Introduction
The oncogenic human papilloma virus (HPV) forms
(HPV16, 18, 31 and 33) are etiological agents in the development of
cervical carcinoma. However, the cell factors engaged in the
process of the cancer development in cervical epithelial cells are
poorly known (1,2).
Using the yeast two-hybrid system and the cDNA
library from normal epithelial tissue, we have previously shown
that the protein CCHCR1 (coiled-coil α-helical rod protein 1)
interacts with the E2 protein of human papillomavirus type 16,
which suggests its important role in the epithelial tissue function
(3).
CCHCR1 is a protein of unknown role in the cell.
This protein shows little homology with known proteins (4). Secondary structure predictions for
the CCHCR1 protein suggests that it contains several segments of
coiled-coil structure. CCHCR1 is either a nuclear or cytoplasmic
protein, but was also found in mitochondria and endosomes (5), and also in keratinocyte pseudopodia
in vitro. Its nuclear localization and possibility for the
dimerization and interactions with DNA (via a leucine zipper motif)
suggests a role of CCHCR1 in regulating cell differentiation or
proliferation (6–8). CCHCR1 was found to be overexpressed
in keratinocytes at psoriatic skin lesions, whereas in paired
samples from normal appearing skin it was barely detectable.
Therefore it was suggested that it could be involved in psoriasis
susceptibility (4), but the
connection of changes in the CCHCR1 gene with psoriasis is
still the subject of investigations (6,9).
Functional analysis in the transgenic mouse model revealed that the
CCHCR1 psoriatic allele is not enough to cause the psoriatic
disease and additional genes or environmental stimulation is
necessary to trigger off the psoriatic phenotype in the mouse
(10,11).
The CCHCR1 gene, encoding a 782 amino acid
protein, is located on chromosome 6 and consists of 18 exons
(5,12). The CCHCR1 gene is highly
polymorphic. It has 2 transcription start sites, which are located
79 and 430 bp upstream of the translation start site (ATG) in exon
2. Alternative transcription of the 5′-untranslated region can be
regulated in a tissue by alternative usage of promoters (5).
The aim of the present study was the analysis of the
CCHCR1 gene in precancerous and cancer lesions and in HPV
positive and negative dysplasia cells.
Materials and methods
Material
The investigations were performed using cervical
cancer specimens obtained from women who were subjected to surgery
during 2004–2008, because of histologically confirmed neoplastic
lesions. The study material consisted of 73 patients that underwent
surgeries due to: squamous cell carcinoma of the cervix or
adenocarcinoma of the cervix and low- and high-grade squamous
intraepithelial lesions. In respect to the differentiation of the
neoplastic cells, the following groups of patients were identified
according to the WHO classification system: G1 (n=10), G2 (n=16)
and G3 (n=13) (Table I).
According to FIGO clinical staging, 14 patients were classified as
stage 0 (carcinoma in situ), 14 as IA, 22 as IB and 3 as
IIA. The patients were between 39 and 61 years of age (mean 54.3).
All patients underwent surgical procedures at the Department of
Gynaecology and Obstetrics of the Medical University of Lublin. The
control group was comprised of normal tissue of the uterine cervix
obtained from 18 patients, 40–72 years of age, who underwent
surgical treatment for uterine myomas.
 | Table IThe WHO and FIGO classification of
patients with tumors of the uterine cervix. |
Table I
The WHO and FIGO classification of
patients with tumors of the uterine cervix.
| N |
|---|
| FIGO
classification |
| 0 | 14 |
| IA | 14 |
| IB | 22 |
| IIA | 3 |
| Total | 53 |
| WHO
classification |
| G1 | 10 |
| G2 | 16 |
| G3 | 13 |
| Total | 39 |
| Squamous cell
carcinoma |
| Keratizing | 22 |
|
Non-keratizing | 10 |
| Basaloid | 4 |
| Total | 36 |
| Adenocarcinoma |
| Endocervical | 1 |
| Endometroid | 1 |
| Clear cell | 1 |
| Total | 3 |
Approvals obtained from the local Ethics Committee
of Medical Universities in Lublin enabled us to collect cervical
cancer specimens.
DNA isolation
Genomic DNA was isolated from tissue samples using
the QIAamp DNA Mini kit (Qiagen) according to the manufacturer’s
indications.
HPV analysis
Genomic DNA was used for amplification by PCR with
two specific primer pairs complementary to the HPV genome,
universal for 33 types of HPV viruses: MY09,
5′-CGTCCMARRGGAWACTGATC-3′ and MY11, 5′-GCMCAGGGWCATAAYAATGG-3′;
for HPV16-E7/16A, 5′-ATAATATAAGGGGTCGGTGG-3′ and E7/16B
5′-CATTTTCGTTCTCGTCATCTG-3′; for HPV18 -ME18A,
5′-CACGGCGACCCTACAAGCTACCTG-3′ and ME18B,
5′-TGCAGCACGAATTGGCACTGGCCTC-3′. PCR reactions were performed in a
total volume of 20 μl. The final mixture contained 1 μM primers,
200 μM dNTPs, 1X PCR buffer (10 mM Tris-HCl, pH 8.8 at 25°C, 1.5 mM
MgCl2, 50 mM KCl, 0.1% Triton X-100) and 1 U/25 μl
mixture of Taq polymerase (Fermentas). The samples were amplified
for 35 cycles. Each cycle consisted of the following steps:
denaturation at 95°C for 1 min (first cycle for 90 sec), annealing
at 50°C for primer pairs MY09/11, 59°C for primer pairs
E7/16A-E7/16B and 55°C for primer pairs ME18A/B, and extension at
72°C for 1 min. The reaction was performed in a DNA thermal cycler
(Biometra). The amplification products were then analyzed on a 2%
agarose gel with the addition of ethidium bromide in a UV
transilluminator.
