Introduction
The 31 kDa protein IL-33 belongs to the IL-1 family
of cytokines (1) and was first
described as a nuclear factor produced by high endothelial venule
cells (NF-HEV) (2). Precursor
IL-33 seems to function as a regulator/repressor of nuclear
transcription (3). This nuclear
function is associated with the homeodomain-like helix-turn-helix
motif in the N-terminal part of IL-33, which mediates DNA binding.
A transcriptional regulatory function of precursor IL-33 is further
supported by the nuclear localization of IL-33 in HUVECs and
IL-33-overexpressing cells, but also in LPS-stimulated murine
astrocytes (2–5). After secretion, mature IL-33 (18 kD
C-terminal fragment) acts as a pro-inflammatory cytokine via a
receptor complex of ST2 and IL-1 receptor accessory protein
(IL-1RacP). As a pro-inflammatory cytokine, mature IL-33 induces
the production of TH2 cytokines by TH2-cells
and the secretion of numerous cytokines by human and murine mast
cells (6,7). The biological effects of IL-33/ST2
are mediated by activation of the p38, ERK, JNK, and NF-κB
signaling pathways (1,8). However, it has been recently shown
that processing of precursor IL-33 results in an inactivation,
rather than an activation of IL-33 (9). In addition, precursor IL-33
activates NF-κB and induces IL-6 synthesis. Therefore, it has been
proposed that IL-33 functions as an endogenous danger signal
(alarmin) to alert cells of the innate immune system to tissue
damage during trauma or infection (9,10).
The IL-33/ST2 system is involved in numerous
diseases and pathological conditions, e.g., fibroproliferative and
cardiovascular diseases, asthma, and rheumatoid arthritis (RA)
(8). An important role of IL-33
in the pathogenesis of RA is suggested by studies in the animal
model of murine collagen-induced arthritis (11–14). Treatment of diseased animals with
soluble ST2 fusion protein or a blocking anti-ST2 receptor reduced
the disease severity compared to non-treated animals (e.g.,
reduction in synovial cellular infiltration, synovial hyperplasia,
joint erosion and serum levels of pro-inflammatory cytokines);
conversely, injection of IL-33 enhanced the signs of
murine-collagen induced arthritis. Regarding human arthritis, IL-33
levels were elevated in the sera and synovial fluid of RA-patients
and showed a positive correlation with the disease activity
(15). IL-33 protein and also its
receptor ST2 were detected in the lining layer and sublining of
synovial membrane of RA patients (12). In addition to the strong
expression of IL-33 in endothelial cells, fibroblasts and
mononuclear inflammatory cells were identified as a potential
source of IL-33 in the inflamed synovial membrane of RA patients
(3,13). IL-33 mRNA and protein expression
was induced in RA synovial fibroblasts (RA-SFs) following
TNF-α/IL-1β stimulation and IL-33 protein was mainly detected in
the nucleus of RA-SFs (12,13).
In addition to the role of mature IL-33 in the
progression of RA (14,16), the nuclear localization of
pro-IL-33 in IL-1β/TNF-α stimulated cells may point to a regulatory
function inside the cells, as it has been described for the other
IL-1 family members IL-1α and IL-1F7b (17,18). Therefore, the present study sought
to analyze the involvement of IL-33 in TNF-α-induced
pro-inflammatory or pro-destructive effector functions of
RA-SFs.
Materials and methods
Patients, tissue digestion and cell
culture
Synovial tissue from RA-patients fulfilling the ARA
criteria was obtained during open joint replacement/arthroscopic
synovectomy from the Clinic of Orthopedics, Eisenberg, Germany
(19). The study was approved by
the Ethics Committee of the University of Jena, Germany, and
patient informed consent was obtained.
RA synovial samples were digested, subsequently
cultured for 7 days, and RA-SFs negatively isolated as previously
described (20,21). RA-SFs were cultured in the virtual
absence of contaminating non-adherent cells and macrophages.
Third-passage cells were used for all the experiments. Mycoplasma
contamination of the cells was excluded by
4′-6-diamidino-2-phenylindole (DAPI) staining.
The stimulation of the cells with different
concentrations of TNF-α (0.1 to 10.0 ng/ml; R&D Systems,
Wiesbaden, Germany) was performed in DMEM/0.2% lactalbumin
hydrolysate. For analysis of signal transduction pathways, cells
were preincubated for 45 min with inhibitors of p38 MAPK (SB203580,
1 μM; Jena Biosciences, Jena, Germany), ERK (U0126, 1 μM, Axxora,
Lörrach, Germany), JNK (SP600125, 20 μM, Jena Biosciences), NFκB
(IκBK inhibitor peptide, cell-permeable, 50 μg/ml; Calbiochem, VWR,
Darmstadt, Germany) (22), PKA
(PKA inhibitor fragment 14–22, myristoylated trifluoroacetate salt,
10 μM, Sigma, Deisenhofen, Germany) (23) or PI3-kinase (wortmannin, 1 μM,
Axxora), followed by TNF-α stimulation for 24 h. For analysis of
the influence of different cytokines/growth factors on IL-33
synthesis, RA-SFs were stimulated with TNF-α, IL-18, PDGF-BB,
TGF-β1 (10 ng/ml each), or IL-1β for 24 h (5 ng/ml; R&D
Systems; Peprotech, London, UK). To analyze the influence of
exogenous IL-33 on signal transduction and functional parameters of
RA-SFs, cells were stimulated with 10 or 100 ng/ml recombinant
human IL-33 (R&D Systems) for 15 min (signal transduction) or
24 h (protein secretion). Viability of the cells was assessed by
ethidium bromide staining.
Analysis of the IL-33 protein expression
and signal transduction by Western blotting
Equal volumes of supernatants or 50 μg cellular
protein were separated by 12% SDS-PAGE using a Laemmli buffer
system as previously described (24). Primary anti-IL-33 antibody was
obtained from R&D Systems. The band intensities were quantified
using the software program Diana III luminescence imaging system
(Raytest, Straubenhardt, Germany) and normalized to β-actin. Signal
transduction in IL-33 and TNF-α stimulated RA-SFs was analyzed as
previously described (25).
Overexpression of IL-33 in RA-SFs
Overexpression of IL-33 in RA-SFs was performed
using the ViraPower™ Lentiviral Expression System according to the
manufacturer’s instructions (Invitrogen, Karlsruhe, Germany). In
brief, the coding sequence of IL-33 was cloned into the plenty6/V5
vector. For producing lentivirus, 296FT cells were transfected with
the ViraPower™ packaging mix, the pLenti IL-33 expression plasmid,
or the empty pLenti expression plasmid using Lipofectamine™ 2000
and Opti-MEM® I medium. After 24 h, the medium was
changed to complete culture medium according to the manufacturer’s
instructions (Invitrogen). Virus-containing supernatants were
collected 72 h post-transfection, centrifuged to remove cell
debris, and stored in aliquots at −80°C. For IL-33 overexpression,
RA-SFs were transduced with the virus-containing supernatant in the
presence of Polybrene™ (6 μg/ml; Sigma, Deisenhofen, Germany).
After 24 h incubation, the virus-containing medium was replaced by
DMEM/10% FCS and blasticidin for selection resulting in cells of
passage 5. Overexpression of IL-33 was analyzed by RT-PCR and
ELISA. As a control, RA-SFs were transduced with empty pLenty6/V5
vector.
Silencing of IL-33 in RA-SFs
Silencing of IL-33 in RA-SFs was performed by the
siRNA technique using reverse transfection according to the
manufacturer’s instructions (Invitrogen). In brief, Stealth™ RNAi
and Lipofectamine™ RNAiMAX (Invitrogen) were mixed in
Opti-MEM® I (Invitrogen) and incubated for 20 min at
room temperature to allow complex formation. Subsequently, cells
suspended in DMEM and 10% FCS without antibiotics were added and
incubated for 24 h. Thereafter, cells were stimulated with 10 ng/ml
TNF-α in DMEM and 0.2% lactalbumin hydrolysate for 24 h.
Supernatants of the cells were collected for the analysis of
cytokine and protease secretion. Cells were washed twice with
ice-cold PBS and subsequently lysed in buffer for RNA-isolation
(Macherey Nagel, Düren, Germany) or ice-cold buffer for protein
extraction (50 mM Tris, 150 mM NaCl, EDTA, pH 7.4, containing 100
mM NP-40, 1 mM phenylmethylsulphonylfluoride, 1 mM
Na3VO4, 2 μg/ml aprotinin, 2 μg/ml pepstatin,
and 2 μg/ml leupeptin). For the analysis of IL-33 silencing, 35 μg
of cellular protein were separated by 15% SDS-PAGE using a Laemmli
buffer system. Transfection efficiency was analyzed using 10 nM
Block-iT™ AlexaFluor® Red Fluorescent Oligo
(Invitrogen).
