysis of glioblastoma stem-like cells targeted by carbon
nanotubes conjugated with CD133 monoclonal antibody. Nanomedicine.
7:69–79. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Yang ZF, Ho DW, Ng MN, et al: Significance
of CD90+ cancer stem cells in human liver cancer. Cancer
Cell. 13:153–166. 2008.
|
|
40.
|
Yang W, Wang C, Lin Y, et al:
OV6+ tumor-initiating cells contribute to tumor
progression and invasion in human hepatocellular carcinoma. J
Hepatol. 57:613–620. 2012.
|
|
41.
|
Uchida N, Sutton RE, Friera AM, et al:
HIV, but not murine leukemia virus, vectors mediated high
efficiency gene transfer into freshly isolated G0/G1 human
hematopoietic stem cells. Proc Natl Acad Sci USA. 95:11939–11944.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Eyler CE and Rich JN: Survival of the
fittest: cancer stem cells in therapeutic resistance and
angiogenesis. J Clin Oncol. 26:2839–2845. 2008. View Article : Google Scholar : PubMed/NCBI
|