CCHCR1 gene polymorphism analysis by
PCR-SSCP and sequencing methods
Genomic DNA was used for amplification by PCR with
two specific primers pairs complementary to fragments of the
CCHCR1 gene (sequence and localization of primers are
provided in Table II). The PCR
reactions were performed in a total volume of 20 μl. The final
mixture contained 1 μM primers, 200 μM dNTPs, 1X PCR buffer (750 mM
Tris-HCl, pH 8.8 at 25°C, 200 mM (NH4)SO4,
0.1% Tween-20), 1.5 mM MgCl2 and 1 unit/25 μl mixture of
Taq polymerase (Fermentas). The samples were amplified for 35
cycles. Each cycle consisted of the following steps: denaturation
at 95°C for 1 min (first cycle for 90 sec), annealing at proper
temperature (Table III) for 30
sec and extension at 72°C for 1 min. The reaction was performed in
a DNA thermal cycler (Biometra). Products of amplification were
analyzed on a 2% agarose gel with addition of ethidium bromide in a
UV transluminator. PCR products longer than 250 nucleotides were
digested with endonucleases (Table
II) before SSCP. Products of each PCR reaction (10 ml reaction
mixture) were denatured chemically with formamide and thermically
at 95°C for 15 min. Subsequently, denaturation samples were placed
on ice. They were electrophoresed on a 10% polyacrylamide gel with
0.5X TBE buffer in 200 V for 12 h, stained with silver salts, and
dried. PCR products were purified with a PCR purification mini kit
(Qiagen) and then sequenced.
| Table IIPrimers used to PCR-SSCP study of the
CCHCR1 gene. |
Table II
Primers used to PCR-SSCP study of the
CCHCR1 gene.
| Exon | Region of
amplification (according to AB088104) | Fragment length
(nt) | Primer sequence
5′→3′ | Annealing
temperature (°C) | Restriction
enzyme |
|---|
| 1a | 91–512 | 422 |
CCACTATGTGTTAGGACTCGAG
GATTCTGGGCAGTGCCTTTACC | 56.0 | PvuII |
| 1b | 765–1174 | 410 |
GTGTCTTTGTTTCTCCTCTTGTCC
GAAAACGGCGTGGATGGATCCC | 57.0 | AluI |
| 2 | 812–1170 | 359 |
CAGAATCTAGAGCCTTCAAATAATGTG
ACGGCGTGGATGGATCCCTA | 55.5 | AluI |
| 3 | 3071–3422 | 372 |
ACCTGCACTAACCTGTCTTTGA
AATCCTTTCTACCCCTGCATTC | 53.0 | PstI |
| 4/5 | 6760–7204 | 445 |
GAGCCCCCTCTTCTTTCCGC
CACAGATACATTCCTGCACCCTC | 57.5 | PstI |
| 6 | 7317–7471 | 155 |
GGCTGCTTTCCTCTGCCCGC
GGGTCTGGGGGTTGGGCTGT | 60.0 | - |
| 7 | 7666–7862 | 197 |
TTCTCCCACTCCTTCTCCCTC
CGGGAGAAAAGAGAGTGCAGTG | 56.5 | - |
| 89 | 9153–9522 | 370 |
GCCCAGCTCTCTCTCCTCC
CCCACCCCTCCATCCCTGAT | 57.5 | AvaII |
| 10/11 | 12065–12473 | 409 |
ATCAGTGACTTGTGCCCTCTC
CACCTCAAAGTGC(AC)CAAACTTC | 54.0 | AvaII |
| 12 | 12569–12765 | 197 |
CTGACTCTTTCTCTTCCCCGT
CTCATCCTCTCCACCCTCTG | 55.5 | - |
| 13/14 | 12815–13240 | 426 |
TCCTTTTAGGGGAGGCAGAG
GAAGGCCCTATCCACCCTG | 54.5 | TaqI |
| 15 | 14462–14656 | 195 |
CTGTGCCTTGGCCTCTCTGT
GTCTGCCCTCCTGTCTCCTA | 55.5 | - |
| 16 | 14725–14964 | 240 |
GGCTCTATCCGGGCTAGG
TCCCTTGTCCCTTTGTGCTTG | 54.5 | - |
| 17 | 15328–15171 | 178 |
CTTTCCCTCCAACTGTCAGC
CTGGTGCTCATCTGCTGTCTT | 54.0 | - |
| Table IIIPrimers used to real-time PCR study
of the CCHCR1 gene. |
Table III
Primers used to real-time PCR study
of the CCHCR1 gene.
| Fragment
length | Primer sequence
(5′→3′) | Annealing
temperature |
|---|
| CCHCR1, F | 204 bp |
TGCGTGCTGCTTTGGCTGG | 60°C |
| CCHCR1, R | |
CCCCTGCTCTTCTGGTTTC | |
| GAPDH, F | 106 bp |
CAATGACCCCTTCATTGACC | 60°C |
| GAPDH, R | |
GACAAGCTTCCCGTTCTCAG | |
| POL II, F | 163 bp |
GCAAATTCACCAAGAGAGAC | 60°C |
| POL II, R | |
ATGTGACCAGGTATGATGAG | |
CCHCR1 gene expression analysis
Cervical carcinoma, precancerous (L-SIL and H-SIL)
and control tissues, collected during planned gynecological
operations, were immediately put in RNAlater™ RNA stabilization
reagent (Qiagen).