Immunohistochemistry for IL-33 in
RA-SFs
For IL-33-immunohistochemistry in RA-SFs,
0.4×105 cells/well (8-chamber slides) were allowed to
adhere for 24 h, stimulated for 24 h with 10 ng/ml TNF-α, 5 ng/ml
IL-1β, 10 ng/ml IL-18, 10 ng/ml PDGF-BB, or 10 ng/ml TGF-β1 in DMEM
with 0.2% lactalbumin hydrolysate, followed by fixation with 3.7%
paraformaldehyde in PBS for 15 min at room temperature (RT) and by
neutralization with 50 mM NH4Cl in PBS for 5 min at RT.
Fixed cells were permeabilized with PBS/0.1% Triton X-100 for 5 min
at RT. To inactivate endogenous peroxidase, cells were incubated
with 0.03% H2O2/PBS for 20 min. Thereafter,
cells were incubated with primary antibody (clone Nessy-1, Axxora)
in PBS/1% bovine serum albumin for 2 h at RT, washed 3 times for 5
min each with PBS/1% BSA, and incubated with
horseradish-peroxidase-coupled goat anti-rabbit IgG (Dako, Hamburg,
Germany) in PBS/1% BSA for 1 h at room temperature. Cells were
washed thoroughly with PBS and stained with DAB.
Analysis of IL-33 mRNA expression and
functional parameters by RT-PCR
Total RNA was isolated from RA-SFs and
reverse-transcribed as previously described (26,27). mRNA expression for IL-33, IL-6,
IL-8, MCP-1, MMP-1, MMP-3, TIMP-1, and the house-keeping gene
aldolase was analyzed by real-time PCR using a RealPlex®
PCR machine (Eppendorf, Hamburg, Germany; (26,27). The primer pairs and PCR conditions
are presented in Table I. The
relative concentrations of IL-33, IL-6, IL-8, MCP-1, MMP-1, MMP-3,
and TIMP-1 mRNA in each sample were calculated by the
RealPlex® software using an external standard curve.
Product specificity of the real-time PCR was confirmed by: i)
melting curve analysis; ii) agarose gel electrophoresis; and iii)
initial cycle sequencing of the PCR products.
 | Table IPrimer sequences and annealing
temperatures used in RT-PCR. |
Table I
Primer sequences and annealing
temperatures used in RT-PCR.
| Gene | | Primer
sequence | Annealing
temperature, °C | Melting
temperature, °C |
|---|
| Aldolase | Sense |
5-tcatcctcttccatgagacactct-3 | 58 | 82 |
| Antisense |
5-attctgctggcagatactggcataa-3 |
| IL-33 | Sense |
5-cacccctcaaatgaatcagg-3 | 60 | 84 |
| Antisense |
5-ggagctccacagagtgttcc-3 |
| IL-6 | Sense |
5-atgaactccttctccacaagcg-3 | 62 | 84 |
| Antisense |
5-ctcctttctcagggctgag-3 |
| IL-8 | Sense |
5-gccaagagaatatccgaact-3 | 60 | 78 |
| Antisense |
5-aggcacagtggaacaaggacttgt-3 |
| MCP-1 | Sense |
5-cagccagatgcaatcaatgcc-3 | 60 | 82 |
| Antisense |
5-tggaatcctgaacccacttct-3 |
| MMP-1 | Sense |
5-gacctggaggaaatcttgc-3 | 58 | 81 |
| Antisense |
5-gttagcttactgtcacacgc-3 |
| MMP-3 | Sense |
5-gaacaatggacaaaggatacaaca-3 | 58 | 81 |
| Antisense |
5-aagattgatgctgtttttgaagaa-3 |
| TIMP-1 | Sense |
5-cttctggcatcctgttgttg-3 | 60 | 82 |
| Antisense |
5-agaaggccgtctgtgggt-3 |
Analysis of functional parameters in
RA-SFs by ELISA
Proliferation was assessed by BrdU incorporation
using a commercially available cell proliferation ELISA (Roche) as
previously described (25). Human
IL-6, IL-8, MCP-1, TIMP-1, and PGE2 were measured in
diluted cell culture supernatants using commercially available
ELISA kits (OptEIA™, BD Biosciences, Heidelberg, Germany; R&D
Systems; GE Healthcare, Freiburg, Germany). Human MMP-1 and MMP-3
were measured in diluted cell culture supernatants as previously
described (27). The resulting
colour was read at 450 nm in microtiter plates spectrophotometer
(Fluostar Optima, BMG LABTECH GmbH, Ortenberg, Germany).
Statistical analysis
Data are presented as means ± standard error of the
mean (SEM). The non-parametric Mann-Whitney U-test was applied to
analyze differences between controls and individual stimuli, or
among different stimuli (software program SPSS 10.0TM;
SPSS Inc., Chicago, IL, USA). Significant differences were accepted
for P≤0.05.
Results
Influence of TNF-α on the expression of
IL-33 mRNA and protein in RA-SFs
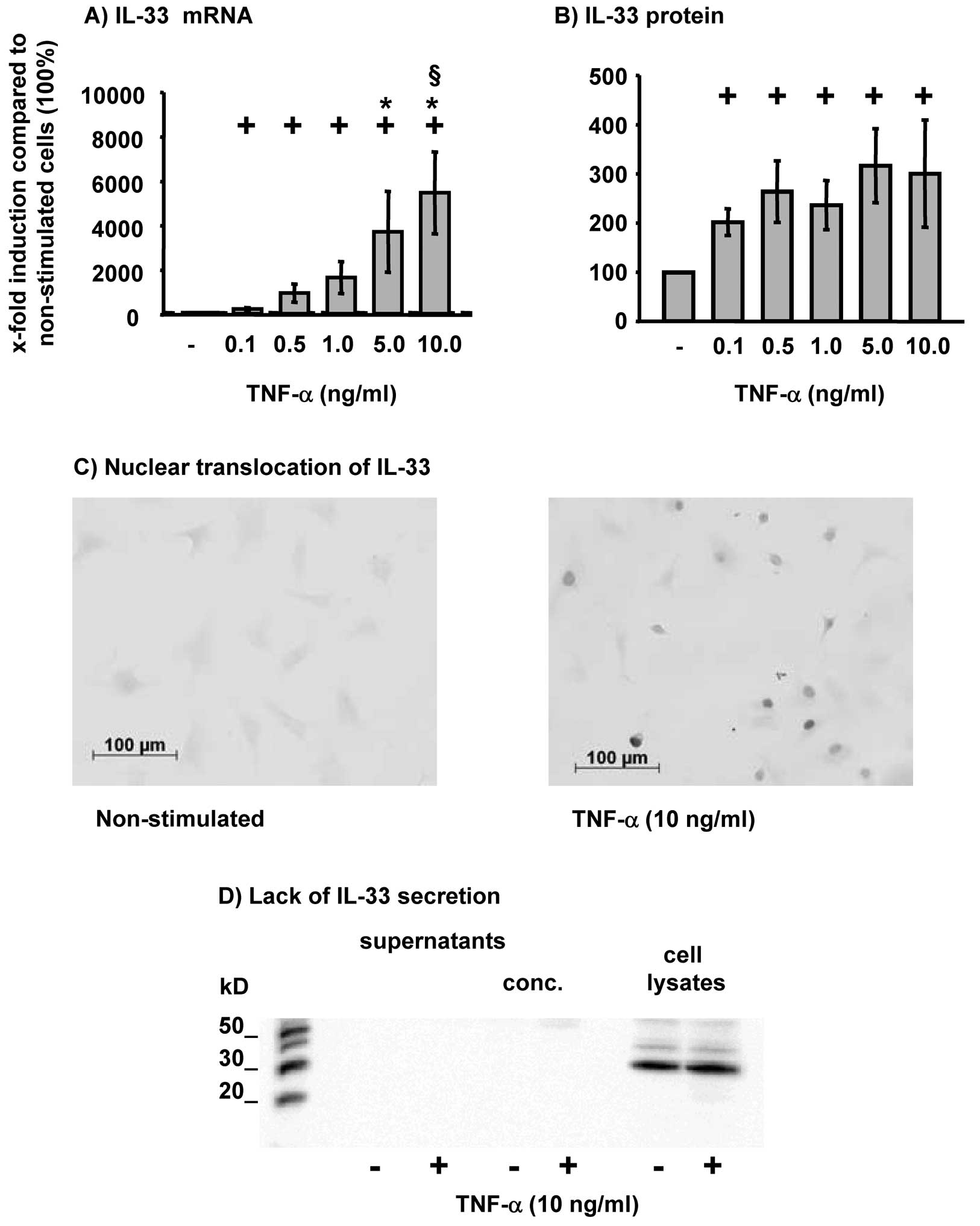
TNF-α significantly and dose-dependently induced the
expression of IL-33 mRNA in RA-SFs (Fig. 1A). The expression of IL-33 was
significantly induced starting at the lowest concentration of TNF-α
(0.1 ng/ml; Fig. 1A) and further
augmented by higher concentrations thereof (plateau at 5.0 or 10
ng/ml TNF-α; Fig. 1A). TNF-α also
significantly induced IL-33 protein in RA-SFs (Fig. 1B); the production of IL-33 reached
a plateau at the lowest concentration of TNF-α (0.1 ng/ml; Fig. 1B).
