Introduction
Reactive oxygen species (ROS) include hydrogen
peroxide, nitric oxide, superoxide anions, hydroxyl radicals and
peroxynitrite. A growing body of evidence suggests that ROS within
cells act as second messengers in the regulation of many important
cellular events, including transcription factor activation, gene
expression and cellular proliferation, differentiation and
senescence (1,2). An excessive production of ROS
frequently leads to serious damage in cells, while mild increases
in ROS can induce cellular senescence (3,4).
ROS can therefore function as antitumorigenic agents (5–7).
Many anticancer therapies, such as the chemotherapeutic agent
doxorubicin, etoposide and cisplatin, in addition to radiotherapy
and photodynamic therapy, can lead to the generation of superoxide
and oxygen radicals within carcinoma cells (8,9).
In this sense, the modulation of oxidative stress could be a target
of anticancer therapies.
A variety of proteins function as ROS scavengers,
such as superoxide dismutase, catalase, glutathione peroxidase,
thioredoxin reductase (TrxR), ascorbate, glutathione. Among them,
TrxR has been suggested as a new target for anticancer drug
development because TrxR is overexpressed in colon, pancreas, lung
and other cancers (10–12) and knockdown of TrxR could
dramatically reduce tumor growth and metastasis in a lung carcinoma
mouse model (13). Developing the
novel TrxR targeting compound is promising in the treatment of
cancer (14).
Oridonin is an ent-kaurane diterpenoid derived from
Rabdosia rubescens (15).
Studies have shown that oridonin induces apoptosis in a variety of
cancer cells including cells from prostate cancer, breast cancer,
non-small cell lung cancer, acute leukemia, glioblastoma multiforme
and melanoma (16–19). In addition, oridonin can also
inhibit cell cycle progression and enhance phagocytosis of
apoptotic cells by macrophages (20). However, the mechanisms underlying
the antitumor activity of oridonin are not fully understood.
Interestingly, exposure of acute promyelocytic leukemia NB4 cells
to oridonin resulted in a significant increase in ROS generation
while the ROS scavenger, N-acetylcysteine (NAC), completely
protected NB4 cells from oridonin-induced apoptosis (16). These findings indicate that ROS
signaling is involved in oridonin-induced apoptosis. Recently, we
demonstrated that oridonin could induce potent growth inhibition,
apoptosis, and senescence of colorectal cancer cells in
vitro and in vivo (21). However, the exact mechanism of
this process remains largely unknown.
In the present study, the role of ROS and
thioredoxin reductase in oridonin-induced cell death and senescence
in human colorectal cancer (SW1116) cells were investigated.
Materials and methods
Cell culture
The colorectal cancer cell line SW1116 was purchased
from the Shanghai Institutes for Biological Sciences. The cells
were maintained in a humidified room air containing 5%
CO2 at 37°C and cultured in DMEM medium (Gibco-BRL)
supplemented with 10% fetal bovine serum and 1%
penicillin-streptomycin (Gibco-BRL). Cells in the logarithmic phase
of growth were used in all experiments.
Reagents
Oridonin (98% purity) provided by Dr Tang Qingjiu
(Shanghai Academy of Agricultural Sciences) was dissolved in DMSO
(Sigma, St. Louis, MO) at a stock concentration of 10 mg/ml and
stored at −20°C. The cell-permeable ROS scavenger NAC was obtained
from Sigma and dissolved in sterile H2O to a stock
concentration of 100 mM. Catalase was obtained from Sigma and
dissolved in 50 mM potassium phosphate buffer at 4,733 U/ml. All
stock solutions were wrapped in foil and maintained at 4°C or
−20°C.
Detection and measurement of
intracellular hydrogen peroxide and superoxide anion
concentrations
Two oxidation-sensitive fluorescent probe dyes,
2′,7′-dichlorodihydrofluorescein diacetate (DCF-DA, Invitrogen,
Molecular Probes, Eugene, OR) and dihydroethidium (DHE, Invitrogen
Molecular Probes), were used to measure the intracellular hydrogen
peroxide and superoxide anion concentrations, respectively. DCF-DA
is deacetylated intracellularly by nonspecific esterases and is
further oxidized by cellular peroxides to the fluorescent compound
2′,7′-dichlorofluorescein. DHE is a fluorogenic probe that detects
superoxide anion radicals with high selectivity. DHE is
cell-permeable and reacts with superoxide anions to form ethidium,
which in turn intercalates deoxyribonucleic acid and exhibits a red
fluorescence. Briefly, cells were treated with oridonin in the
presence or absence of NAC or catalase for the indicated time
periods. After washing with phosphate-buffered saline (PBS), cells
were incubated with 20 μM DCF-DA or 5 μM DHE at 37°C for 30 min
according to the manufacturer’s instructions. The fluorescence
signals were detected by a FACStar flow cytometer (Beckman
Coulter). For each sample, 5,000 or 10,000 events were collected.
Hydrogen peroxide and superoxide anion levels were expressed in
terms of mean fluorescence intensity.
Detection of intracellular glutathione
(GSH)
Cellular GSH levels were analyzed using
5-chloromethylfluorescein diacetate (CMFDA, Invitrogen, Molecular
Probes). Cytoplasmic esterases convert nonfluorescent CMFDA to
fluorescent 5-chloromethylfluorescein, which can then react with
the glutathione. CMFDA is a useful membrane-permeable dye for
determining levels of intracellular glutathione (22–25). Briefly, cells were treated with
oridonin in the presence or absence of ROS scavengers, or catalase
for the indicated time periods. After washing with PBS, the cells
were incubated with 5 μM CMFDA at 37°C for 30 min according to the
manufacturer’s instructions. CMF fluorescence was detected by the
FACStar flow cytometer (Beckman Coulter). For each sample, 5,000 or
10,000 events were collected.
