Introduction
X-linked retinoschisis (XLRS, MIM 312700) is a
hereditary retinal disease characterized by a splitting of the
neurosensory retina, with a prevalence of 1:5,000 to 1:25,000 males
worldwide (1). Typical fundus
changes include radiating cysteic maculopathy in most cases and
peripheral retinoschisis in half of the cases (2). However, the disease has a high
degree of phenotypic variability (3–6),
in which genetic testing is of value in confirming the diagnosis
(4).
XLRS accounts for most congenital retinoschisis
(2,7) and is due to mutations in the
retinoschisin gene (RS1, OMIM 312700) localized on Xp22.13
(8,9). The encoded protein, retinoschisin,
is secreted from photoreceptors and bipolar cells as a functional
homo-octameric complex that is thought to play a role in cellular
adhesion and cell-to-cell interaction (10).
Gene transference to mouse models of X-linked
juvenile retinoschisis, which suggest gene replacement may be a
possible future therapy for patients (11–13). Genetic diagnosis is the basis for
gene transference in the future. Therefore, we have to fully
understand the molecular basis of XLRS. To date, more than 160
different RS1 mutations have been identified in patients
with XLRS (http://www.dmd.nl/rs), including small
intragenic deletions, nonsense and missense mutations, frame shift
insertions and deletions, and splice site mutations. However, there
are still some RS1 mutations that remain unknown.
In this study, we analyzed the coding exons and the
adjacent regions of RS1 in patients from 20 unrelated
Chinese families with XLRS. Ten hemizygous mutations, including 4
novel mutations, were detected in 14 families.
Subjects and methods
Probands with XLRS from 20 unrelated families were
enrolled in this study. Written informed consent was obtained from
the participating individuals or their guardians prior to the
collection of clinical data and genomic samples. This study was
approved by the Internal Review Board of the Zhongshan Ophthalmic
Center.
Mutation detection
Genomic DNA was prepared from venous leukocytes. Six
pairs of primers (Table I) were
used to amplify the six coding exons and the adjacent intronic
sequence of RS1 (NCBI human genome build 37.2, NG_008659.1
for genomic DNA, NM_000330.3 for mRNA, and NP_000321.1 for
protein). Touchdown polymerase chain reaction (PCR) was performed
with decreasing 0.5°C per cycle from 64°C for the first 15 cycles
then down to 57°C (the annealing temperature) for the remaining 21
cycles. GC buffer was used. DNA sequences of the amplicons were
identified with ABI BigDye Terminator cycle sequencing kit version
3.1 (Applied Biosystems, Foster City, CA) on an ABI 3130 Genetic
Analyzer (Applied Biosystems). Sequencing results and consensus
sequences from the NCBI human genome database were compared by
using the SeqMan II program of the Lasergene package (DNA Star,
Inc., Madison, WI) and then aligned to identify variations. Each
variation was confirmed by bidirectional sequencing. Mutation
description followed the recommendation of the Human Genomic
Variation Society (HGVS). Variations detected in patients were
further evaluated in controls by sequencing 176 normal
individuals.
 | Table IPrimers used for the amplification
and sequencing of RS1. |
Table I
Primers used for the amplification
and sequencing of RS1.
| Exon | Direction | Primer sequence
(5′-3′) | Size of amplified
fragment (bp) | Annealing
temperature (°C) |
|---|
| 1 | F |
GGTTAACTTGATGGGGCTCA | 374 | 57 |
| R |
AACTGGAAAGCCATCCACAC |
| 2 | F |
TCTATTTCACTTTTCCATGTAACGA | 243 | 57 |
| R |
ACCATGCCCAGCCAAAATA |
| 3 | F |
GACGATGCATAAGGACTGAGTG | 296 | 57 |
| R |
AGCGTTCAGGGGGTTAATTC |
| 4 | F |
GCAAAGCAGATGGGTTTGTT | 359 | 57 |
| R |
CCACCACGCCAGTTAATTTT |
| 5 | F |
CAGGGGGCTCTTTGGATG | 389 | 57 |
| R |
ACAGAGGGCAGTGACAGGAG |
| 6 | F |
CACCCGCAAACTGCTTTAAC | 384 | 57 |
| R |
TGCGAAATATAGCCCTGTCC |
The Sorting Intolerant From Tolerant (SIFT) program
and the Polymorphism Phenotyping (PolyPhen-2) were used to predict
whether an amino acid substitution was likely to affect the protein
function (14,15).
