Introduction
Percutaneous transluminal coronary angioplasty
(PTCA) is a procedure used to open stenotic coronary arteries due
to cholesterol-laden atherosclerotic plaques (1). During PTCA, a cardiologist inserts a
catheter carrying a deflated balloon in the femoral artery and up
to the blocked site in the heart. The balloon is then inflated,
allowing the blood to flow again (1). A long-term analysis showed that 10-
and 20-year survival in PTCA patients was similar to survival in
coronary artery bypass graft patients, but more interventions had
to be repeated during the first year in PTCA patients (2). Indeed, restenosis, or blood vessel
re-narrowing leading to restricted blood flow, still occurs in
approximately 30% of patients within 6 months after undergoing
PTCA. This complication represents a major disadvantage of PTCA
therapies. Modern use of drug-eluting stents (DESs) has improved
the outcome in these patients; however, 5–10% of patients are still
affected by restenosis after DES placement (3).
Reactive oxygen species (ROS) and oxidative stress
are involved in the pathogenesis of intimal thickening in
atherosclerosis and restenosis (4,5).
Ohsawa et al (6)
demonstrated that hydrogen is a potent anti-oxidant, able to
selectively scavenge and neutralize hydroxyl radicals (such as
•OH and ONOO−) and able to protect the brain
against ischemiareperfusion (I/R) injury and stroke. Hydrogen
treatment is able to reduce a number of markers of oxidative stress
(7) and a number of clinical and
experimental studies demonstrated that hydrogen acts as a
scavenging agent, selectively neutralizing ROS in vivo and
exerting potent cellular protective effects (8–11).
Hydrogen has been shown to protect against restenosis in models of
myocardial I/R injury (12,13). Such results underline the use of
hydrogen (H2) gas in therapeutic medical
applications.
Although the use of H2 gas may pose
certain safety issues, due to its inflammability,
hydrogen-saturated saline (HRSS) has been shown to be safe, easy to
administer and effective in carotid I/R injury (14), renal I/R injury (15,16), acute liver injury (9), carbon monoxide intoxication
(17) and atherosclerotic lesions
(18). The exact mechanisms by
which hydrogen exerts its effects on the vascular wall remain
unknown. Aside from its anti-oxidant effects, there are certain
indications that hydrogen may directly interact with some specific
pathways (14,19).
The present study examined the anti-proliferative
effects of HRSS on abnormal vascular smooth muscle cells (VSMC)
proliferation and investigated the mechanisms responsible for these
effects.
Materials and methods
Preparation of hydrogen-rich saline
and medium
HRSS and media (HRM) were produced as previously
described (20). Briefly,
hydrogen gas was dissolved in saline and in Dulbecco’s modified
Eagle’s serum (DMEM; Gibco, USA) supplemented with 10% fetal bovine
serum (FBS). We obtained saturation using a pressure of 0.4 MPa for
6 h. HRSS and HRM were maintained at 4°C and fresh solutions were
prepared each week. Gas chromatography was used to confirm the
hydrogen levels by the method described by Ohsawa et al
(6). Cells were cultured in
closed culture flasks.
Rat balloon injury model
All experimental procedures involving animals were
approved by the Institutional Animal Care and Use Committee of the
Hebei Medical University (Shijiazhuang, China). A total of 30 male
Sprague-Dawley rats (weight, 280–320 g) were purchased from the
animal center of the University and were divided into five groups:
the sham group (no angioplasty) and the balloon-injured groups
including the control group and 3 groups administered with HRSS at
2.5, 5, 10 ml/kg, intraperitoneally, respectively.
Rats were anaesthetized with 3.6% (w/v)
chlorohydrate (1 ml/100 g, intraperitoneally). Left common carotid
artery (CCA) was balloon-denuded as previously described (21,22). In brief, a median incision was
performed on the anterior neck and left carotid arteries were
isolated. A ligature in external carotid artery (ECA) was distally
placed and the proximal ends of the CCA and internal carotid
arteries (ICA) were clamped. An incision was performed in the ECA.
Following blood removal, a 0.13 mm balloon catheter was delicately
introduced into the CCA using the incision in the ECA. The balloon
was inflated using saline to distend the CCA and pulled back to the
ECA. The catheter was removed and the ECA’s proximal end was
ligated. Clamps were removed to re-establish blood flow.