Total-RNA was isolated from cervical precancerous
and cancer tissues and control cells using the RNeasy Mini kit
(Qiagen) according to the manufacturer’s indications. RNA samples
were treated with DNase I (Promega) and 1 μg of RNA (of each
sample) was reverse-transcribed with SuperScript™ II
RNaseH− reverse transcriptase (Invitrogen) into cDNA
using oligo(dT) primers. Real-time PCR was conducted in a Light
Cycler Real-Time detection system (Roche Diagnostics) using
SYBR®-Green I as the detection dye. Target cDNA was
quantified using the relative quantification method. The quantity
of the CCHCR1 transcripts in each sample was standaridized
by either glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or RNA
polymerase II (POL II) transcript levels. The RT-PCR reactions were
performed in total volume of 20 μl. cDNA of 2 μl was added to an 18
μl mixture of LC-FastStart DNA Master SYBR-Green I, 1.5 mM
MgCl2 and primers (sequence and localization of primers
are given in Table III).
Statistical analysis
The results obtained were analyzed statistically.
Values of the analyzed parameters, due to the quotient scale of
measurement, were characterized by average value, standard
deviation and median, with the lower and higher quartile providing
the changeability range. Due to the diagonal distribution of the
studied parameters evaluated using the Shapiro-Wilk W test for the
analyses of existence of differences, non-parametric tests were
used. To discover differences between the compared groups, the
Kruskal-Wallis H test was used to compare more than two groups and
the Mann-Whitney U test to compare two independent groups. The 5%
error in concluding was assumed, and P<0.05 indicated
statistically significant differences.
Methylation level analysis
Analysis of the DNA methylation level was made using
the EZ DNA Methylation kit™ (Zymo Research) according to the
manufacturer’s indications. Genomic DNA (0.5–1 μg) was used for the
reaction of deamination. Deaminated DNA was used for the PCR
reaction, with starters complementary to deaminated DNA: CCHCR1-BF
5′-TTTAAGTAGTGTTAGTTTGTG-3′ and CCHCR1-BR
5′-TCTTCATCTATCCCTTCACC-3′. The PCR reactions were performed in a
total volume of 10 μl. The final mixture contained 1 μM primers,
200 μM dNTPs, 1X PCR buffer, 2 mM MgCl2 and 1 unit
FastStart TaqDNA Polymerase (Roche Diagnostics) and 2 μl of
deaminated DNA. The samples were amplified for 40 cycles. Each
cycle consisted of the following steps: preliminary denaturation at
95°C for 5 min, denaturation at 95°C for 35 sec, annealing at 56°C
for 45 sec and extension at 72°C for 1 min. Reaction was performed
in a DNA thermal cycler (Biometra). Products of amplification were
analyzed on a 2% agarose gel with addition of ethidium bromide in
UV transluminator. The products 675 bp long were cut out of the
agarose gel, eluted with MinElute Gel Extraction kit (Qiagen),
according to manufacturer’s instructions and cloned into
pGEM® T-Easy Vector (Promega). DH5α E. coli were
transformed with recombinant pGEM® T-Easy. Clones with
recombinant plasmid were selected, plasmids were isolated with
QIAprep Spin Miniprep kit (Qiagen), according to the manufacturer’s
instructions and then sequenced.
Computer analysis
Programs MPromDb (http://bioinformatics.med.ohio-state.edu/MPromDb/index.jsp)
and Cpgplot (http://www.ebi.ac.uk/emboss/Cpgplot) were used for
computer analysis of the region located upstream the first
transcription start site of the CCHCR1 gene.
Results
In DNA probes isolated from cervical cells, HPV was
detected in 32 of 36 patients with squamous cell carcinoma, 11 of
14 with H-SIL, in all of those with adenocarcinoma and none of the
control group women (Table IV).
In the same DNA probes the CCHCR1 gene was analyzed with
PCR, SSCP and sequencing methods. The changes detected in the study
region are present in Table V.