Annexin V/PI staining
Apoptosis was determined using Annexin V-fluorescein
isothiocyanate (FITC) staining and PI labeling. Annexin V can
identify the externalization of phosphatidylserine during apoptotic
progression and therefore can detect cells in early apoptosis.
Briefly, cells were treated with oridonin in the presence or
absence of NAC or catalase for the indicated time periods. After
washing twice with cold PBS, cells were resuspended in 500 μl of
binding buffer [10 mM HEPES/NaOH (pH 7.4), 140 mM NaCl, 2.5 mM
CaCl2] at a concentration of 1×106 cells/ml.
Next, 5 μl of Annexin V-FITC (Pharmingen, San Diego, CA) and 10 μl
of 20 μg/ml PI were added to these cells, which were analyzed with
a FACStar flow cytometer (Beckman Coulter). Viable cells were
negative for both PI and Annexin V, apoptotic cells were positive
for Annexin V and negative for PI, and late apoptotic dead cells
displayed both high Annexin V and PI labeling. Non-viable cells
that had undergone necrosis were positive for PI and negative for
Annexin V.
Cell senescence assay
Senescence-associated β-galactosidase activity was
determined with a senescence detection kit (Biovision, Mountain
View, CA) using fixed cells (26). The development of a blue color in
the cytoplasm was detected and photographed using a Nikon (Nikon
Instruments Inc., Lewisville, TX) inverted microscope equipped with
a color CCD camera. Four pictures were taken of each well.
β-galactosidase-stained cells and unstained cells were counted and
used to calculate a final average ratio of the number of stained to
unstained cells in each well.
Western blot analysis
Cells were incubated with oridonin and/or NAC for
the indicated time periods. Cells were then washed in PBS and
suspended in five volumes of lysis buffer [20 mM HEPES (pH 7.9),
20% glycerol, 200 mM KCl, 0.5 mM EDTA, 0.5% NP-40, 0.5 mM DTT, 1%
protease inhibitor cocktail (Sigma)]. The lysates were then
collected and stored at −20°C. Protein concentrations in the
supernatants were determined by the Bradford method. Supernatant
samples containing 20 μg of total protein were resolved on 10–15%
SDS-PAGE gels and transferred onto Immobilon-P PVDF membranes
(Millipore, MA) by electroblotting; membranes were then probed with
anti-PARP, p16, c-Myc and p53 (Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA) primary antibodies and subsequently with
horseradish peroxidase-conjugated secondary antibodies. The
membrane blots were developed using the ECL kit (Amersham,
Arlington Heights, IL).
Determination of TrxR activity by the
dithionitrobenzene reduction assay
All experiments were performed in 96-well plates.
Recombinant active rat TrxR (0.05 units) was incubated with various
concentrations of oridonin for 60 min. TrxR activity was assayed by
the dithionitrobenzene method in a solution containing 50 mM
Tris-HCl (pH 7.5), 200 μM NADPH, 5 mM DTNB, and 1 mM EDTA. The
absorbance at 412 nm was measured with a Synergy H4 Hybrid
Microplate Reader (Bio-Tek, Winooski, VT). A blank reading without
TrxR was subtracted from every sample. TrxR enzyme activity was
calculated as a percentage of the activity of the DMSO
vehicle-treated sample.
Determination of TrxR activity in cell
lysates
The activity of TrxR in cell lysates was measured
using the TrxR assay kit (Cayman Chemical, Ann Arbor, MI) according
to the manufacturer’s instructions. Briefly, cells were collected
by scraping and were homogenized on ice in cold buffer (50 mM
potassium phosphate and 1 mM EDTA, pH 7.4). The protein
concentration in the supernatant of the lysate was determined using
the Bradford method. Protein samples, NADPH, and DTNB were added to
the assay buffer in 96-well plates and gently mixed. The absorbance
at 412 nm was measured with a Synergy H4 Hybrid Microplate Reader.
A blank reading without protein was subtracted from every sample.
TrxR enzyme activity was calculated as a percentage of the activity
of the DMSO vehicle-treated control sample.
Statistical analysis
The Student’s t-test was used to evaluate the
differences between two different groups. A P-value <0.05 was
considered statistically significant.