Rats were then housed (12 h light cycles) and fed
for 2 weeks (free access to food and water), and were then
sacrificed using a pentobarbital overdose. Sections from carotid
arteries from both sides were excised and fixed using 4%
paraformaldehyde. Histologically stained sections were used to
assess the extent of neointimal formation using computed
planimetry. The intima-to-media (I/M) area ratio was calculated as
the mean of these determinations (23).
Cell culture
VSMCs were isolated from the thoracic aorta of
10–12-week-old male Sprague-Dawley rats (23–25) and were cultured (37°C in a
humidified 5% CO2 incubator) in DMEM with 10% FBS,
penicillin 100 U/ml and streptomycin 100 μg/ml. Purity was
confirmed by immunocytochemical localization of α-smooth muscle
actin. Upon confluence, cells were sub-cultured using 0.5% (w/v)
trypsin. Media were changed every 3 days and experiments were
performed between 3 to 6 passages.
Cell proliferation assay
MTT assay
VSMCs (4×103 cells/well) were placed in a
96-well plate. Following incubation, sterile MTT (20 μl of 5 mg/ml)
was added, and incubated for 4 h at 37°C; 150 μl of dimethyl
sulfoxide (DMSO) was then added. Absorbance (490 nm) was measured
using a microplate reader (26).
BrdU assay
VSMCs were synchronized by serum deprivation and
incubated with 10% FBS for 24 h with or without HRM. BrdU labeling
mixture was then added and cultures were incubated for 12 h at
37°C. Cell numbers were measured using a cell proliferation ELISA
(Roche Molecular Biochemicals, Germany) (9,27).
Wound healing assay
The migration of VSMCs was evaluated by performing a
cell-wounding assay according to a previously described method
(28). Cells grown to 100%
confluence on glass slides were scraped off the slides using a cell
scraper. These cells were used to create a 3-mm-wide wound and were
then incubated at 37°C for 24 h in DMEM containing 10% FBS. Cells
were then fixed with methanol and stained with hexamethyl
pararosaniline. Cell migration activity was expressed as the number
of cells that migrated into the wound area in each field.
Flow cytometric analysis of the cell
cycle
VSMCs were synchronized at the G0-phase by serum
depletion for 48 h. After replenishment with fresh DMEM, the cells
were preincubated with hydrogen-rich medium, and 10% FBS was added
to allow progression of cell cycle. After 48 h, cells were
trypsinized, centrifuged at 1,250 x g for 5 min. DNA was stained
with propidium iodide (PI) (50 μg/ml) for 30 min at 37°C, and
5×103 cells were then analyzed by flow cytometry.
Flow cytometric analysis of cell
apoptosis
To assess apoptosis, we suspended HRM-cultured cells
in buffer (400 μl; 10 mM HEPES/NaOH; 140 mM NaCl; 2.5 mM
CaCl2; pH 7.4) after a careful wash. FITC-conjugated
Annexin V (5 μl; BD Biosciences, San Diego, CA, USA) was added and
cells were incubated for 15 min at 4–8°C in the dark. PI (10 μl)
was added. After 5 min, cells were washed again and 400 μl of
buffer was added. Cells were counted using a FACSCalibur (BD
Biosciences).
RT-PCR for Bax/Bcl-2
Total RNA was extracted using the TRIzol reagent
method and cDNA was obtained using a cDNA synthesis kit
(TransScript first-strand) according to the manufacturer’s
instructions. Bcl-2 primers were: ATGGGGTGAAC TGGGGGAGGATTG
(forward) and TTTCATATTTGTTT GGGGCAGGTC (reverse). Bax primers
were: GAGAGGAT GGCTGGGGAGAC (forward) and GGTGAGCGAGGCGG TGAGGACT
(reverse). DNA fragments were analyzed on a 1.5% agarose gel.
Quantitative RNA data were expressed by normalizing band density to
GAPDH (internal control).