Results
Mutation analysis
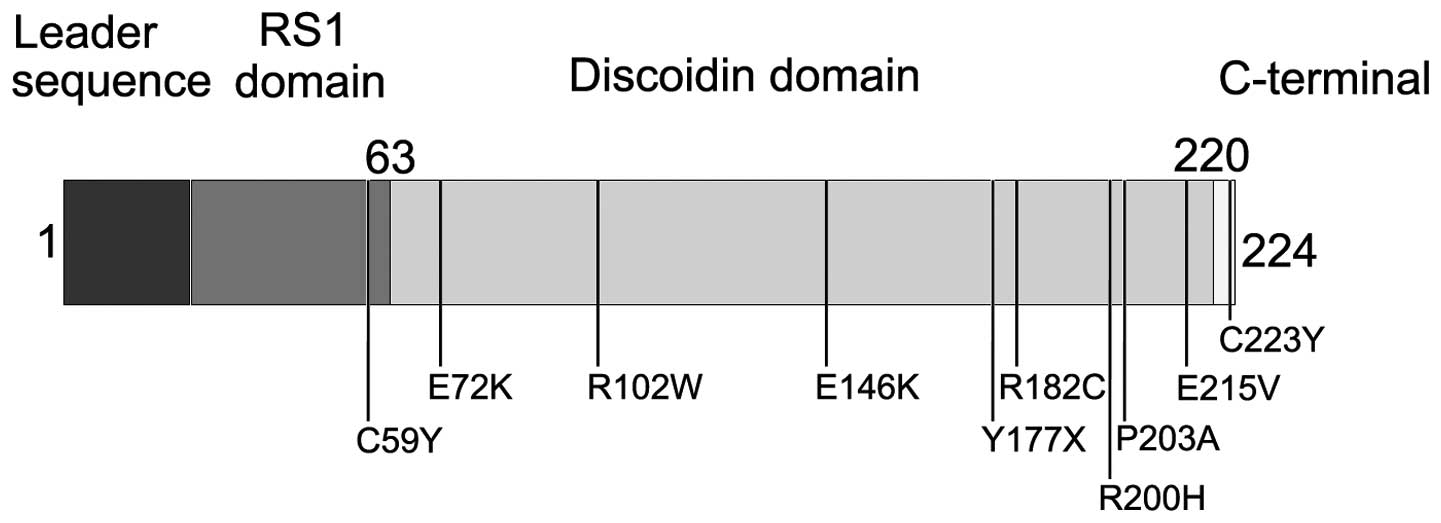
Ten hemizygous mutations in RS1 were detected
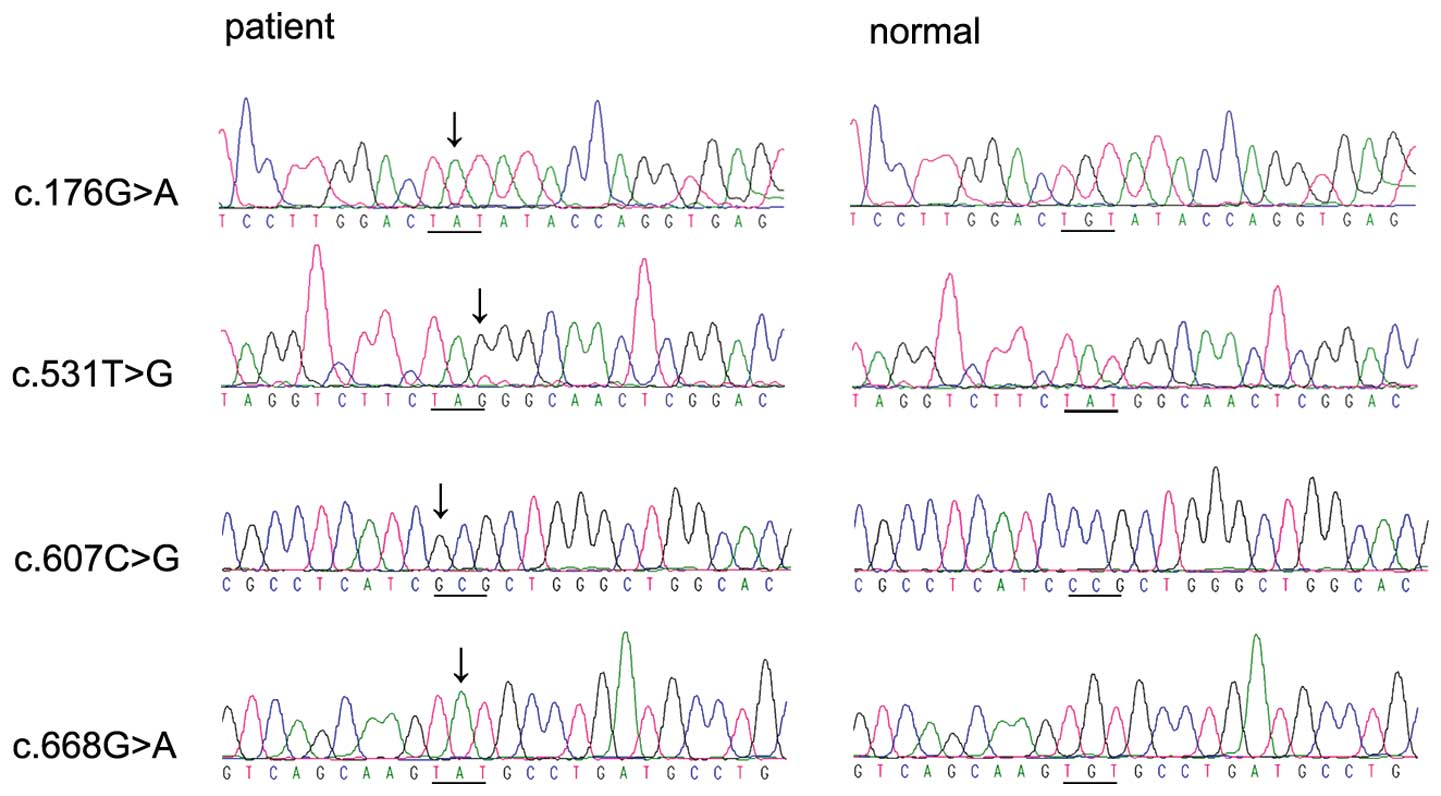
in patients from 14 of the 20 families with retinoschisis (Table II and Fig. 1), including c:176G>A
(p:Cys59Tyr) in exon 3, c:214G>A (p:Glu72Lys) and c:304C>T
(p:Arg102Trp) in exon 4, c:436G>A (p:Glu146Lys) in exon 5,
c.531T>G (p:Tyr177X), c:544C>T (p:Arg182Cys), c:599G>A
(p:Arg200His), c:607C>G (p:Pro203Ala), c:644A>T (p:Glu215Val)
and c:668G>A (p:Cys223Tyr) in exon 6. Of the 10, the
c:176G>A, c:531T>G, c:607C>G and c:668G>A were novel.