Results
Oridonin increases hydrogen peroxide and
decreases superoxide anion and glutathione levels in SW1116
cells
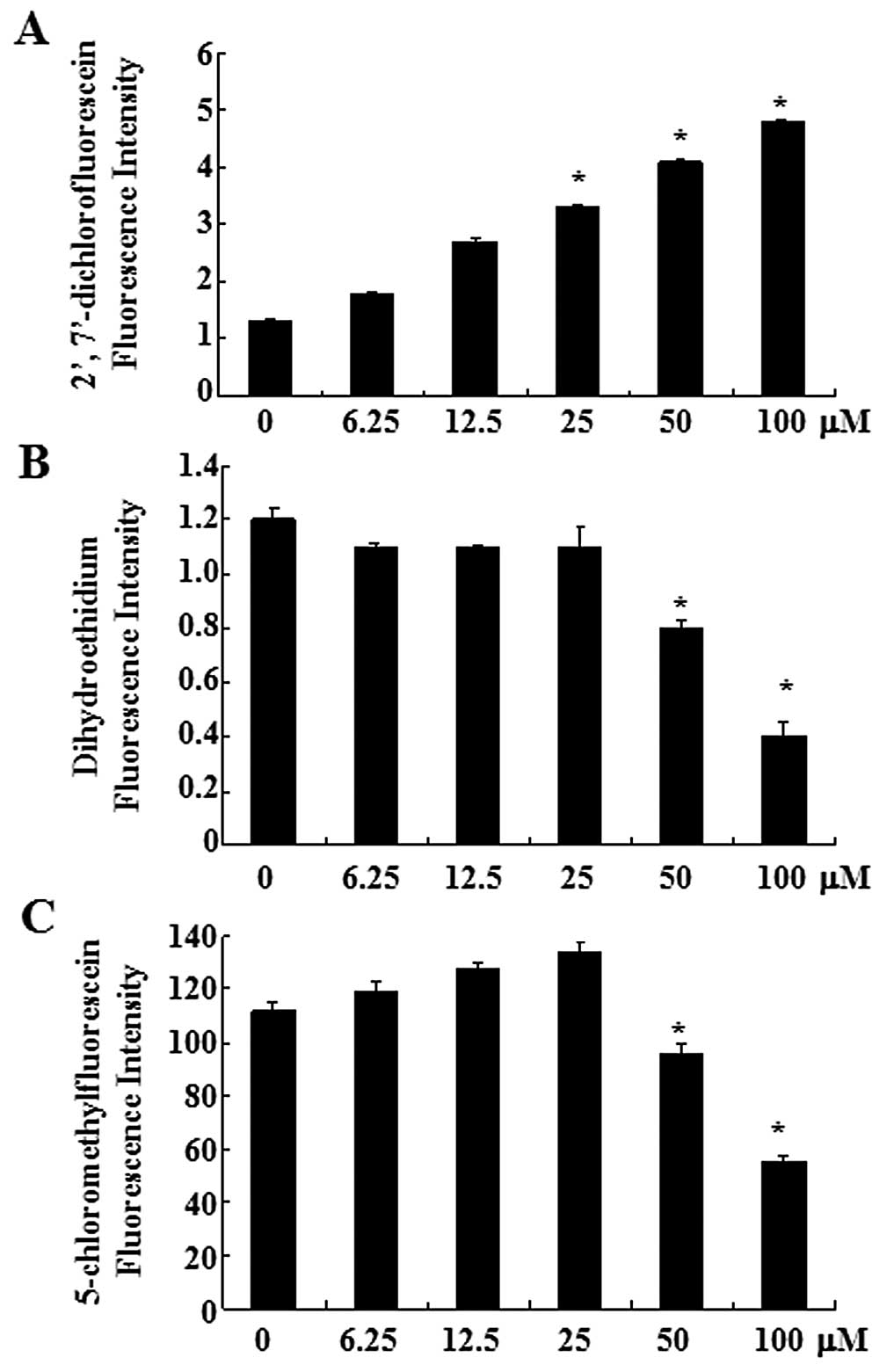
First, the production of intracellular hydrogen
peroxide was assessed in oridonin-treated SW1116 cells using the
DCF-DA fluorescence dye. As shown in Fig. 1A, oridonin increased the
intracellular hydrogen peroxide level in a dose-dependent manner.
Treatment with oridonin at 6.25 μM for 2 h increased the
intracellular hydrogen peroxide levels, compared with those in the
untreated control cells. The intracellular superoxide anion level
in oridonin-treated SW1116 cells was assessed using the DHE
fluorescence dye. Red fluorescence derived from DHE, which reflects
superoxide anion accumulation, decreased significantly in SW1116
cells treated with 50 and 100 μM oridonin, compared with the
untreated control cells (Fig.
1B).
As an important component of the antioxidant system,
cellular GSH has been shown to be crucial for the regulation of
cell proliferation, cell cycle progression, apoptosis and
senescence. We therefore analyzed the changes of GSH level in
oridonin-treated SW1116 cells by using the CMF fluorescence probe.
Oridonin at 50 or 100 μM significantly decreased the level of
intracellular GSH content at 2 h (Fig. 1C), indicating the depletion of
intracellular GSH content of SW1116 cells. The decrease of CMF
fluorescence was observed within 120 min at the exposure to
oridonin (50 and 100 μM) (Fig.
1C). However, at a lower concentration (6.25–25 μM), a slight
increase of the CMF fluorescence was detected at 2 h.