Non-stimulated RA-SFs showed a weak staining for
IL-33 (Fig. 1C). However,
following stimulation with TNF-α RA-SFs showed a strong positive
staining predominantly localized in the nucleus (Fig. 1C). To analyze whether
TNF-α-induced IL-33 protein is secreted, IL-33 protein was analyzed
in the supernatants and cell lysates of RA-SFs following TNF-α
stimulation. It was clearly shown that IL-33 was not secreted by
RA-SFs, but remained exclusively in the cell lysates (Fig. 1D).
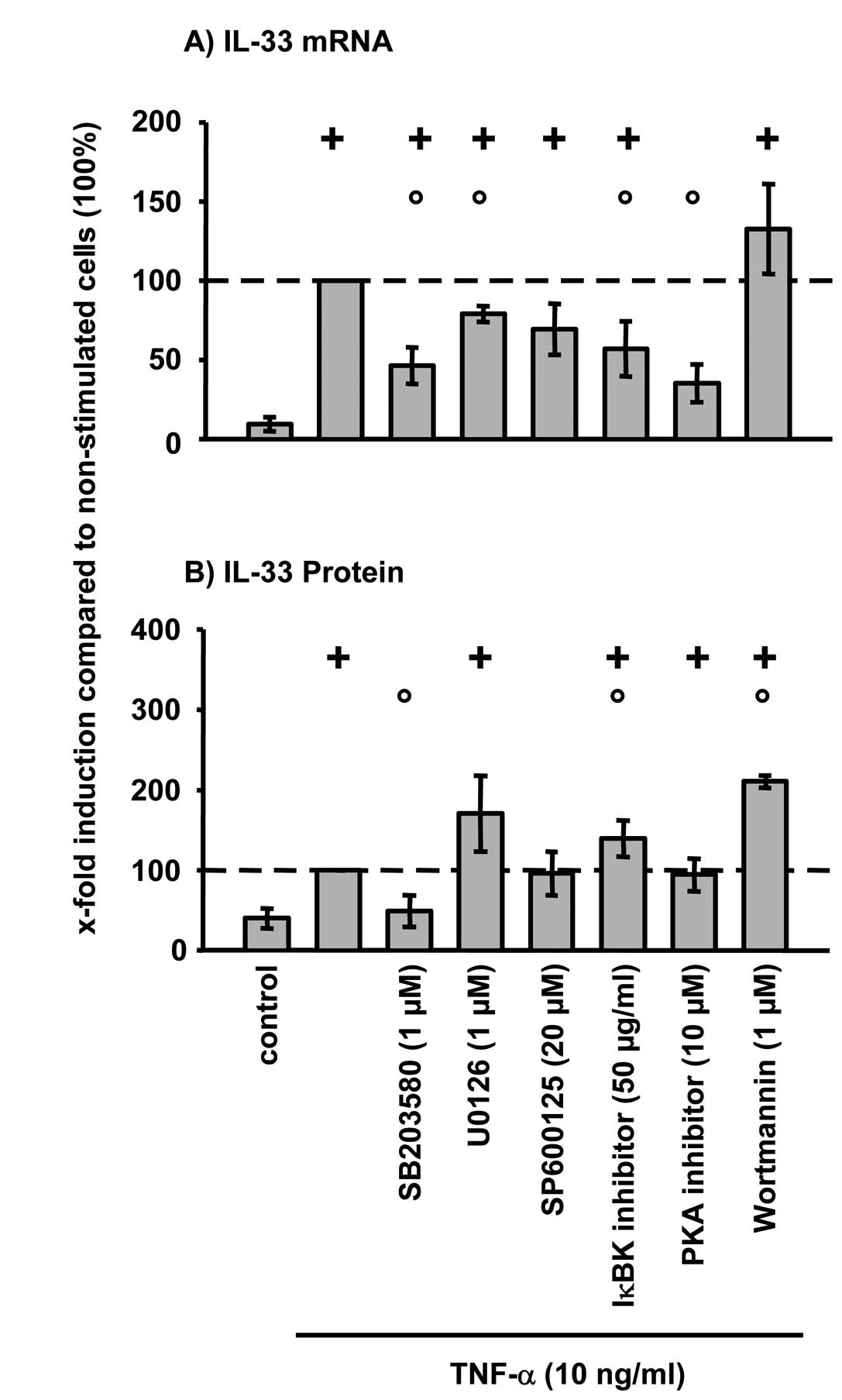
TNF-α-induced IL-33 mRNA expression in RA-SFs was
significantly reduced by inhibition of p38 MAPK (SB203580), ERK
(U0126), NFκB (IκBK inhibitor peptide), and PKA (PKA inhibitor
fragment 14–22) (Fig. 2A).
Inhibition of JNK (SP600125) and PI3-kinase (wortmannin) had no
significant influence on the TNF-α-induced IL-33 mRNA expression.
However, at the protein level inhibition of p38 significantly
reduced TNF-α-induced IL-33 (Fig.
2B). In contrast, inhibition of the NFκB pathway and PI3-kinase
significantly induced IL-33 protein in RA-SFs. No significant
viability differences were observed between non-stimulated cells
(control) and TNF-α-stimulated cells with/without inhibitors
(viability in all cases: >97%; data not shown).