Changes in the sequence of the CCHCR1 gene were detected in
the non-coding and coding region as well. Changes in the non-coding
region were found in 20.3% of the examined probes from women with
cervical cancer or precancerous lesions and in 16.67% of the
control probes. Most of the detected changes were SNPs. Changes in
the coding region were found in 22.8% of the probes with cervical
cancer and in 16.67% of the control probes. These changes were
detected in exons 3, 9, 12 and 15. All of identified changes were
SNPs. Six types of changes (at sites 3169, 3171, 3189 and 3356 in
exon 3; 9436 and 9437 in exon 9) were connected with the amino acid
change and two others changes (at sites 12622 in exon 12 and 14494
in exon 15) were not.
| Table IVStudy groups and frequency of human
papilloma virus (HPV) type 16 and/or 18 DNA occurrence. |
Table IV
Study groups and frequency of human
papilloma virus (HPV) type 16 and/or 18 DNA occurrence.
| Group | No. of cases | HPV oncogenic types
16/18 | % HPV positive |
|---|
| L-SIL | 6 | 3 | 66.67 |
| H-SIL | 14 | 8 | 78.57 |
| Squamous cell
carcinoma | 36 | 26 | 88.89 |
| Adenocarcinoma | 3 | 1 | 100 |
| Control | 18 | 0 | 0 |
| Table VChanges detected in the coding
sequence of the CCHCR1 gene. |
Table V
Changes detected in the coding
sequence of the CCHCR1 gene.
| Change in protein
sequencea | | |
|---|
|
| | |
|---|
| Change in DNA
sequenceb | Change | Amino acid
position | Position in
codon | | Number of
probesc | Recognition |
|---|
| 246 (c→t) exon
1a | | Non-coding
sequence | | SNP | 3 | Carcinoma
planoepitheliale |
| | | | | 2 | CIN3 |
| | | | | 2 | Control |
| 251 (g→c) exon
1a | | Non-coding
sequence | | - | 1 | Carcinoma
planoepitheliale |
| 394 (g→t) exon
1a | | Non-coding
sequence | | - | 1 | Carcinoma
planoepitheliale |
| 808 (a→g) exon
1b | | Non-coding
sequence | | SNP | 4 | Carcinoma
planoepitheliale |
| | | | | 1 | CIN3 |
| | | | | 1 | Control |
| 3169 (g→a) exon
3 |
CGG(Arg)→CAG(Gln) | 102 | 2 | SNP | 1 | Carcinoma
planoepitheliale |
| | | | | 1 | CIN3 |
| | | | | 1 | Control |
| 3171 (c→t) exon
3 |
CGG(Arg)→TGG(Trp) | 103 | 1 | SNP | 2 | Carcinoma
planoepitheliale |
| | | | | 1 | CIN3 |
| | | | | 3 | Control |
| 3189 (c→t) exon
3 |
AGG(Arg)→AGC(Ser) | 109 | 3 | SNP | 2 | Carcinoma
planoepitheliale |
| | | | | 1 | CIN3 |
| | | | | 2 | Control |