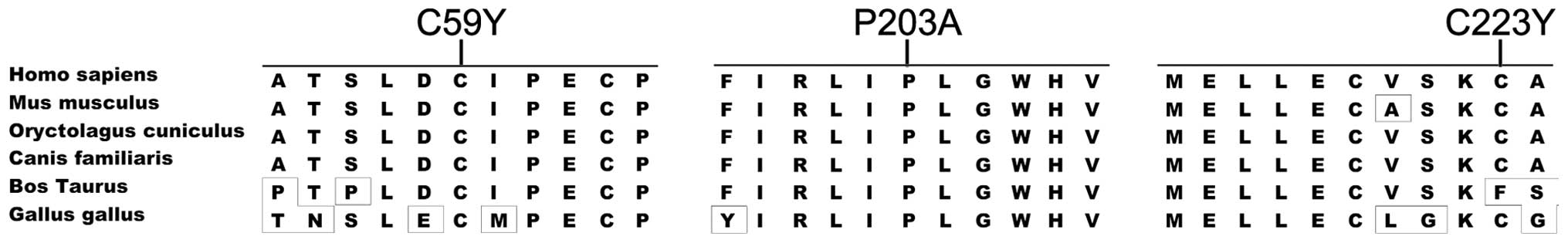
These novel mutations occurred in highly conserved regions
(Fig. 2) and were predicted to be
pathogenic (Table II). They were
absent in 176 normal individuals.
| Table IIThe mutations of the RS1 gene
in XLRS. |
Table II
The mutations of the RS1 gene
in XLRS.
| | | | Computational
prediction | Frequency | | |
|---|
| | | |
|
| | |
|---|
| Exon | Patient ID | Nucleotide
change | Amino acid
change | Blosum62 | PolyPhen | SIFT | Patients | Controls | Note | Ref |
|---|
| 3 | QT42, QT335 | c:176G>A | p:Cys59Tyr | 9→-2 | 0.996 | 0 | 2/20 | 0/176 | Novel | |
| 4 | QT221, QT232,
QT653 | c:214G>A | p:Glu72Lys | 5→1 | 0.998 | 0 | 3/20 | | Reported | (19) |
| 4 | MD15 | c:304C>T | p:Arg102Trp | 5→-3 | 1 | 0 | 1/20 | | Reported | (20) |
| 5 | RP6 | c:436G>A | p:Glu146Lys | 5→1 | 0.961 | 0.17 | 1/20 | | Reported | (21) |
| 6 | MD30 | c:531T>G | p:Tyr177X | | | | 1/20 | 0/176 | Novel | |
| 6 | QT417, QT212 | c:544C>T | p:Arg182Cys | 5→-3 | 1 | 0.01 | 2/20 | | Reported | (22) |
| 6 | QT848 | c:599G>A | p:Arg200His | 5→0 | 1 | 0 | 1/20 | | Reported | (23) |
| 6 | QT911 | c:607C>G | p:Pro203Ala | 7→-1 | 1 | 0.13 | 1/20 | 0/176 | Novel | |
| 6 | QT219 | c:644A>T | p:Glu215Val | 5→-3 | 1 | 0 | 1/20 | | Reported | (31) |
| 6 | QT758 | c:668G>A | p:Cys223Tyr | 9→-2 | 0.996 | 0.01 | 1/20 | 0/176 | Novel | |
All 10 probands with hemizygous RS1 mutations
(the clinical data of 4 probands were not available) had clinical
symptoms and signs of retinoschisis (Table III). The four probands with
novel mutations showed macular and peripheral retinoschisis.
| Table IIIClinical information on individuals
with RS1 variations. |
Table III
Clinical information on individuals
with RS1 variations.