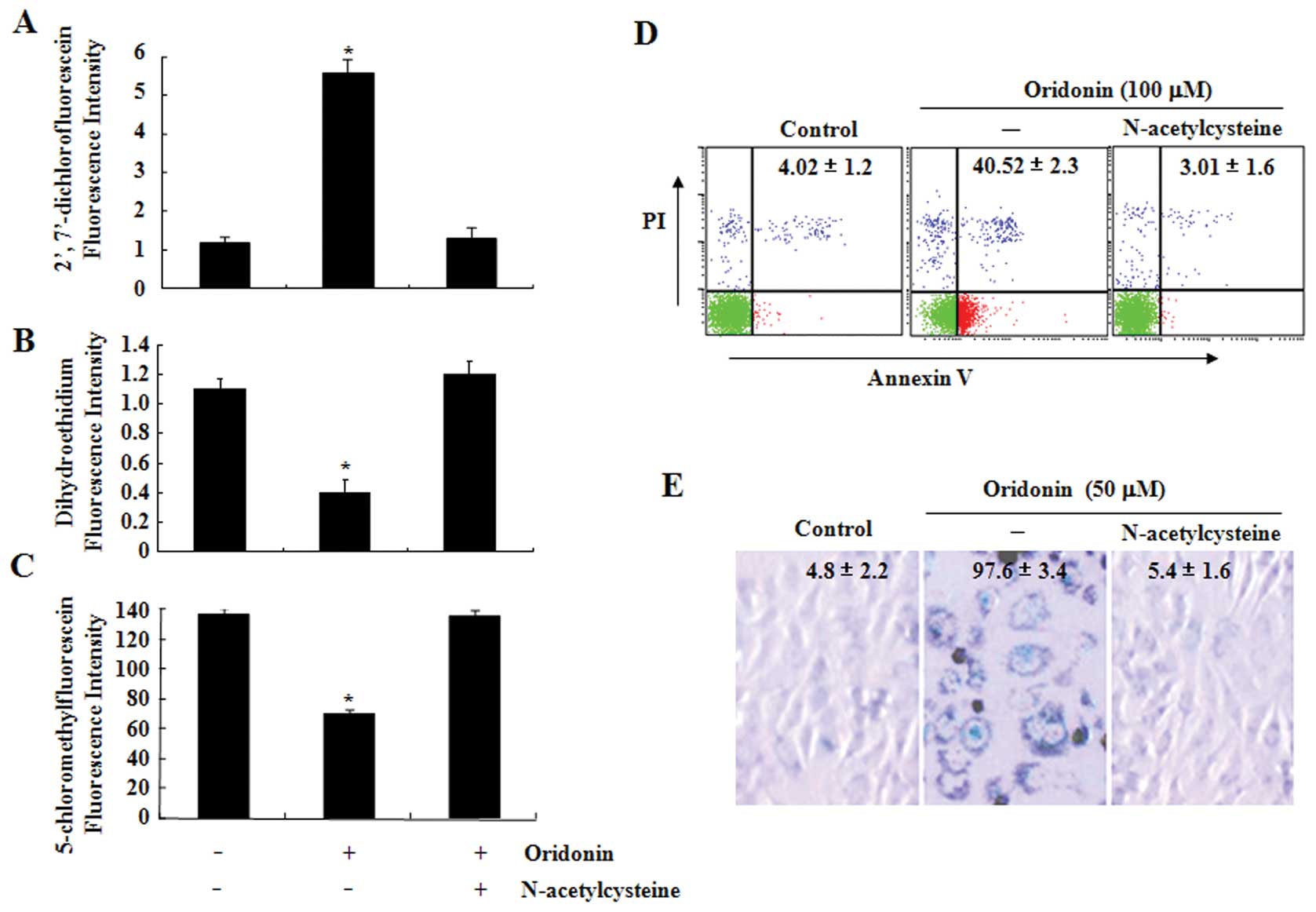
The ROS scavenger NAC protects SW1116
cells from oridonin-induced apoptosis or senescence
To determine the role of ROS production in
oridonin-induced apoptosis and senescence, SW1116 cells were
treated with oridonin in the presence or absence of NAC, a well
known ROS scavenger, for 2 h. As expected, NAC fully reversed the
oridonin-induced increase in hydrogen peroxide as well as the
decrease in superoxide anion and GSH levels (Fig. 2A–C). Next, effects of NAC on
oridonin-induced cell death and senescence in SW1116 cells were
also examined. As shown in Fig. 2D
and E, co-treatment with NAC and oridonin completely protected
the cells from oridonin-induced cell death and senescence.
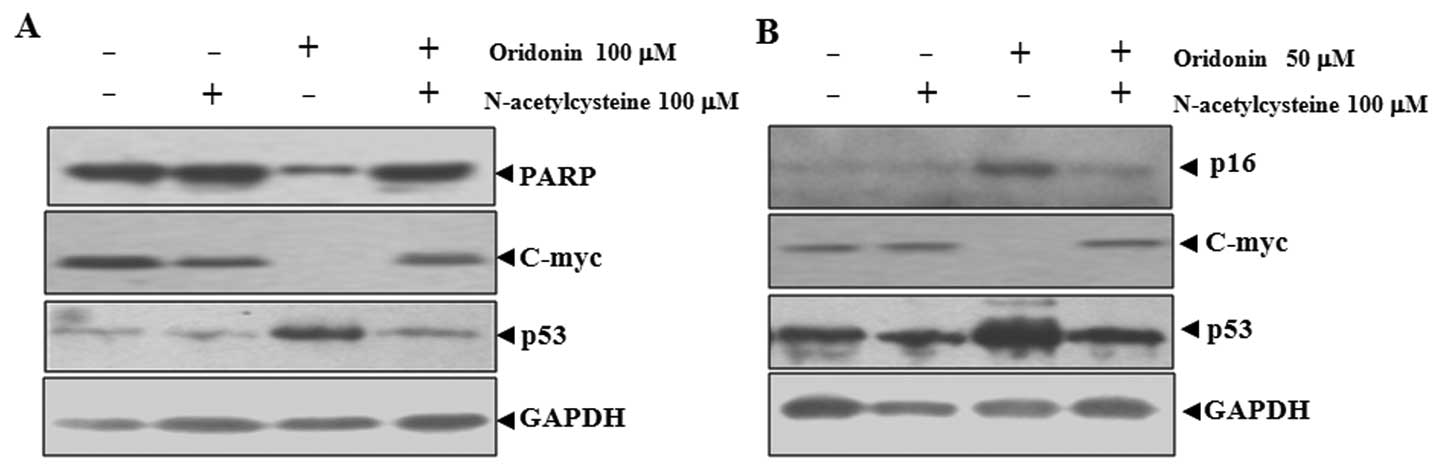
NAC abrogates oridonin-induced changes in
poly(ADP-ribose) polymerase (PARP), p16, p53 and c-Myc in SW1116
cells
Oridonin-induced apoptosis and senescence were
related to the upregulation of p53 and p16 and downregulation of
c-Myc (21). Because NAC can
inhibit oridonin-induced apoptosis and senescence, the effect of
NAC on the expression of these proteins was examined. As shown in
Fig. 3A, the application of
oridonin at 100 μM mainly led to apoptosis in SW1116 cells, as
indicated by the degradation of PARP, which is a major substrate
for caspases and a marker for apoptosis (27). Co-treatment with NAC inhibited the
degradation of PARP. At 50 μM, oridonin mainly induced senescence
in SW1116 cells, as reflected by the induction of p16, a hallmark
of both replicative and accelerated senescence (28). In this case, co-treatment with NAC
inhibited the induction of p16 (Fig.