| 3356 (g→c) exon
3 |
AGG(Arg)→AGC(Ser) | 164 | 3 | SNP | 2 | Carcinoma
planoepitheliale |
| | | | | 3 | CIN3 |
| | | | | 3 | Control |
| 9436 (c→t) exon
9 |
CGT(Arg)→TGT(Trp) | 416 | 1 | SNP | 2 | Carcinoma
planoepitheliale |
| | | | | 1 | CIN3 |
| 9437 (g→a) exon
9 |
CGG(Arg)→CAG(Gln) | 417 | 2 | SNP | 1 | Carcinoma
planoepitheliale |
| 12622 (t→c) exon
12 |
GAT(Asp)→GAC(Asp) | 500 | 3 | SNP | 1 | Carcinoma
planoepitheliale |
| | | | | 1 | Adenocarcinoma |
| | | | | 1 | CIN3 |
| 14494 (g→a) exon
15 |
TTG(Leu)→TTA(Leu) | 637 | 3 | SNP | 1 | Carcinoma
planoepitheliale |
The mRNA CCHCR1 levels were determined by a
real-time PCR method with RNA probes isolated from cervical
precancerous, cancer and control cells. RT-PCR results are
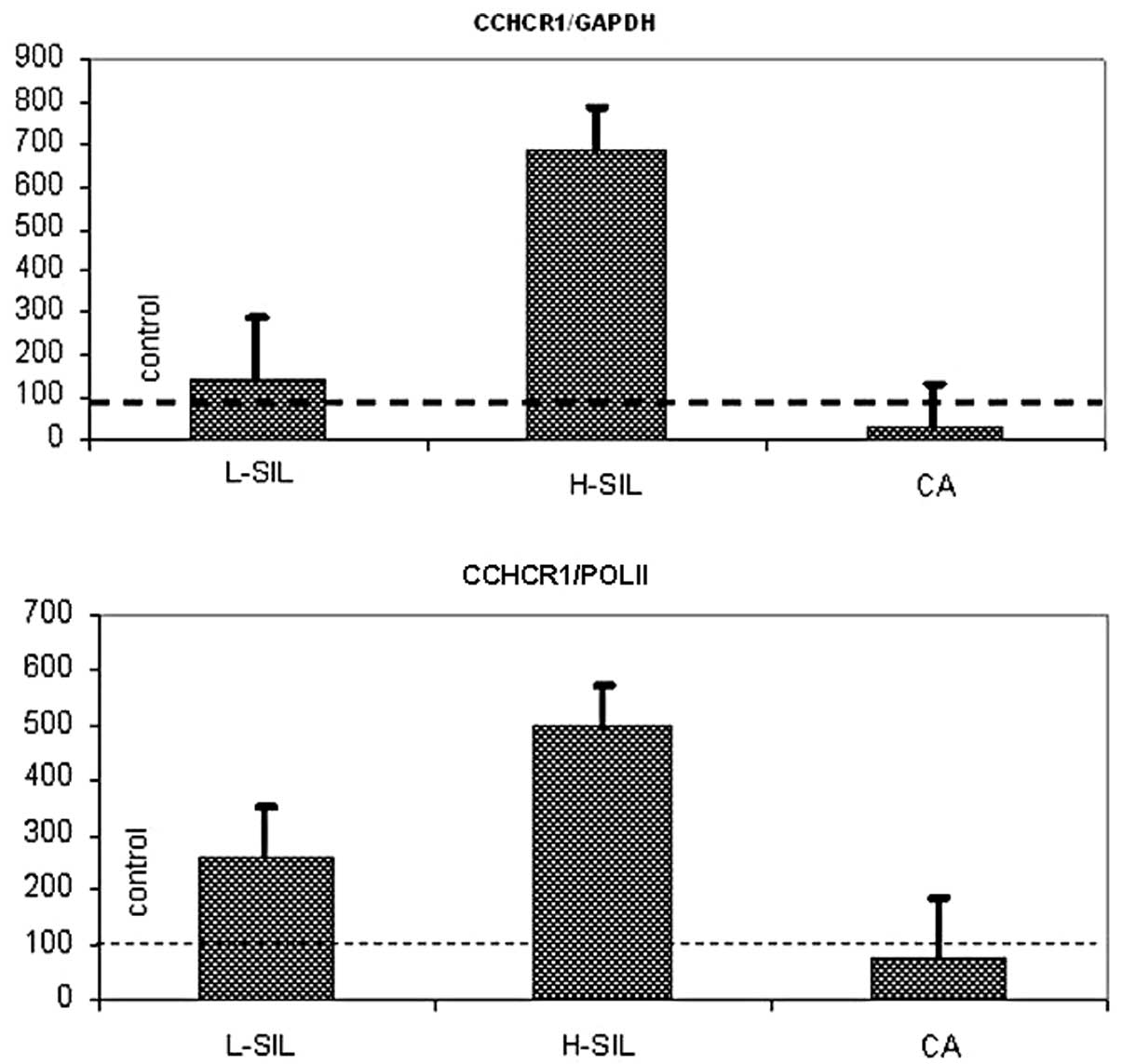
presented as a percentage of their controls at Fig. 1 and in Table VI. The highest CCHCR1
transcripts values were detected in the H-SIL group. Interestingly,
CCHCR1 gene expression was slightly decreased in cervical
carcinoma cells, compared with control probes.
| Table VIExpression of CCHCR1 gene,
real-time PCR data. |
Table VI
Expression of CCHCR1 gene,
real-time PCR data.
A. Comparison of
the CCHCR1/GAPDH values in the control, L-SIL, H-SIL and CA groups
|
|---|
| Group | Mean | Median | 25th
percentile | 75th
percentile | Range |
|---|
| Control group | 0.0061 | 0.00048 | 0.00033 | 0.00066 | 0.00032–0.034 |
| L-SIL | 0.009 | 0.0036 | 0.00034 | 0.0123 | 0.00034–0.034 |
| H-SIL | 0.042 | 0.036 | 0.0075 | 0.076 | 0.0065–0.090 |
| CA | 0.0021 | 0.0015 | 0.00047 | 0.0029 | 0.00034–0.0065 |
|
| B. Comparison of
the CCHCR1/POL II values in the control, L-SIL, H-SIL and CA
groups |
|
| Group | Mean | Median | 25th
percentile | 75th
percentile | Range |
|
| Control group | 0.104 | 0.0162 | 0.012 | 0.047 | 0.0104–0.524 |
| L-SIL | 0.270 | 0.233 | 0.026 | 0.456 | 0.0245–0.649 |
| H-SIL | 0.522 | 0.509 | 0.221 | 0.823 | 0.101–0.968 |
| CA | 0.082 | 0.0506 | 0.022 | 0.110 | 0.00625–0.284 |
According to the Kruskal-Wallis H test,
statistically significant differences were found in CCHCR1
transcripts levels between the compared groups (H=9.06; P=0.03 for
CCHCR1/GAPDH and H=9.23; P=0.03 for CCHCR1/POL II). Intergroup
analysis revealed differences between the control group and H-SIL
and H-SIL and CA (P=0.005 for CCHCR1/GAPDH and P=0.01 for
CCHCR1/POL II).