Effects of IL-33 overexpression on the
TNF-α-induced functions of RA-SFs
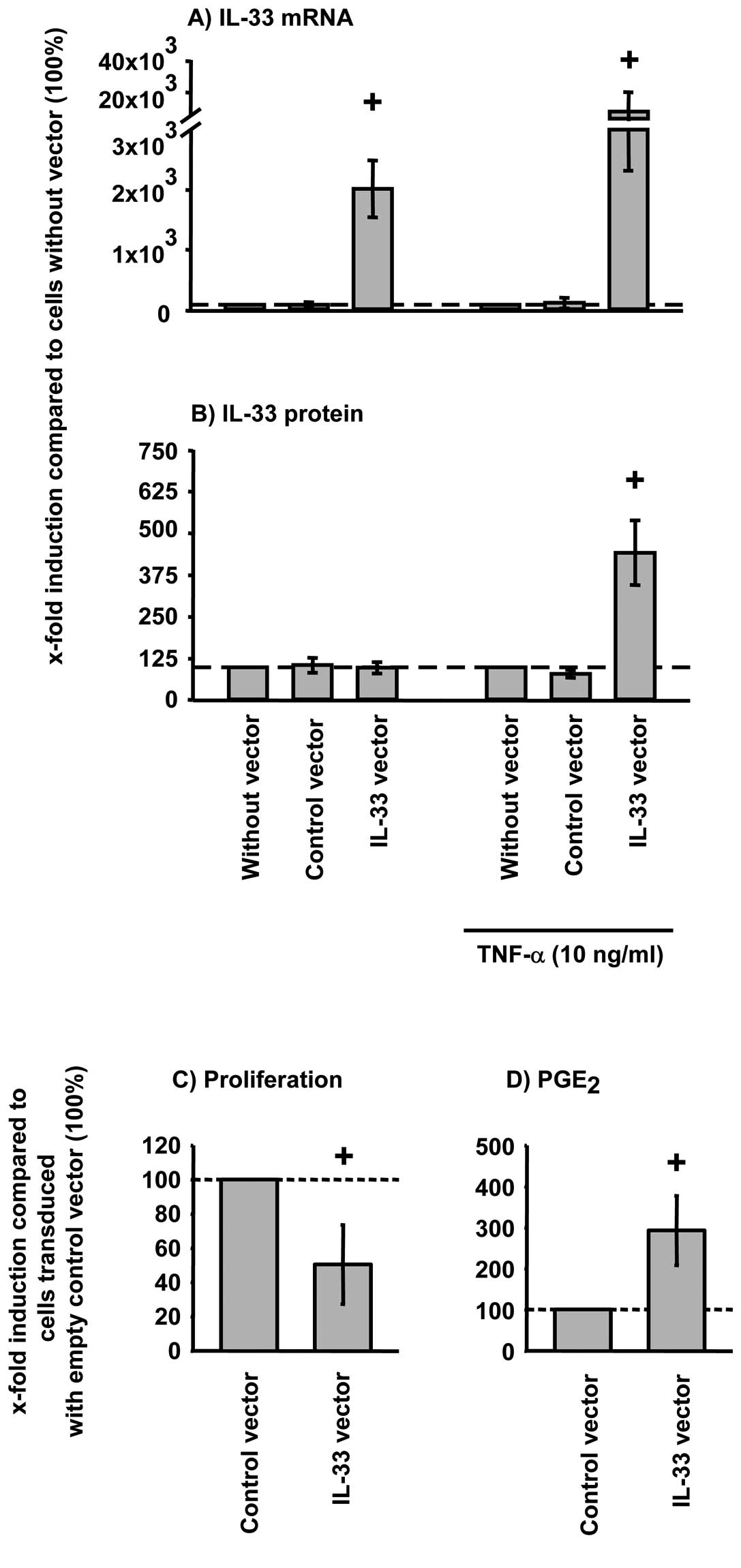
Lentiviral overexpression of IL-33 mRNA
significantly augmented the TNF-α induced IL-33 mRNA and protein
expression in RA-SFs (Fig. 3A and
B).
TNF-α significantly downregulated the proliferation
of RA-SFs (data not shown) Overexpression of IL-33 further
augmented the TNF-α induced downregulation of RA-SFs proliferation
(Fig. 3C). The TNF-α-induced
PGE2 secretion in RA-SFs, in contrast, is further
enhanced by IL-33 overexpression (Fig. 3D).
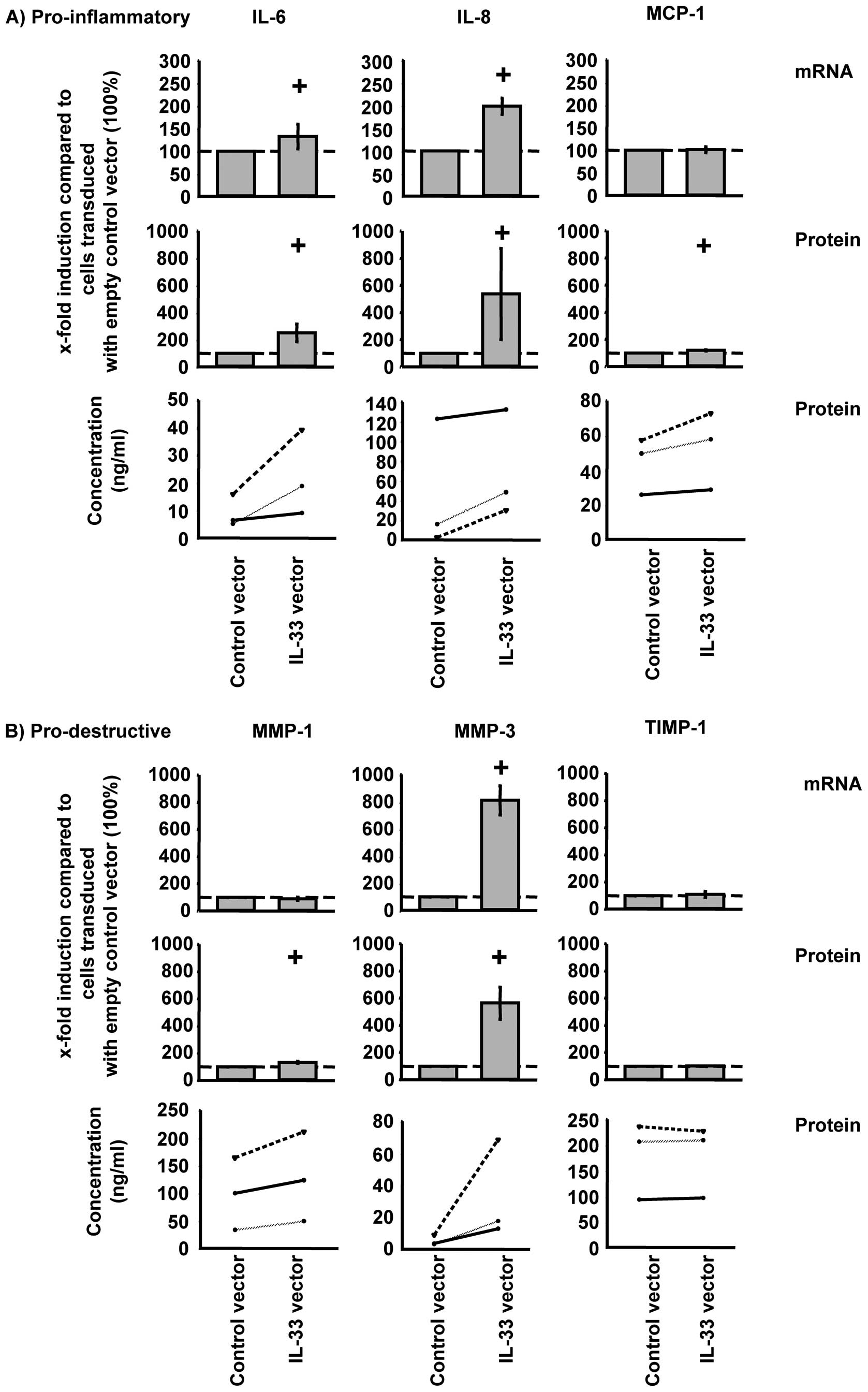
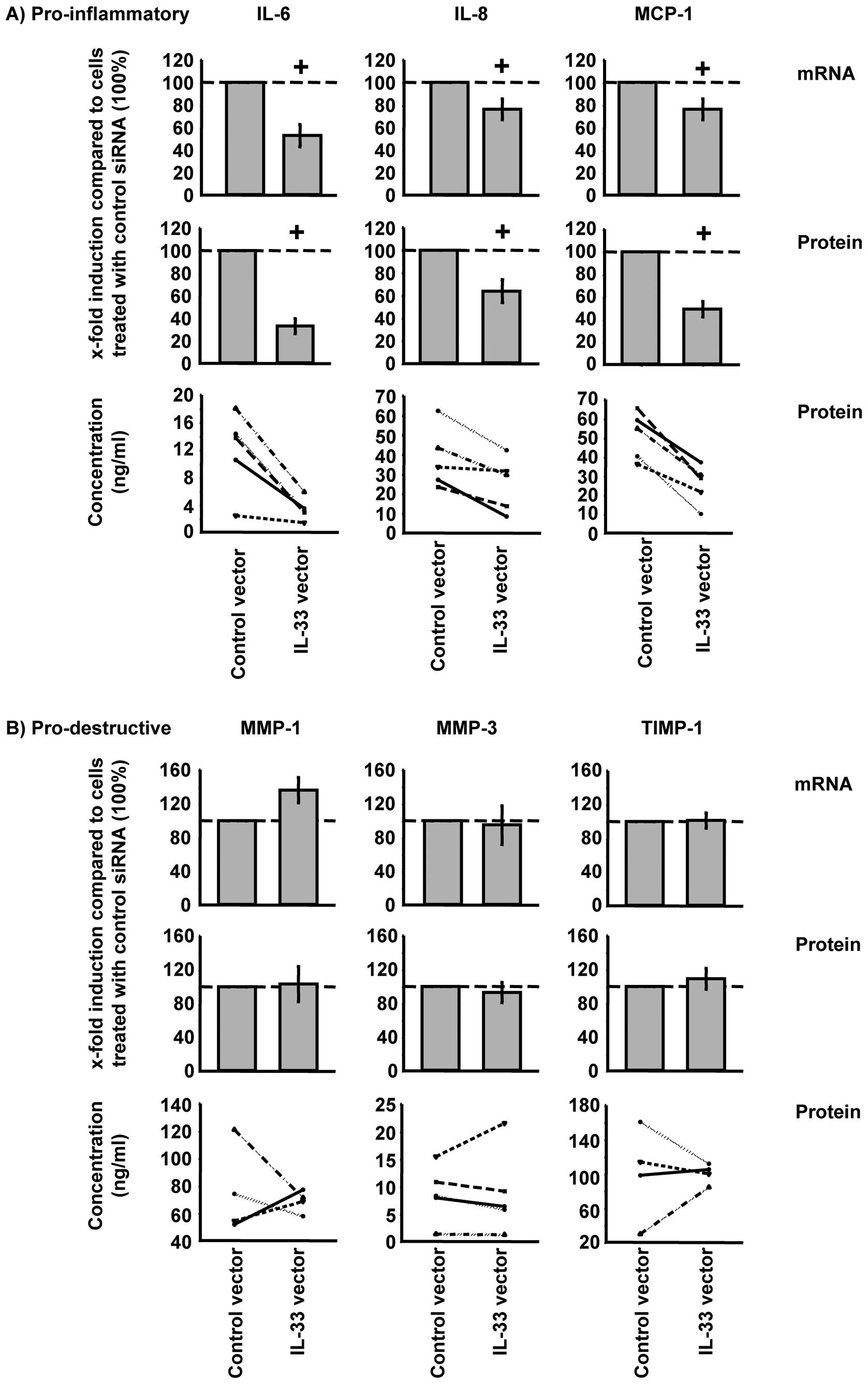
At the mRNA level, overexpression of IL-33
significantly increased TNF-α-induced IL-6, IL-8, and MMP-3 in
RA-SFs (Fig. 4; shown as relative
and absolute values). However, at the protein level, overexpression
of IL-33 significantly induced the secretion of IL-6, IL-8, MCP-1,
MMP-1, and MMP-3 (Fig. 4).