| Mutations | Age (years) | | BCVA | | | | | | |
|---|
|
|
| |
| | | | | | |
|---|
| Patient ID | Nucleotide | Protein | Exam | Onset | Family history | OD | OS | Macular change | Peripheral
change | Retinal hole | Strabismus | OCT | ERG(b/a) |
|---|
| QT042 | 176G>A | Cys59Tyr | N/A | N/A | No | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| QT335 | 176G>A | Cys59Tyr | 11 | 6 | No | 0.4 | 0.2 | mRS | pRS | No | No | RS | N/A |
| QT221 | 214G>A | Glu72Lys | 19 | EC | Yes | 0.1 | 0.2 | mRS | PD | No | No | N/A | N/A |
| QT232 | 214G>A | Glu72Lys | 18 | 8 | No | 0.4 | 0.2 | mRS | Degenenation | No | No | N/A | N/A |
| QT653 | 214G>A | Glu72Lys | 5 | 3 | No | 0.3 | 0.7 | mRS | pRS | Yes | No | N/A | Reduced |
| MD015 | 304C>T | Arg102Trp | N/A | 7 | No | 0.2 | 0.3 | PD, FRB | No | No | No | N/A | N/A |
| RP006 | 436G>A | Glu146Lys | 5 | 4 | No | FC | 0.03 | PD, FRB | No | No | No | N/A | Reduced |
| MD030 | 531T>G | Tyr177X | 6 | 5 | No | 0.3 | FC | mRS | pRS | No | Yes | N/A | Reduced |
| QT212 | 544C>T | Arg182Cys | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| QT417 | 544C>T | Arg182Cys | 12 | EC | No | 0.3 | 0.03 | No | pRS | Yes | No | N/A | N/A |
| QT848 | 599G>A | Arg200His | 21 | EC | No | 0.6 | 0.4 | mRS | No | No | No | N/A | Reduced |
| QT911 | 607C>G | Pro203Ala | 22 | EC | No | 0.2 | 0.4 | mRS | pRS | No | Yes | N/A | N/A |
| QT219 | 644A>T | Glu215Val | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| QT758 | 668G>A | Cys223Tyr | 9 | 6 | No | 0.4 | 0.3 | mRS | pRS | Yes | No | RS | N/A |
Discussion
In this study, ten different hemizygous mutations in
RS1 were identified in 14 families with XLRS. These
mutations are predicted to be pathogenic. All patients with
mutations demonstrated typical signs of XLRS. The ten mutations
affected different domains of retinoschisin, including the RS1
domain (1 mutation), discoidin domain (8 mutations) and C-terminal
segment (1 mutation). These mutations were not randomly distributed
over the gene (Fig. 3) because
80% of mutations were clustered in the discoidin domain (16). The two novel mutations, Tyr177X
and Pro203Ala in the discoidin domain, may cause a shorter
retinoschisin form or protein misfolding (13). The cysteine mutations in the
RS1 domain (Cys59Tyr) and C-terminal segment (Cys223Tyr) may
cause failure of the discoidin domain to assemble into a normal
multisubunit complex (17,18).
Most of RS1 mutation loci were hot mutation
spots, while the Cys59, Glu72, Arg102, Glu146, Arg182, Arg200,
Pro203, Glu215 and Cys223 could be substituted by 1–2 other kinds
of amino acids and be reported more frequently (19–30). However, the mutations in the
present study also differed from those reported previously. The
RS1 mutations accounts for 70% of the Chinese retinoschisis
(14/20) cases in our study. The Cys59Tyr, Tyr177X, Pro203Ala,
Glu215Val and Cys223Tyr mutations only are present in the Chinese
population (31), and the
Cys59Tyr mutation was more common (10% frequency in our
retinoschisis cases). The Glu72Lys mutation is the most common
among Chinese (15%) as well as other populations (19,32), while another very common mutation,
Pro192Ser (33), which was
reported from people of different ethnic backgrounds was not found.
We do not know whether the spectrum and frequency of RS1
gene in the Chinese is different from others. Our study contributes
to the current state of knowledge.
In summary, we identified ten mutations in 14 of 20
families with XLRS. Our results expand the mutation spectrum of
RS1 that might enrich our understanding of the molecular
basis of XLRS in the Chinese population.
Acknowledgements
The authors thank all of the patients and controls
subjects for their participation. This study was supported by the
Open Research Fund Program of State Key Laboratory of
Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-Sen University,
and in part by grant 30725044 from the National Science Fund for
Distinguished Young Scholars.