Because there is no information in the literature
about the structure and function of the regulatory region of the
CCHCR1 gene we decided to perform computer analysis of the
region located upstream of the first transcription start site using
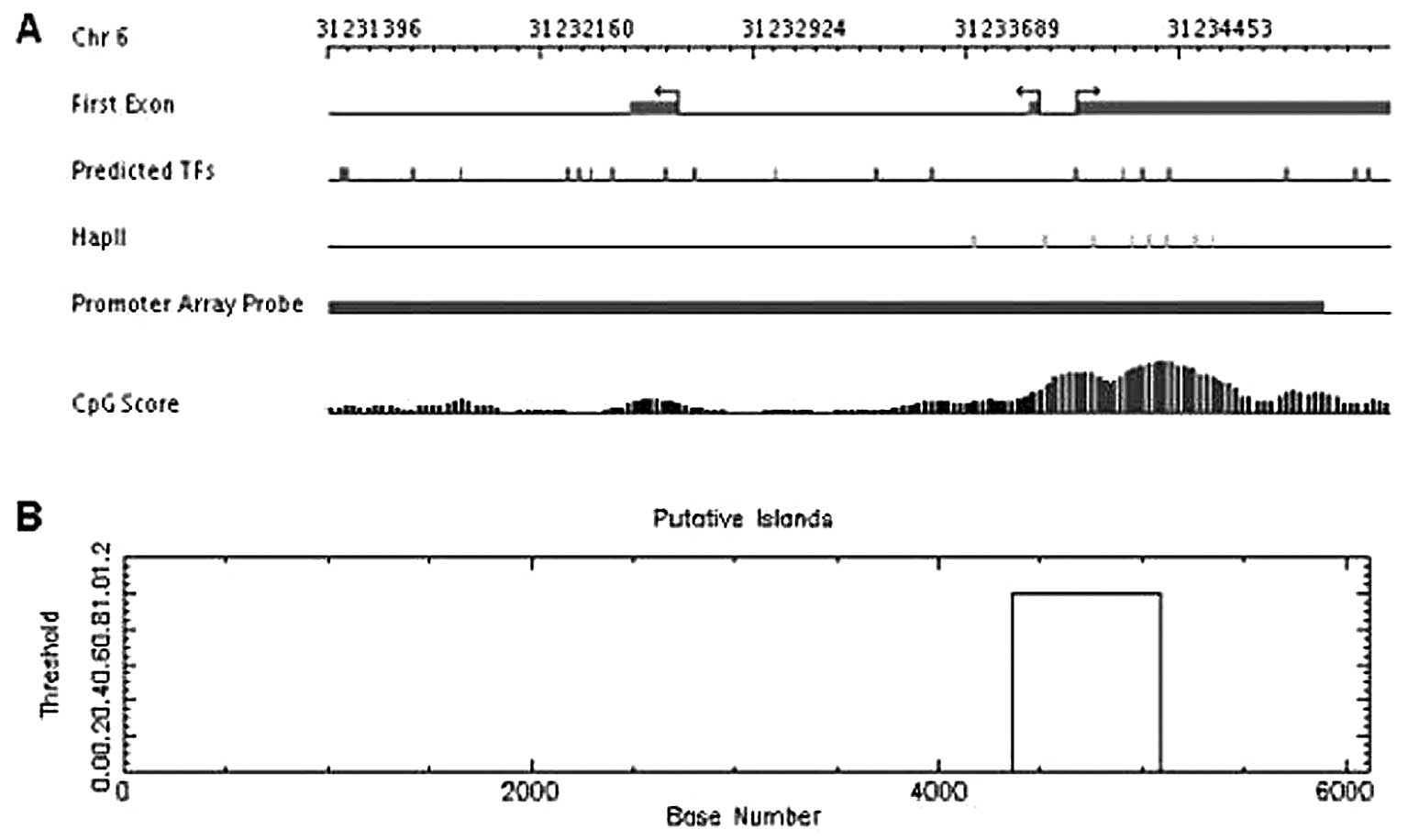
the programs MPromDb and Cpgplot. We analyzed a DNA fragment about
6 kbp long and found a CpG island, about 1 kbp long, located just
above first transcription start site of the CCHCR1 gene
(Fig. 2).
Therefore, we performed the preliminary analysis of
methylation level of the 647 bp DNA fragment with 55 CG pairs
within. For this analysis we used DNA isolated from cervical
epithelial cells collected from 3 healthy women (control probes)
and from 3 women with carcinoma planoepitheliale. We did not notice
any differences in the DNA methylation profile in the control and
cervical probes. In the analyzed DNA fragment all CG pairs were not
methylated.
Discussion
The function of the CCHCR1 protein, is poorly known.
It is most probably engaged in the control of epithelial cell
proliferation (4,8,13).
It is also involved in processes like tissue-specific gene
transcription (14),
steroidogenesis (5) and probably
metabolism of steroids (13).
Suomela et al (15) have
suggested that CCHCR1 plays a role in keratinocyte transformation.
Our previous analysis revealed that CCHCR1 interacts with the
protein E2 of the human papillomavirus type16 (HPV16) in normal
cervical epithelial cells (3). E2
is the main viral regulatory protein. It plays a significant role,
among other things, in regulating the expression of other viral
proteins (16,17).
The CCHCR1 gene is highly polymorphic
(4,6,10,12). In the genome of psoriatic persons
some mutations were identified, which could probably change the
secondary structure of this protein and influence its biochemical
properties and antigenicity (7,8).
We also demonstrated that the CCHCR1 gene in the non tumor
and tumor cells of the cervical epithelium is highly polymorphic.
Almost all detected changes, in the non-coding as well as in the
coding region, were single nucleotide polymorphisms (SNPs)
characterized in databases (www.ncbi.nlm.nih.gov/SNP/). Obtained results did not
allow us to identify changes which were characteristic of the
cervical cancer patients. Our analyses were performed in a
relatively small research group and did not allow for the reliable
evaluation the CCHCR1 genotype frequency. It would be
intentional to expand the research group to confirm or exclude if a
particular genotype has a higher frequency in a population of ill
women compared to a control group.