Overexpression of IL-33 did not significantly influence TIMP-1
expression at the mRNA or protein levels (Fig. 4B).
Effects of IL-33 silencing on the
TNF-α-induced functions of RA-SFs
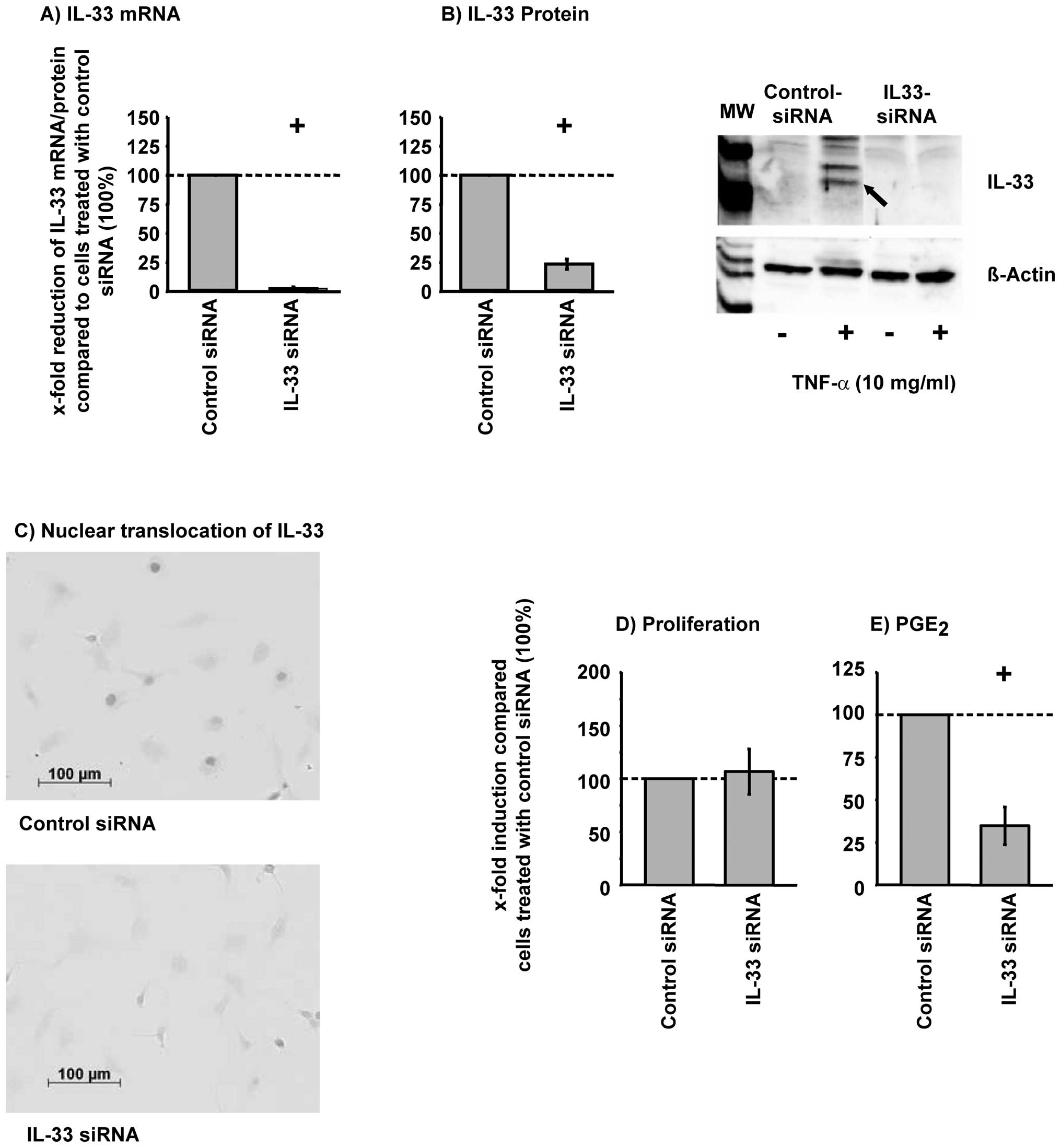
Silencing of IL-33 significantly reduced the
TNF-α-induced IL-33 mRNA and protein expression in RA-SFs (Fig. 5A and B). Transfection efficiency
was greater than 85% using Block-iT™ AlexaFluor® Red
Fluorescent Oligo (85.4% ±2.7 positive cells). In addition,
immunohistochemical analysis of IL-33 siRNA showed that IL-33 siRNA
abolished the TNF-α-induced nuclear translocation of IL-33
(Fig. 5C). The viability of the
cells was not significantly influenced by silencing of IL-33
(viability, >97%; data not shown). The TNF-α-regulated
proliferation of RA-SFs was not significantly influenced by IL-33
silencing (Fig. 5D). In contrast,
silencing of IL-33 significantly reduced the TNF-α-induced
PGE2 secretion (Fig.
5E).
Silencing of IL-33 significantly reduced the TNF-α
induced synthesis of IL-6, IL-8, and MCP-1 in RA-SFs at the mRNA
and protein levels (Fig. 6A). In
contrast to the pro-inflammatory mediators, the synthesis of the
pro-destructive mediators was not significantly influenced by IL-33
silencing in RA-SFs (Fig.
6B).
Influence of different cytokines and
growth factors on the IL-33 protein expression in RA-SFs
In addition to TNF-α, the pro-inflammatory cytokine
IL-1β significantly induced IL-33 synthesis (fold-induction
compared to control cells: TNF-α, 7.1-fold; IL-1β, 15.7-fold;
Fig. 7A). In contrast, the
pro-inflammatory cytokine IL-18 or the growth factors PDGF-BB and
TGF-β1 had no significant influence on IL-33 synthesis. In
agreement with these data, nuclear translocation of IL-33 was
solely observed in TNF-α or IL-1β-stimulated cells (Fig. 7B; results for IL-1β and PDGF-BB
are shown).