3B).
The inactivation of c-Myc and the activation of p53
are important for both apoptosis and senescence (29–32). NAC co-treatment abrogated
oridonin-induced downregualtion of c-Myc and upregulation of p53 in
parallel with the inhibition of apoptosis and senescence (Fig. 3). These results indicated that
c-Myc and p53 are involved in oridonin-induced senescence and
apoptosis.
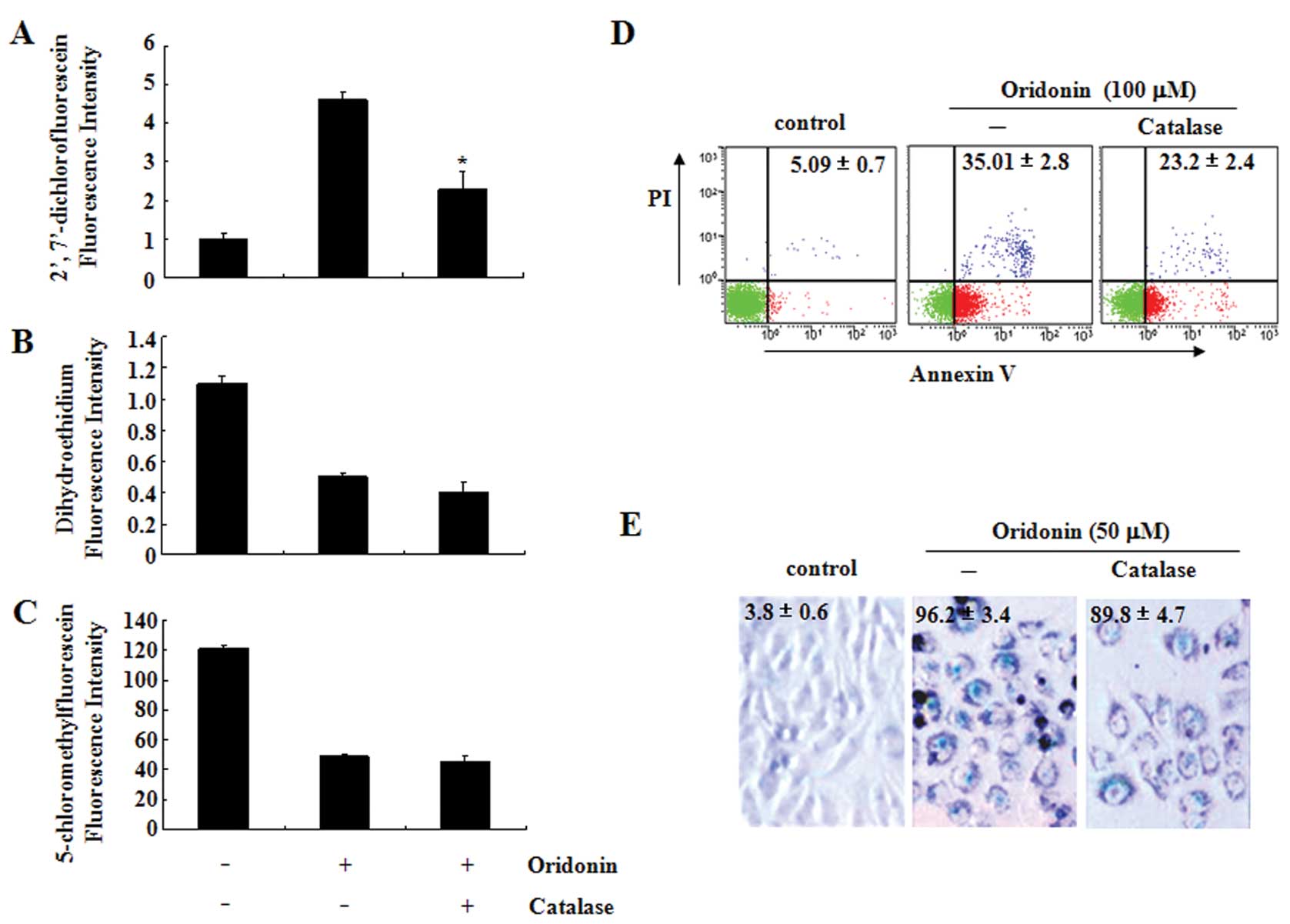
Effects of catalase on ROS production,
apoptosis and senescence in oridonin-treated SW1116 cells
To further determine the role of hydrogen peroxide
in oridonin-induced apoptosis and senescence, SW1116 cells were
treated with oridonin in the presence or absence of catalase (30
U/ml), a hydrogen peroxide-scavenging enzyme (33). Catalase partially reversed an
oridonin-induced increase in hydrogen peroxide (Fig. 4A) but not the decrease of
superoxide anion and GSH (Fig. 4B and
C). Interestingly, catalase significantly decreased the
percentage of Annexin V-positive cells (Fig. 4D) but not the
senescence-associated β-galactosidase-positive cells induced by
oridonin (Fig. 4E).