Tiala et al (13) suggest, that for the pathogenesis
of such diseases like psoriasis the amount of protein produced in
the cell could also be important. Very small differences in the
experssion of the CCHCR1 gene in basal kerationcytes could
influence the local production of steroids (13,18). SNPs in the CCHCR1 gene
could have an impact on the amount of produced protein, through an
influence on the mRNA stability, gene expression or different
regulation of this gene in response to environmental stress
(13). In this context nucleotide
changes in the non-coding region may have an influence on the
regulation of CCHCR1 gene expression, but this suggestion
need to be experimentally confirmed.
CCHCR1 gene expression analysis revealed that
CCHCR1 mRNA levels were higher in L-SIL and H-SIL probes
compared to those in normal cervical cells. The highest
CCHCR1 mRNA levels were observed in H-SIL probes, whereas
the levels were lower in cervical carcinoma compared to control
probes. The differences were statistically significant.
Overexpression of the CCHCR1 gene could
stimulate the proliferation of cervical carcinoma cells at the
early stages of neoplasia. Suomela et al (15) found that proliferative cells of
cutaneous squamous cell cancer (SCC) at the invasive front
expressed CCHCR1, whereas the cohesive cancer cells in the middle
were CCHCR1-negative. CCHCR1 expression was associated with
positive EGFR staining. Cells positive for the hyperproliferation
marker Ki67 were located mostly in the same areas as
CCHCR1-positive cells. However, in grade III SCCs Ki67 was more
abundantly present in cohesive tumor areas that were devoid of
CCHCR1 expression. CCHCR1 expression was not induced in
vitro in the most aggressive and metastatic SCC cell lines. It
was also found that with ascending tumorigenicity and proliferative
state, CCHCR1 mRNA was downregulated in HaCaT cells. In
contrast to benign KC hyperproliferation in psoriasis, the
hyperproliferation marker Ki67 expresion in vivo and in
vitro was associated with CCHCR1 expression in malignant
transformation. CCHCR1 may participate in regulating cell
proliferation only to a certain point in oncogenesis (15).
Little is known about the regulation of the
CCHCR1 gene expression and the structure of its regulatory
region. For that reason we performed computer analysis of the
region located upstream of the first transcription start site which
is probably of great importance as far as the regulation of
CCHCR1 expression is concerned. The analysis revealed that
the CpG island, about 1 kbp long, is located just above the first
transcription start site of the CCHCR1 gene.
CpG islands are regions of higher frequency of CpG
compared to the rest of the genome. CpG islands are protected from
DNA methylation in normal cells, at all stages of development and
in all types of tissues by mechanisms, which are poorly understood
(19,20). These regions function as strong
promoters and can initiate DNA replication as well (19).
A lot of changes occurring in cancerous cells are
caused by genetic and epigenetic abnormalities. The genome can
undergo simultaneous general hypomethylation and regional
hypermethylation of CpG islands, which can result in the occurrence
of conditions leading to tumor development. This phenomenon can
result in silencing of tumor suppression genes, loss of gene
imprinting, and less probably, oncogene activation through
demethylation, increase of the frequency of point mutations in
methylated CpGs and instability of microsatellite DNA (20–23). However, there is no consensus as
to the direct role of DNA methylation in carcinogenesis.
We analyzed the region located upstream of the first
transcription start site of the CCHCR1 gene. We observed no
differences in the methylation level between carcinoma
planoepitheliale and the normal probes. In the analyzed region all
CG pairs were not methylated. Even though the analysis was carried
out in few probes, we can assume that mechanisms other than
methylation of the region with the probable regulatory function are
responsible for the changes in CCHCR1 mRNA levels.
Continuation of research on this subject seems to be
very interesting and important, particularly in relation to the
CCHCR1 and the HPV16 E2 protein interactions. Recent reports
suggest that CCHCR1 can act as the negative regulator of
keratinocyte proliferation and that inappropriate function of
CCHCR1 protein can lead to incorrect keratinocyte proliferation
(11) and transformation
(15).
Acknowledgements
The authors would like to thank Mr. Michal Luczak,
from the Department of Biochemistry and Molecular Biology, Poznan
University of Medical Sciences, for his great help in the analysis
of the methylation level of the CCHCR1 gene. This study was
supported by the Interdisciplinary Research Grant from the Adam
Mickiewicz University and Poznan University of Medical Sciences
(no. 512 00 035) and a research grant from the Dean of Faculty of
Biology, Adam Mickiewicz University.