Influence of exogenous IL-33 on signal
transduction and protein expression of pro-inflammatory and
pro-destructive mediators in RA-SFs
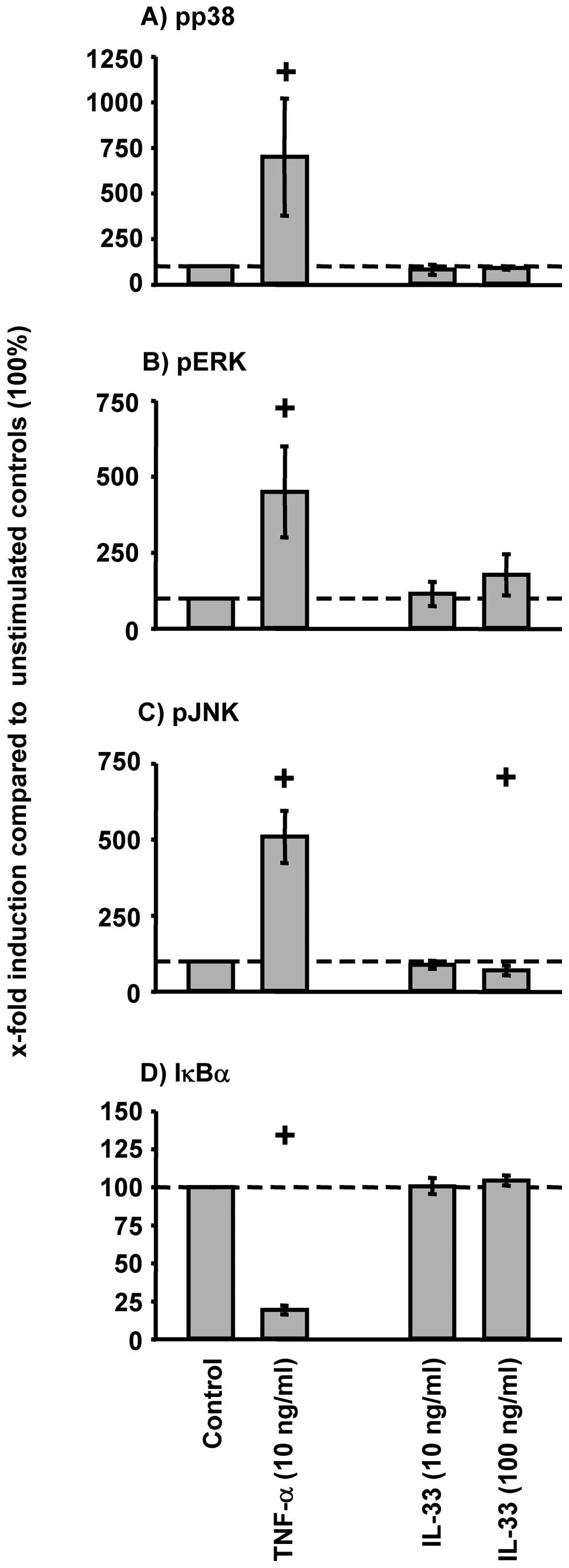
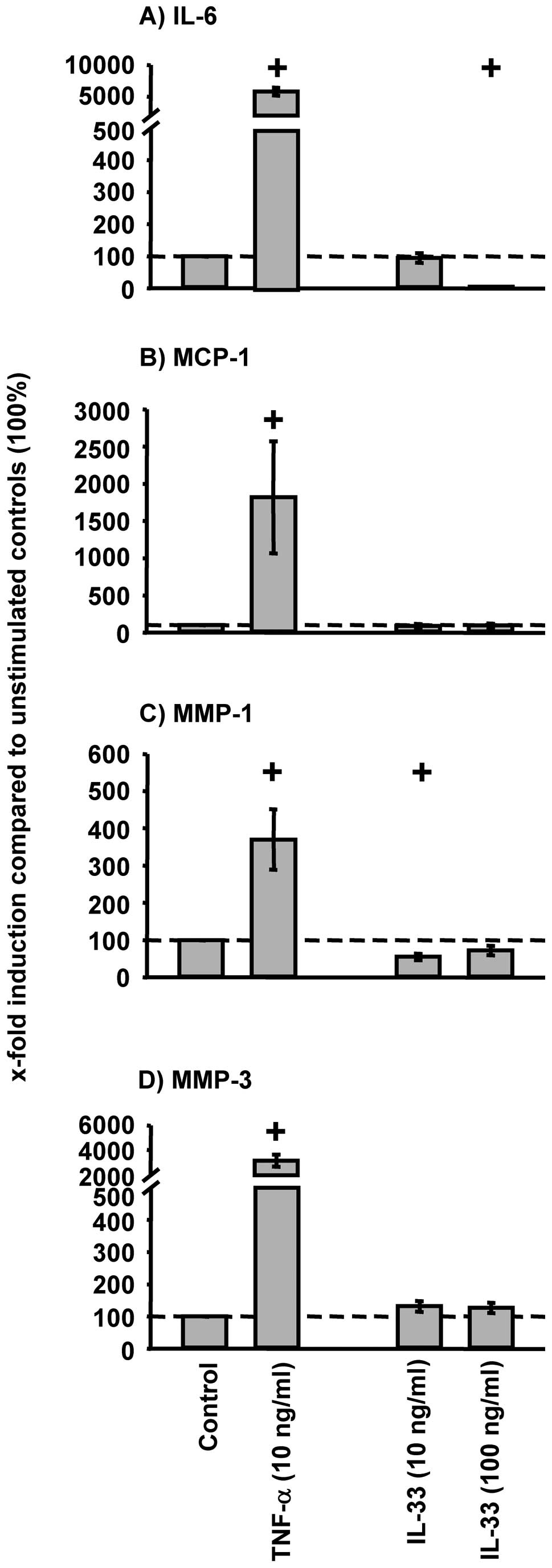
External IL-33 stimulation (10 or 100 ng/ml) of
RA-SFs did not induce a significant increase in the phosphorylation
of p38, ERK, or JNK (Fig. 8A-C).
Also, no significant decrease of IκBα was observed following IL-33
stimulation of RA-SFs (Fig. 8D).
Accordingly, IL-33 stimulation did not significantly induce the
protein secretion of IL-6, MCP-1, MMP-1, and MMP-3 (Fig. 9). In all cases, TNF-α induced a
significant activation of the analyzed signaling pathways and a
significant increase of the secretion of pro-inflammatory and
pro-destructive mediators (Figs.
8 and 9).
Discussion
The present study demonstrates for the first time,
that i) TNF-α induced IL-33 expression is regulated via p38; ii)
the pro-inflammatory cytokines TNF-α and IL-1β are potent inducers
of IL-33 synthesis; and iii) IL-33 is involved in the regulation of
TNF-α-induced synthesis of pro-inflammatory and pro-destructive
molecules. Therefore, IL-33 is a critical regulator of
TNF-α-induced functions in RA-SFs, pointing to a central role of
this cytokine in the perpetuation of pro-inflammatory and
pro-destructive processes in RA and other inflammatory and
degenerative diseases.
In agreement with previously published data, the
present study shows that IL-33 mRNA/protein expression is induced
in RA-SFs following TNF-α stimulation (12,13). Interestingly, IL-33 was only
detected in the nucleus of TNF-α-stimulated RA-SFs, but not or only
barely in the supernatant of stimulated RA-SFs [present
publication, unpublished data and (13)]. This nuclear localization has also
been observed in HUVECs and glia cells (2,4,5)
and supports the proposed function of IL-33 as a transcriptional
regulator (3). Nuclear
translocation of IL-33 following TNF-α/IL-1β stimulation of RA-SFs
therefore indicates that IL-33 may function as a transcriptional
regulator rather than as an external cytokine. This view is further
supported by the observation that the long signaling ST2 receptor
is not expressed on the cells (13). In addition, RA-SFs do not respond
to IL-33 stimulation with an activation of signal transduction
molecules (p38, Erk, JNK, and NF-κB) or IL-6, MCP-1, MMP-1, and
MMP-3 expression (present publication; (13). The incapability of RA-SFs to
secrete mature IL-33 may be based on an inability of the cells to
efficiently cleave pro-IL-33. Indeed, IL-1α, another member of the
IL-1 family sharing several properties with IL-33, is only cleaved
and secreted by monocytes and macrophage cell lines, but not by
fibroblast cell lines (28,29). In addition, only detergent-damaged
RA-SFs release the 30 kDa IL-33 precursor into the supernatant
(15) further supporting the view
that IL-33 is not actively secreted by RA-SFs.
Currently nothing is known about the signaling
pathways involved in the TNF-α-induced IL-33 synthesis in RA-SFs.
The present study shows for the first time that TNF-α-induced IL-33
mRNA expression is mainly regulated via p38, underlining its
central pro-inflammatory role and identifying IL-33 as a new
potential target for anti-rheumatic therapy with inhibitors of this
signaling pathway.