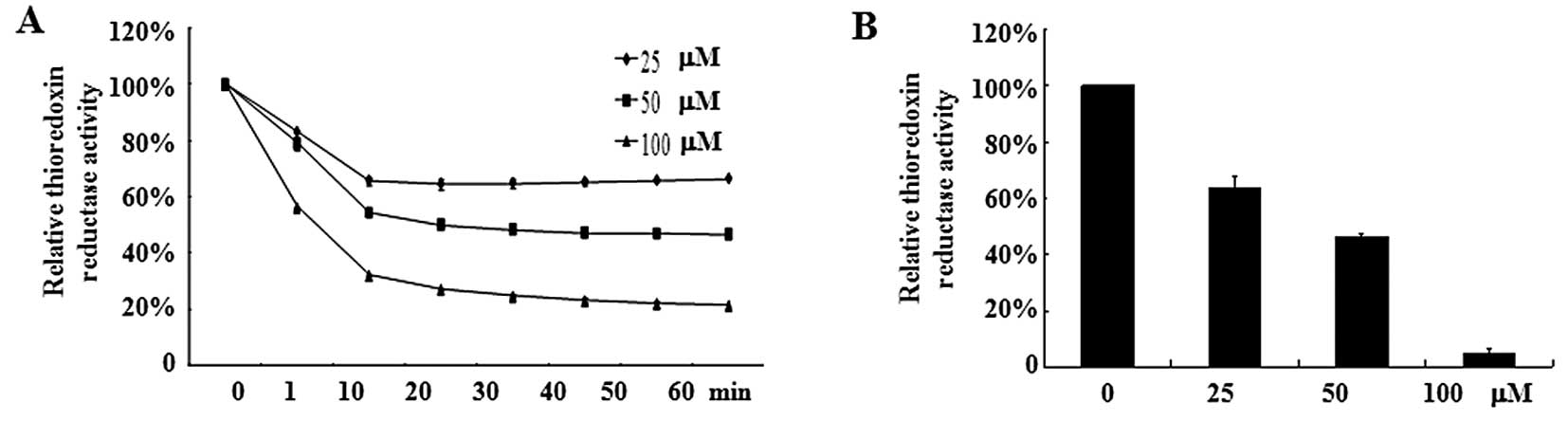
Oridonin inhibits TrxR activity
TrxR, an oxidoreductase that reduces oxidized
proteins, represents an important valid target for cancer therapy.
Because TrxR is easily targeted by electrophilic compounds and
oridonin is a Michael acceptor with electrophilic propensity, we
assumed that oridonin may directly inhibit TrxR activity, which
causes an increase of hydrogen peroxide. As shown in Fig. 5A, oridonin caused a dose-dependent
reduction of TrxR activity in vitro. Incubation of TrxR with
100 μM oridonin led to an 80% decrease in TrxR activity compared
with DMSO treatment. In line with this result, SW1116 cells treated
with oridonin at 50 or 100 μM for 2 h exhibited a marked decrease
in TrxR activity (Fig. 5B). These
results indicate that TrxR is one of the targets for oridonin.
Discussion
We previously reported that oridonin induces potent
growth inhibition, apoptosis, and senescence of colorectal cancer
cells in vitro and in vivo (21). However, the underlying mechanism
is largely unknown. In this study, we demonstrated that the
antitumor activity of oridonin was attributed to its ROS increasing
activity. Furthermore, TrxR inhibition is involved in this
process.
ROS serve many cellular functions including actins
as growth stimulants, second messengers, aging accelerants, cell
death inducers and antibacterial agents. Based on our previous
results and those of others (16,34), we hypothesized that oridonin may
induce apoptosis and senescence in colon cancer cells via
increasing ROS. Although it is currently not possible to precisely
quantify the amount of subcellular ROS, tools to detect several
kinds of ROS in the cells are available. With these chemical
probes, we found that exposure to oridonin led to the increase of
hydrogen peroxide and decrease of superoxide anions and GSH in
SW116 cells and that the effects of oridonin on ROS level were
dose-dependent. In the cellular environment, hydrogen peroxide can
be derived from superoxide anions; this derivation can either occur
spontaneously or can be catalyzed by superoxide dismutase (35). Whether oridonin could enhance the
conversion of superoxide anions to hydrogen peroxide or inhibit the
production of superoxide anions is currently unknown.
Many studies have demonstrated that hydrogen
peroxide plays an important role in chemotherapy-induced apoptosis
and senescence in cells (6–9).