References
|
1
|
IM MacDonaldR SasiMolecular genetics of
inherited eye disordersClin Invest Med1747449819947867253
|
|
2
|
SK SikkinkS BiswasNR ParryPE StangaD
TrumpX-linked retinoschisis: an updateJ Med
Genet44225232200710.1136/jmg.2006.04734017172462
|
|
3
|
MA ApushkinGA FishmanAS RajagopalanFundus
findings and longitudinal study of visual acuity loss in patients
with X-linked
retinoschisisRetina25612618200510.1097/00006982-200507000-0001216077359
|
|
4
|
A TantriTR VrabecA Cu-UnjiengA FrostWH
Annesley JrLA DonosoX-linked retinoschisis: a clinical and
molecular genetic reviewSurv
Ophthalmol49214230200410.1016/j.survophthal.2003.12.00714998693
|
|
5
|
D ShuklaA RajendranD GibbsB
SuganthalakshmiK ZhangP SundaresanUnusual manifestations of
X-linked retinoschisis: clinical profile and diagnostic
evaluationAm J
Ophthalmol144419423200710.1016/j.ajo.2007.05.01617631851
|
|
6
|
JE KimMS RuttumMJ KoeberlEL HassemerDJ
SidjaninGenetic and clinical evaluation of juvenile retinoschisisJ
AAPOS13215217200910.1016/j.jaapos.2008.11.00519393523
|
|
7
|
CG SauerA GehrigR
Warneke-WittstockPositional cloning of the gene associated with
X-linked juvenile retinoschisisNat
Genet17164170199710.1038/ng1097-1649326935
|
|
8
|
R Mendoza-LondonoKT HiriyannaEL BinghamA
Colombian family with X-linked juvenile retinoschisis with three
affected females finding of a frameshift mutationOphthalmic
Genet203743199910.1076/opge.20.1.37.229910415464
|
|
9
|
L HuopaniemiA RantalaE TahvanainenA de la
ChapelleT AlitaloLinkage disequilibrium and physical mapping of
X-linked juvenile retinoschisisAm J Hum
Genet601139114919979150161
|
|
10
|
D BeschG RudolphGenetic diseases of the
eyeKlin Monbl Augenheilkd2229559712005(In German)
|
|
11
|
SH MinLL MoldayMW SeeligerProlonged
recovery of retinal structure/function after gene therapy in an
Rs1h-deficient mouse model of X-linked juvenile retinoschisisMol
Ther12644651200510.1016/j.ymthe.2005.06.00216027044
|
|
12
|
FM DykaRS MoldayCoexpression and
interaction of wild-type and missense RS1 mutants associated with
X-linked retinoschisis: its relevance to gene therapyInvest
Ophthalmol Vis Sci4824912497200710.1167/iovs.06-146517525175
|
|
13
|
RS MoldayFocus on molecules: retinoschisin
(RS1)Exp Eye Res84227228200710.1016/j.exer.2005.12.01316600216
|
|
14
|
PC NgS HenikoffPredicting deleterious
amino acid substitutionsGenome
Res11863874200110.1101/gr.17660111337480
|
|
15
|
S SunyaevV RamenskyI KochW Lathe IIIAS
KondrashovP BorkPrediction of deleterious human allelesHum Mol
Genet10591597200110.1093/hmg/10.6.59111230178
|
|
16
|
LL MoldayD HicksCG SauerBH WeberRS
MoldayExpression of X-linked retinoschisis protein RS1 in
photoreceptor and bipolar cellsInvest Ophthalmol Vis
Sci42816825200111222545
|
|
17
|
WW WuJP WongJ KastRS MoldayRS1, a
discoidin domain-containing retinal cell adhesion protein
associated with X-linked retinoschisis, exists as a novel
disulfide-linked octamerJ Biol
Chem2801072110730200510.1074/jbc.M41311720015644328
|
|
18
|
WW WuRS MoldayDefective discoidin domain
structure, subunit assembly, and endoplasmic reticulum processing