IL-33 synthesis is differentially induced by
cytokines and growth factors in RA-SFs. The pro-inflammatory
cytokines TNF-α and, in particular, IL-1β strongly induce IL-33
synthesis. In contrast, the pro-inflammatory cytokine IL-18 or the
growth factors PDGF-BB and TGF-β1 did not significantly stimulate
IL-33 synthesis. In addition, a nuclear localization of IL-33 was
only observed in IL-1β or TNF-α-stimulated cells, further
emphasizing the prominent role of these 2 cytokines in regulating
the expression/functional effects of IL-33. The induction of IL-33
by IL-1β and TNF-α also further underlines the central role of
these cytokines in the pathogenesis of RA for the
induction/regulation of disease-relevant molecules (30). Strong induction of IL-33 by IL-1β
is in good agreement with previously published data in CNS glial
cultures showing that IL-1β induces IL-33 more strongly than
pathogen-associated molecular patterns, e.g., dsRNA or LPS
(4). In contrast, superconfluent
HUVECs responded with a down-regulation of IL-33 protein following
IL-1β or TNF-α stimulation (5).
Therefore, the regulation of IL-33 by the pro-inflammatory
cytokines IL-1β and TNF-α seems to be cell type- and/or
pathway-specific.
This study shows for the first time that IL-33 is
involved in the regulation of TNF-α-induced functions in RA-SFs.
Overexpression of IL-33 enhanced the TNF-α-induced reduction of
proliferation in RA-SFs. Interestingly, the TNF-α-mediated
downregulation of proliferation is solely dependent on the p38
signal pathway in RA-SFs (25).
This is in good agreement with the regulation of TNF-α-induced
IL-33 synthesis via p38 (Fig. 2)
suggesting a downregulation of RA-SFs proliferation by TNF-α via a
p38-IL-33 axis. The TNF-α/IL-33-induced downregulation of RA-SFs
proliferation is in sharp contrast with previous reports suggesting
induction of SFs proliferation by TNF-α (31,32). This discrepancy may be explained
by the usage of differentially purified RA-SFs (anti-CD14-purified
cells in the present publication vs. cells purified by passaging)
and differential usage of serum-free or serum-containing
medium.
Overexpression of IL-33 further augmented
TNF-α-induced pro-inflammatory and pro-destructive functions in
RA-SFs. Although this result was generally confirmed by IL-33
silencing, TNF-α-induced pro-destructive mediators were less
strongly downregulated (MMP-1, MMP-3, TIMP-1). The difference in
the effects of IL-33 overexpression and silencing on the regulation
of TNF-α-induced pro-destructive functions may be based on the
involvement of additional signaling pathways, e.g. p38 and NFκB
(25,33). In fact, the inhibition/silencing
of one signaling molecule can increase the activity of other
signaling pathways influencing cell functions (34).
The present data indicate that IL-33 has an
enhancing effect on TNF-α-induced pro-inflammatory and
pro-destructive functions in RA-SFs. This is in apparent contrast
to the initially proposed function of IL-33 as a transcriptional
repressor (3). However, the
evidence for this function of IL-33 is predominantly based on in
vitro assays using reporter vectors exclusively driven by
multiple galactose 4 binding sites (3,35).
Also, there was no influence on the expression of selected genes by
IL-33 overexpression or silencing in HUVECs (5,35).
In addition, it has been proposed that IL-33 may function as a
potentiator of gene expression by decreasing the local
concentration of transcriptional repressors on specific promoters
and thereby allowing activators to bind more efficiently (3). Thus, the precise transcriptional
influence of IL-33 on the expression of individual genes in
specific cell types will have to be further analyzed.
In the present study, overexpression and silencing
of IL-33 influenced mRNA and protein expression of selected target
genes to a comparable degree. This indicates that in RA-SFs IL-33
exerts its enhancing influence at the transcriptional level, either
exclusively or in combination with other mechanisms. This is
supported by recent data showing that a short motif of IL-33 binds
with to the acidic pocket formed by the histone H2A-H2B dimer at
the surface of the nucleosome, a region important for chromatin
compaction and subsequent transcriptional activity (35) . However, the chromatin-binding
motif of IL-33 induced a higher order structure of chromatin and
mutations of the motif reduced its transcriptional repressor
properties. It therefore remains the subject of future studies how
the differential influence of IL-33 on gene transcription can be
mechanistically explained.
The regulation of pro-inflammatory mediators by
nuclear IL-33 is in good agreement with previously published data
using inflammatory cells. Stimulation of these cells with IL-33
induced the synthesis of several pro-inflammatory mediators, e.g.,
IL-6, IL-8, and MCP-1 (7,36,37). However, in contrast to RA-SFs,
these cells express ST2 and therefore responded to external IL-33
stimulation (37,38). Thus, pro-inflammatory mediators
may be enhanced by nuclear IL-33 in cells not expressing the IL-33
receptor ST2.
Interestingly, lentiviral IL-33 overexpression
enhanced IL-33 mRNA and protein expression only in TNF-α-stimulated
RA-SFs, pointing to a stabilization of IL-33 by TNF-α. A similar
effect has been reported for another member of the IL-1 family,
IL-1F7b (39). In agreement with
our observation for IL-33, IL-1F7b overexpressing RAW264.7 cells
showed high intracellular IL-1F7b level only after LPS stimulation.
Therefore, the mRNA of different members of the IL-1 family may be
stabilized by pro-inflammatory stimuli, resulting in an increased
protein synthesis.
The present study identifies IL-33 as a critical
regulator of TNF-α-induced pro-inflammatory and pro-destructive
functions in RA-SFs (likely at the transcriptional level) and
raises interesting questions concerning cell type- or gene-specific
effects and/or the exact molecular mechanism of gene
regulation.
Acknowledgements
B. Ukena (Experimental Rheumatology Unit, University
Hospital Jena, Germany) is gratefully acknowledged for technical
assistance and E. Palombo-Kinne, for critical revision of the
manuscript. The study was supported by the German Federal Ministry
of Education and Research [(BMBF; grants FKZ 01ZZ9602, 01ZZ0105,
and 010405 to R.W.K., Interdisciplinary Center for Clinical
Research (IZKF) Jena, including a grant for junior researchers to
E.K.; grants FKZ 0312704B and 0313652B to R.W.K., Jena Centre for
Bioinformatics and grant 01GS0413, NGFN-2 to R.W.K.), the German
Research Foundation (DFG; grants KI 439/7-1 and KI 439/6-1 to
R.W.K.), and a grant for the advancement of female scientists to
E.K. (LUBOM Thuringia)].