Abbreviations:
|
CCHCR1
|
coiled-coil α-helical rod protein
1
|
|
HPV
|
human papilloma virus
|
|
H-SIL
|
high-grade squamous intraepithelial
lesions
|
|
L-SIL
|
low-grade squamous intraepithelial
lesions
|
|
SNP
|
single nucleotide polymorphism
|
|
SSCP
|
single stranded conformation
polymorphism
|
References
|
1
|
H zur HausenPapillomaviruses and cancer:
from basic studies to clinical applicationNat Rev
Cancer2342350200212044010
|
|
2
|
FX BoschA LorinczN MunozCJ MeijerKV
ShahThe causal relation between human papillomavirus and cervical
cancerJ Clin Pathol55244265200210.1136/jcp.55.4.24411919208
|
|
3
|
AK Olejnik-SchmidtMT SchmidtW KedziaA
Gozdzicka-JozefiakSearch for cellular partners of human
papillomavirus type 16 E2 proteinArch
Virol153983990200810.1007/s00705-008-0061-618305892
|
|
4
|
K AsumalahtiT LaitinenR Itkonen-VatjusML
LokkiS SuomelaE SnellmanA candidate gene for psoriasis near HLA-C,
HCR (Pg8), is highly polymorphic with a disease-associated
susceptibility alleleHum Mol
Genet915331542200010.1093/hmg/9.10.153310888604
|
|
5
|
T SugawaraH ShimizuN HoshiA NakajimaS
FujimotoSteroidogenic acute regulatory protein-binding protein
cloned by a yeast two-hybrid systemJ Biol
Chem2784248742494200310.1074/jbc.M30229120012909641
|
|
6
|
K O’BrienS HolmS NilssonL CarlénT
RosenmüllerC EnerbäckThe HCR gene on 6p21 is unlikely to be
psoriasis susceptibility geneJ Invest
Dermatol116750754200111348465
|
|
7
|
K AsumalahtiC VealT LaitinenS SuomelaM
AllenO ElomaaCoding haplotype analysis supports HCR as the putative
susceptibility gene for psoriasis at the MHC PSORS1 locusHum Mol
Genet11589597200210.1093/hmg/11.5.58911875053
|
|
8
|
S SuomelaO ElomaaK AsumalahtiL ArjaSL
KarvonenJ PeltonenHCR, a candidate gene for psoriasis, is expressed
differently in psoriasis and other hyperproliferative skin
disorders and is downregulated by interferon-γ in keratinocytesJ
Invest Dermatol12113601364200314675183
|
|
9
|
NVC ChiaP StuartRP NairT HanselerS
JenischHW LimVariations in the HCR (Pg8) gene are unlikely to be
casual for familiar psoriasisJ Invest
Dermatol116823200110.1046/j.1523-1747.2001.01245-2.x11348479
|
|
10
|
O ElomaaI MajuriS SuomelaK AsumalahtiH
JiaoZ MirzaelTransgenic mouse models support HCR as an effector
gene in PSORS1 locusHum Mol
Genet1315511561200410.1093/hmg/ddh17815190014
|
|
11
|
I TialaJ WakkinenS SuomelaP PuolakkainenR
TammiS ForsbergThe PSORS1 locus gene CCHCR1 affects keratinocyte
proliferation in transgenic miceHum Mol
Genet1710431051200810.1093/hmg/ddm37718174193
|
|
12
|
YT ChangYM ShiaoPJ ChinYL LiuFC ChouS
WuGenetic polymorphism of the HCR gene and a genomic segment in
close proximity to HLA-C are associated with patients with
psoriasis in TaiwanBr J
Dermatol15011041111200410.1111/j.1365-2133.2004.05972.x15214895
|
|
13
|
I TialaS SuomelaJ HuuhtanenJ WakkinenM
Holtta-VuoriK KainuThe CCHCR1 (HCR) gene is relevant for skin
steroidognesis and downregulated in cultured psoriatic
keratinocytesJ Mol Med
(Berl)85589601200710.1007/s00109-006-0155-017221218
|
|
14
|
N CorbiT BrunoR De AngelisM Di PadovaV
LibriMG Di CertoRNA polymerase II subunit 3 is retained in the
cytoplasm by its interaction with HCR, the psoriasis vulgaris
candidate gene productJ Cell
Science11842534260200510.1242/jcs.0254516141233
|
|
15
|
S SuomelaO ElomaaT SkoogR Ala-ahoL
JeskanenJ PärssinenCCHCR1 is up-regulated in skin cancer and
associated with EGFR expressionPloS
One4e6030200910.1371/journal.pone.000603019551138
|
|
16
|
G DellK GastonHuman papillomaviruses and
their role in cervical cancerCell Mol Life
Sci5819231942200110.1007/PL0000082711766888
|
|
17
|
J DoorbarThe papilloma life cycleJ Cin
Virol32SS7S15200510.1016/j.jcv.2004.12.006
|
|
18
|
W ChenSJ TsaiCY LiaoRY TsaiYJ ChenBJ
PanHigher levels of steroidogenic acute regulatory protein and type
I 3-beta-hydroxysrteroid dehydrogenase in the scalp of men with
androgenetic alopeciaJ Invest
Dermatol12623322335200610.1038/sj.jid.570044216778788
|
|
19
|
PA JonesD TakaiThe role of DNA methylation
in mammalian
epigeneticsScience29310681070200110.1126/science.106385211498573
|
|
20
|
R JaenischA BirdEpigenetic regulation of
gene expression: how the genome integrates intrinsic and
environmental signalsNat
Genet33SupplS245S254200310.1038/ng108912610534
|
|
21
|
S KalariGP PfeiferIdentification of driver
and passenger DNA methylation in cancer by epigenetic analysisAdv
Genet70277308201010.1016/B978-0-12-380866-0.60010-120920752
|
|
22
|
Y KondoJP IssaDNA methylation profiling in
cancerExpert Rev Mol
Med12e23201010.1017/S146239941000155920663272
|
|
23
|
M KulisM EstellerDNA methylation and
cancerAdv Genet702756201010.1016/B978-0-12-380866-0.60002-2
|