of retinoschisin are primary mechanisms responsible for X-linked
retinoschisisJ Biol Chem2782813928146200310.1074/jbc.M302464200
|
|
19
|
Y HottaK FujikiM HayakawaJapanese juvenile
retinoschisis is caused by mutations of the XLRS1 geneHum
Genet10314214419989760195
|
|
20
|
JA DoddsAK SrivastavaKR HoldenUnusual
phenotypic expression of an XLRS1 mutation in X-linked juvenile
retinoschisisJ Child
Neurol21331333200610.1177/0883073806021004190116900931
|
|
21
|
NW KhanJA JamisonJA KempPA SievingAnalysis
of photoreceptor function and inner retinal activity in juvenile
X-linked retinoschisisVision
Res4139313942200110.1016/S0042-6989(01)00188-211738458
|
|
22
|
Y MashimaK ShinodaS IshidaIdentification
of four novel mutations of the XLRS1 gene in Japanese patients with
X-linked juvenile retinoschisis. Mutation in brief no 234Online Hum
Mutat13338199910.1002/(SICI)1098-1004(1999)13:4%3C338::AID-HUMU16%3E3.0.CO;2-010220153
|
|
23
|
B LledoJ TenD Rodriguez-ArnedoJ LlacerR
BernabeuPreimplantation genetic diagnosis of X-linked
retinoschisisReprod Biomed
Online16886892200810.1016/S1472-6483(10)60157-518549702
|
|
24
|
D TuvdendorjY IsashikiN OhbaS SonodaS
IzumoTwo Japanese patients with mutations in the XLRS1
geneRetina22354357200210.1097/00006982-200206000-0001712055472
|
|
25
|
Y InoueS YamamotoT InoueTwo novel point
mutations of the XLRS1 gene in patients with X-linked juvenile
retinoschisisAm J
Ophthalmol134622624200210.1016/S0002-9394(02)01592-112383832
|
|
26
|
F SimonelliG CennamoC ZivielloClinical
features of X linked juvenile retinoschisis associated with new
mutations in the XLRS1 gene in Italian familiesBr J
Ophthalmol8711301134200310.1136/bjo.87.9.113012928282
|
|
27
|
S WaliaGA FishmanRS MoldayRelation of
response to treatment with dorzolamide in X-linked retinoschisis to
the mechanism of functional loss in retinoschisinAm J
Ophthalmol147111115200910.1016/j.ajo.2008.07.04118834580
|
|
28
|
T WangA ZhouCT WatersE O’ConnorRJ ReadD
TrumpMolecular pathology of X linked retinoschisis: mutations
interfere with retinoschisin secretion and oligomerisationBr J
Ophthalmol908186200610.1136/bjo.2005.07804816361673
|
|
29
|
FM DykaWW WuTA PfeiferLL MoldayTA
GrigliattiRS MoldayCharacterization and purification of the
discoidin domain-containing protein retinoschisin and its
interaction with
galactoseBiochemistry4790989106200810.1021/bi800938g18690710
|
|
30
|
X MaX LiL WangNovel XLRS1 gene mutations
cause X-linked juvenile retinoschisis in Chinese familiesJpn J
Ophthalmol524851200810.1007/s10384-007-0488-418369700
|
|
31
|
M ZengC YiX GuoIdentification of novel
mutations in the XLRS1 gene in Chinese patients with X-linked
juvenile retinoschisisCurr Eye Res32685691200717852193
|
|
32
|
B LeschV SzaboM KanyaClinical and genetic
findings in Hungarian patients with X-linked juvenile
retinoschisisMol Vis14232123322008
|
|
33
|
LC EksandhV PonjavicR AyyagariPhenotypic
expression of juvenile X-linked retinoschisis in Swedish families
with different mutations in the XLRS1 geneArch
Ophthalmol11810981104200010.1001/archopht.118.8.109810922205
|