At low or mild concentrations, ROS seem to protect the cells or
induce cell senescence, while at high concentration, ROS can
initiate cell death by damaging many biological molecules. To
reveal the role of hydrogen peroxide in oridonin-induced apoptosis,
NAC, an ROS scavenger, were used in combination with oridonin. NAC
completely abrogated the oridonin-induced increases of hydrogen
peroxide levels and decreases of superoxide anion levels.
Accordingly, oridonin induced apoptosis and senescence and the
relevant changes of p53, p16 and c-Myc were also abrogated. These
data indicate that hydrogen peroxide plays a key role in
oridonin-induced apoptosis and senescence. Interestingly, when the
levels of hydrogen peroxide were partially decreased by catalase,
only the apoptosis but not the senescence was significantly
reversed, indicating that the concentration of hydrogen peroxide
determined the cell fate. These data suggest that hydrogen peroxide
may play a major role in oridonin-induced apoptosis and senescence,
although we cannot rule out tht other ROS, such as nitric oxide, or
hydroxyl radicals may also be involved.
The mammalian thioredoxin system, which consists of
NADPH, thioredoxin and thioredoxin reductase, plays an important
role in protecting cells from oxidative stress-induced apoptosis
(36). As overexpression of TrxR
has been reported in several human cancers and is associated with
decreased survival and resistance to chemotherapy, TrxR has been
proposed as a target for anticancer drug development. Considering
the extensive involvement of TrxR in the redox regulations and its
close link with cancer and that it is easily attacked by
electrophilic compound, we investigated the effect of oridonin on
TrxR activity. Interestingly, oridonin dose-dependently inhibited
the activity of TrxR in vitro and in cells. The trend was
correlated to that of oridonin-induced hydrogen peroxide increase.
Thus, oridonin may induce the accumulation of hydrogen peroxide by
targeting TrxR.
In summary, we show here that hydrogen peroxide
plays an important role in oridonin-induced apoptosis and
senescence in colon cancer cells. We also provided evidence for the
first time that oridonin could inhibit TrxR activity.
Acknowledgements
This study was supported by the National Basic
Research Program of China (no. 2010CB912104), the National Natural
Science Foundation of China (nos. 81070433, 91013008 and 81172322),
the Science and Technology Committee of Baoshan District (08-E-13),
and the Number 3 People’s Hospital affiliated with the Shanghai
Jiao-Tong University School of Medicine (syz07-04).
References
|
1
|
C GonzalezG Sanz-AlfayateMT AgapitoA
Gomez-NinoA RocherA ObesoSignificance of ROS in oxygen sensing in
cell systems with sensitivity to physiological hypoxiaRespir
Physiol
Neurobiol1321741200210.1016/S1569-9048(02)00047-212126693
|
|
2
|
CP BaranMM ZeiglerS TridandapaniCB
MarshThe role of ROS and RNS in regulating life and death of blood
monocytesCurr Pharm
Des10855866200410.2174/138161204345286615032689
|
|
3
|
J YamadaS YoshimuraH YamakawaCell
permeable ROS scavengers, Tiron and Tempol, rescue PC12 cell death
caused by pyrogallol or hypoxia/reoxygenationNeurosci
Res4518200310.1016/S0168-0102(02)00196-712507718
|
|
4
|
A TakahashiN OhtaniK YamakoshiMitogenic
signalling and the p16INK4a-Rb pathway cooperate to enforce
irreversible cellular senescenceNat Cell
Biol812911297200610.1038/ncb149117028578
|
|
5
|
S MenaA OrtegaJM EstrelaOxidative stress
in environmental-induced carcinogenesisMutat
Res6743644200910.1016/j.mrgentox.2008.09.01718977455
|
|
6
|
AT ShawMM WinslowM MagendantzSelective
killing of K-ras mutant cancer cells by small molecule inducers of
oxidative stressProc Natl Acad Sci
USA10887738778201110.1073/pnas.110594110821555567
|
|
7
|
L RajT IdeAU GurkarSelective killing of
cancer cells by a small molecule targeting the stress response to
ROSNature475231234201110.1038/nature1016721753854
|
|
8
|
RH EngelAM EvensOxidative stress and
apoptosis: a new treatment paradigm in cancerFront
Biosci11300312200610.2741/179816146732
|
|
9
|
P StorzReactive oxygen species in tumor
progressionFront Biosci1018811896200510.2741/166715769673
|
|
10
|
Y SunB RigasThe thioredoxin system
mediates redox-induced cell death in human colon cancer cells:
implications for the mechanism of action of anticancer agentsCancer
Res6882698277200810.1158/0008-5472.CAN-08-201018922898
|
|
11
|
H HanDJ BearssLW BrowneR CalaluceRB
NagleDD Von HoffIdentification of differentially expressed genes in
pancreatic cancer cells using cDNA microarrayCancer
Res6228902896200212019169
|
|
12
|
Y SoiniK KahlosU NapankangasWidespread
expression of thioredoxin and thioredoxin reductase in non-small
cell lung carcinomaClin Cancer Res717501757200111410516
|
|
13
|
MH YooXM XuBA CarlsonVN GladyshevDL