Abbreviations:
|
RA
|
rheumatoid arthritis
|
|
RA-SFs
|
RA synovial fibroblasts
|
|
OA
|
osteoarthritis
|
|
MCP-1
|
monocyte chemotactic protein-1
|
|
MMP
|
matrix metalloproteinase
|
References
|
1
|
J SchmitzA OwyangE OldhamIL-33, an
interleukin-1-like cytokine that signals via the IL-1
receptor-related protein ST2 and induces T helper type 2-associated
cytokinesImmunity23479490200510.1016/j.immuni.2005.09.01516286016
|
|
2
|
ES BaekkevoldM RoussigneT
YamanakaMolecular characterization of NF-HEV, a nuclear factor
preferentially expressed in human high endothelial venulesAm J
Pathol1636979200310.1016/S0002-9440(10)63631-012819012
|
|
3
|
V CarriereL RousselN OrtegaIL-33, the
IL-1-like cytokine ligand for ST2 receptor, is a
chromatin-associated nuclear factor in vivoProc Natl Acad Sci
USA104282287200710.1073/pnas.060685410417185418
|
|
4
|
CA HudsonGP ChristophiRC GruberJR
WilmoreDA LawrencePT MassaInduction of IL-33 expression and
activity in central nervous system gliaJ Leukoc
Biol84631643200810.1189/jlb.120783018552204
|
|
5
|
AM KuchlerJ PollheimerJ BaloghNuclear
interleukin-33 is generally expressed in resting endothelium but
rapidly lost upon angiogenic or proinflammatory activationAm J
Pathol17312291242200810.2353/ajpath.2008.08001418787100
|
|
6
|
M IikuraH SutoN KajiwaraIL-33 can promote
survival, adhesion and cytokine production in human mast cellsLab
Invest87971978200710.1038/labinvest.370066317700564
|
|
7
|
D MoulinO DonzeD Talabot-AyerF MezinG
PalmerC GabayInterleukin (IL)-33 induces the release of
pro-inflammatory mediators by mast
cellsCytokine40216225200710.1016/j.cyto.2007.09.01318023358
|
|
8
|
R KakkarRT LeeThe IL-33/ST2 pathway:
therapeutic target and novel biomarkerNat Rev Drug
Discov7827840200810.1038/nrd266018827826
|
|
9
|
C CayrolJP GirardThe IL-1-like cytokine
IL-33 is inactivated after maturation by caspase-1Proc Natl Acad
Sci USA10690219026200910.1073/pnas.081269010619439663
|
|
10
|
C MoussionN OrtegaJP GirardThe IL-1-like
cytokine IL-33 is constitutively expressed in the nucleus of
endothelial cells and epithelial cells in vivo: a novel
‘alarmin’?PLoS One3e3331200818836528
|
|
11
|
BP LeungD XuS CulshawIB McInnesFY LiewA
novel therapy of murine collagen-induced arthritis with soluble
T1/ST2J Immunol173145150200410.4049/jimmunol.173.1.14515210768
|
|
12
|
D XuHR JiangP KewinIL-33 exacerbates
antigen-induced arthritis by activating mast cellsProc Natl Acad
Sci USA1051091310918200810.1073/pnas.080189810518667700
|
|
13
|
G PalmerD Talabot-AyerC
LamacchiaInhibition of interleukin-33 signaling attenuates the
severity of experimental arthritisArthritis
Rheum60738749200910.1002/art.2430519248109
|
|
14
|
WA Verri JrFO SoutoSM VieiraIL-33 induces
neutrophil migration in rheumatoid arthritis and is a target of
anti-TNF therapyAnn Rheum
Dis6916971703201010.1136/ard.2009.12265520472598
|
|
15
|
Y MatsuyamaH OkazakiH TamemotoIncreased
levels of interleukin 33 in sera and synovial fluid from patients
with active rheumatoid arthritisJ
Rheumatol371825201010.3899/jrheum.09049219918048
|
|
16
|
S KaiedaK ShinPA NigrovicSynovial
fibroblasts promote the expression and granule accumulation of
tryptase via interleukin-33 and its receptor ST-2 (IL1RL1)J Biol
Chem2852147821486201010.1074/jbc.M110.11499120427273
|
|
17
|
G PalmerS TrollietD Talabot-AyerF MezinD
MagneC GabayPre-interleukin-1alpha expression reduces cell growth
and increases interleukin-6 production in SaOS-2 osteosarcoma
cells: differential inhibitory effect of interleukin-1 receptor
antagonist
(icIL-1Ra1)Cytokine31153160200510.1016/j.cyto.2005.03.007
|
|
18
|
S SharmaN KulkMF NoldThe IL-1 family
member 7b translocates to the nucleus and down-regulates
proinflammatory cytokinesJ
Immunol18054775482200810.4049/jimmunol.180.8.547718390730
|
|
19
|
FC ArnettSM EdworthyDA BlochThe American
Rheumatism Association 1987 revised criteria for the classification
of rheumatoid arthritisArthritis
Rheum31315324198810.1002/art.17803103023358796
|
|
20
|
T ZimmermannE KunischR PfeifferIsolation
and characterization of rheumatoid arthritis synovial fibroblasts
from primary culture - primary culture cells markedly differ from
fourth-passage cellsArthritis Res37276200110.1186/ar142
|
|
21
|
A HirthA SkapenkoRW KinneF EmmrichH
Schulze-KoopsU SackCytokine mRNA and protein expression in
primary-culture and repeated-passage synovial fibroblasts from
patients with rheumatoid arthritisArthritis
Res4117125200210.1186/ar39111879547
|
|
22
|
N SwaroopF ChenL WangS DokkaD ToledoY
RojanasakulInhibition of nuclear transcription factor-kappaB by
specific IkappaB kinase peptide inhibitorPharm
Res1816311633200110.1023/A:101305101909811758774
|
|
23
|
DB GlassHC ChengL Mende-MuellerJ ReedDA
WalshPrimary structural determinants essential for potent
inhibition of cAMP-dependent protein kinase by inhibitory peptides
corresponding to the active portion of the heat-stable inhibitor
proteinJ Biol Chem264880288101989
|
|
24
|
S AlsalamehRJ AminE KunischHE JasinRW
KinnePreferential induction of prodestructive matrix
metalloproteinase-1 and proinflammatory interleukin 6 and
prostaglandin E2 in rheumatoid arthritis synovial fibroblasts via
tumor necrosis factor receptor-55J Rheumatol30168016902003
|
|
25
|
E KunischM GandesiriR FuhrmannA RothR
WinterRW KinnePredominant activation of MAP kinases and
pro-destructive/pro-inflammatory features by TNF alpha in
early-passage synovial fibroblasts via TNF receptor-1: failure of
p38 inhibition to suppress matrix metalloproteinase-1 in rheumatoid
arthritisAnn Rheum Dis6610431051200710.1136/ard.2006.062521
|
|
26
|
D PohlersA BeyerD KoczanT WilhelmHJ
ThiesenRW KinneConstitutive upregulation of the transforming growth
factor-beta pathway in rheumatoid arthritis synovial
fibroblastsArthritis Res Ther9R59200710.1186/ar221717594488
|
|
27
|
D PretzelD PohlersS WeinertRW KinneIn
vitro model for the analysis of synovial fibroblast-mediated
degradation of intact cartilageArthritis Res
Ther11R25200910.1186/ar261819226472
|
|
28
|
LM CarruthS DemczukSB MizelInvolvement of
a calpain-like protease in the processing of the murine interleukin
1 alpha precursorJ Biol Chem266121621216719912061304
|
|
29
|
WM SidersJC KlimovitzSB
MizelCharacterization of the structural requirements and cell type
specificity of IL-1 alpha and IL-1 beta secretionJ Biol
Chem268221702217419938408078
|
|
30
|
FM BrennanIB McInnesEvidence that
cytokines play a role in rheumatoid arthritisJ Clin
Invest11835373545200810.1172/JCI3638918982160
|
|
31
|
B GrimbacherWK AicherHH PeterH
EibelTNF-alpha induces the transcription factor Egr-1,
pro-inflammatory cytokines and cell proliferation in human skin
fibroblasts and synovial lining cellsRheumatol
Int17185192199810.1007/s0029600500329542779
|
|
32
|
H KitasatoM NodaT AkahoshiActivated Ras
modifies the proliferative response of rheumatoid synovial cells to
TNF-alpha and TGF-alphaInflamm
Res50592597200110.1007/PL0000023911822784
|
|
33
|
K AupperleB BennettZ HanD BoyleA ManningG
FiresteinNF-kappa B regulation by I kappa B kinase-2 in rheumatoid
arthritis synoviocytesJ
Immunol16627052711200110.4049/jimmunol.166.4.270511160335
|
|
34
|
Y TakadaH IchikawaA PataerS SwisherBB
AggarwalGenetic deletion of PKR abrogates TNF-induced activation of
IkappaBalpha kinase, JNK, Akt and cell proliferation but
potentiates p44/p42 MAPK and p38 MAPK
activationOncogene2612011212200710.1038/sj.onc.120990616924232
|
|
35
|
L RousselM ErardC CayrolJP GirardMolecular
mimicry between IL-33 and KSHV for attachment to chromatin through
the H2A-H2B acidic pocketEMBO
Rep910061012200810.1038/embor.2008.14518688256
|
|
36
|
M Funakoshi-TagoK TagoM HayakawaTRAF6 is a
critical signal transducer in IL-33 signaling pathwayCell
Signal2016791686200810.1016/j.cellsig.2008.05.01318603409
|
|
37
|
T Pecaric-PetkovicSA DidichenkoS KaempferN
SpieglCA DahindenHuman basophils and eosinophils are the direct
target leukocytes of the novel IL-1 family member
IL-33Blood11315261534200910.1182/blood-2008-05-15781818955562
|
|
38
|
M SuzukawaM IikuraR KoketsuAn IL-1
cytokine member, IL-33, induces human basophil activation via its
ST2 receptorJ
Immunol18159815989200810.4049/jimmunol.181.9.598118941187
|
|
39
|
P BuflerF Gamboni-RobertsonT AzamSH KimCA
DinarelloInterleukin-1 homologues IL-1F7b and IL-18 contain
functional mRNA instability elements within the coding region
responsive to lipopolysaccharideBiochem
J381503510200410.1042/BJ2004021715046617
|