HatfieldThioredoxin reductase 1 deficiency reverses tumor phenotype
and tumorigenicity of lung carcinoma cellsJ Biol
Chem2811300513008200610.1074/jbc.C60001220016565519
|
|
14
|
A MukherjeeSG MartinThe thioredoxin
system: a key target in tumour and endothelial cellsBr J
Radiol81S57S68200810.1259/bjr/3418043518819999
|
|
15
|
KK RenHZ WangLP XieThe effects of oridonin
on cell growth, cell cycle, cell migration and differentiation in
melanoma cellsJ
Ethnopharmacol103176180200610.1016/j.jep.2005.07.02016169170
|
|
16
|
F GaoQ TangP YangY FangW LiY WuApoptosis
inducing and differentiation enhancement effect of oridonin on the
all-trans-retinoic acid-sensitive and -resistant acute
promyelocytic leukemia cellsInt J Lab
Hematol32e114e122201010.1111/j.1751-553X.2009.01147.x
|
|
17
|
GB ZhouH KangL WangOridonin, a diterpenoid
extracted from medicinal herbs, targets AML1-ETO fusion protein and
shows potent antitumor activity with low adverse effects on t(8;21)
leukemia in vitro and in
vivoBlood10934413450200710.1182/blood-2006-06-03225017197433
|
|
18
|
T IkezoeSS ChenXJ TongD HeberH TaguchiHP
KoefflerOridonin induces growth inhibition and apoptosis of a
variety of human cancer cellsInt J Oncol2311871193200312964003
|
|
19
|
LS MarksRS DiPaolaP NelsonPC-SPES: herbal
formulation for prostate
cancerUrology60369377200210.1016/S0090-4295(02)01913-112350462
|
|
20
|
TC HsiehEK WijeratneJY LiangAL
GunatilakaJM WuDifferential control of growth, cell cycle
progression, and expression of NF-kappaB in human breast cancer
cells MCF-7, MCF-10A, and MDA-MB-231 by ponicidin and oridonin,
diterpenoids from the chinese herb Rabdosia rubescensBiochem
Biophys Res
Commun337224231200510.1016/j.bbrc.2005.09.04016176802
|
|
21
|
FH GaoXH HuW LiOridonin induces apoptosis
and senescence in colorectal cancer cells by increasing histone
hyperacetylation and regulation of p16, p21, p27 and c-mycBMC
Cancer10610201010.1186/1471-2407-10-61021054888
|
|
22
|
M PootTJ KavanaghHC KangRP HauglandPS
RabinovitchFlow cytometric analysis of cell cycle-dependent changes
in cell thiol level by combining a new laser dye with Hoechst
33342Cytometry12184187199110.1002/cyto.9901202141710962
|
|
23
|
A MachoT HirschI MarzoGlutathione
depletion is an early and calcium elevation is a late event of
thymocyte apoptosisJ Immunol1584612461919979144473
|
|
24
|
DW HedleyS ChowEvaluation of methods for
measuring cellular glutathione content using flow
cytometryCytometry15349358199410.1002/cyto.9901504118026225
|
|
25
|
MA PapiezM DybalaM Sowa-KucmaEvaluation of
oxidative status and depression-like responses in Brown Norway rats
with acute myeloid leukemiaProg Neuropsychopharmacol Biol
Psychiatry33596604200910.1016/j.pnpbp.2009.02.01519268504
|
|
26
|
CA SchmittSenescence, apoptosis and
therapy - cutting the lifelines of cancerNat Rev
Cancer3286295200310.1038/nrc104412671667
|
|
27
|
P DeckerD IsenbergS MullerInhibition of
caspase-3-mediated poly(ADP-ribose) polymerase (PARP) apoptotic
cleavage by human PARP autoantibodies and effect on cells
undergoing apoptosisJ Biol
Chem27590439046200010.1074/jbc.275.12.904310722754
|
|
28
|
DA GewirtzSE HoltLW ElmoreAccelerated
senescence: an emerging role in tumor cell response to chemotherapy
and radiationBiochem
Pharmacol76947957200810.1016/j.bcp.2008.06.02418657518
|
|
29
|
X DiRP ShiuIF NewshamDA GewirtzApoptosis,
autophagy, accelerated senescence and reactive oxygen in the
response of human breast tumor cells to adriamycinBiochem
Pharmacol7711391150200910.1016/j.bcp.2008.12.01619185564
|
|
30
|
KR JonesLW ElmoreC
Jackson-Cookp53-Dependent accelerated senescence induced by
ionizing radiation in breast tumour cellsInt J Radiat
Biol81445458200510.1080/0955300050016854916308915
|
|
31
|
JC BarrettLA AnnabD AlcortaG PrestonP
VojtaY YinCellular senescence and cancerCold Spring Harb Symp Quant
Biol59411418199410.1101/SQB.1994.059.01.0467587095
|
|
32
|
QA QuickDA GewirtzAn accelerated
senescence response to radiation in wild-type p53 glioblastoma
multiforme cellsJ
Neurosurg105111118200610.3171/jns.2006.105.1.11116871885
|
|
33
|
T SousaD PinhoM MoratoRole of superoxide
and hydrogen peroxide in hypertension induced by an antagonist of
adenosine receptorsEur J
Pharmacol588267276200810.1016/j.ejphar.2008.04.04418519134
|
|
34
|
YH ZhangYL WuSI TashiroS OnoderaT
IkejimaReactive oxygen species contribute to oridonin-induced
apoptosis and autophagy in human cervical carcinoma HeLa cellsActa
Pharmacol Sin3212661275201110.1038/aps.2011.9221892202
|
|
35
|
M RethHydrogen peroxide as second
messenger in lymphocyte activationNat
Immunol311291134200210.1038/ni1202-112912447370
|
|
36
|
A HolmgrenJ LuThioredoxin and thioredoxin
reductase: current research with special reference to human
diseaseBiochem Biophys Res
Commun396120124201010.1016/j.bbrc.2010.03.08320494123
|