Western blot analysis
The cells and arterial tissue were solubilized or
homogenized in lysate buffer on ice. Homogenates were centrifuged
at 12,000 x g for 30 min at 4°C, yielding a cell protein
supernatant. Proteins were subjected to SDS-PAGE and transferred to
polyvinylidene difluoride membranes (PVDF; Millipore, Billerica,
MA, USA). Samples were then probed with antibodies against: Ras,
phosphorylated and nonphosphorylated ERK1/2, MEK1/2, proliferative
cell nuclear antigen (PCNA), Akt and β-actin. Non-specific binding
was blocked with 1.5% (w/v) evaporated skimmed milk (Difco,
Franklin Lakes, NJ, USA) in TBS (154 mM NaCl, 10 mM Tris base).
Anti-rabbit or anti-mouse secondary antibodies conjugated to
IRDYe700DX and IRDYe800 (1:5,000; Rockland Immunochemicals,
Gilbertsville, PA, USA) were used to probe primary antibodies.
Protein bands were detected and quantified on an Odyssey 2-color
infrared imaging system (LI-COR Biosciences, Lincoln, NE, USA). The
integrated signal densities were normalized first to β-actin (the
loading control) and subsequently expressed in terms of the
fraction abundance relative to control cells or arterial tissues.
These experiments were performed in triplicate.
Immunohistochemistry and image
analysis
Immunohistochemistry was performed as previously
described (29). The control and
sham groups were treated with anti-PCNA (1:100) antibody on Day 14
following balloon injury. Sections were counterstained with
hematoxylin. Staining intensities were determined by measurement of
the integrated optical density (IOD) with light microscopy using a
computer-based Image-Pro Morphometric system. Measurements were
conducted by 2 independent observers in a double-blind manner.
Determination of reactive oxygen
species
DCFH-DA assay for intracellular
reactive oxygen species
VSMCs (1×105 cells) were incubated with
10% FBS in the presence or absence of HRM for 12 h. Cells were
stained with 10 μM of DCFH-DA for 30 min at 37°C, detached with
trypsin/EDTA, washed, re-suspended in PBS, and immediately analyzed
by flow cytometry using a FACScan by fluorescence intensity
(FL-1,525 nm) of 10,000 cells (23).
Determination of MDA concentration in
VSMCs and hyperplasia neointima using TBARS production
Following incubation with HRM for 24 h, cells were
collected and washed with cold PBS, and hyperplastic neointimal
tissues were homogenized. Tissue samples (0.1 ml), 8.1% SDS (0.2
ml), 20% acetic acid buffer solution (1.5 ml, pH 3.5), 1% total
bile acid (TBA) thiobarbituric solution (1.5 ml), and distilled
water (1 ml) were added. The solution was heated in a 95°C water
bath for 40 min and then allowed to cool; it was then centrifuged
at 3,500 rpm/min for 15 min and measured at 532 nm using a
spectrophotometer (14,30).
Determination of 8-OHdG in VSMCs and
hyperplasia neointima
The 8-OHdG amounts were determined in VSMCs and
hyperplastic neointimal tissues using a Bioxytech 8-OHdG-EIA kit.
DNA was isolated using the DNAzol reagent and quantified. DNA (400
μg) was resuspended in 50 μl reaction mixture containing 100 mmol/l
sodium acetate (pH 5.0) and 5 mmol/l MgCl2 and digested
using DNaseI. Assay was performed according to the manufacturer’s
instructions (Shanghai Lanji Biological Institute, China).
Statistical analysis
Data are provided as the means ± SEM. One-way
analysis of variance (ANOVA) followed by the LSD-t-test were
performed using SPSS 19.0 software. P<0.05 was considered to
indicate a statistically significant difference.
Results
ROS and Ras signaling pathways are
involved in the effects of HRSS on neointimal hyperplasia
Balloon-induced intimal hyperplasia was evident in
treated rats compared with rats of the control group (Fig. 1A). Results showed that 3 doses of
HRSS were effective for prevention of neointimal formation in a
dose-dependent manner (Fig. 1A).
These findings showed neointimal area ratio reductions of 36.9,
52.2 and 71.8% (P<0.01) in HRSS-treated groups using 2.5, 5 ml
and 10 ml/kg doses, respectively, compared with the balloon-injured
control group (Fig. 1B).
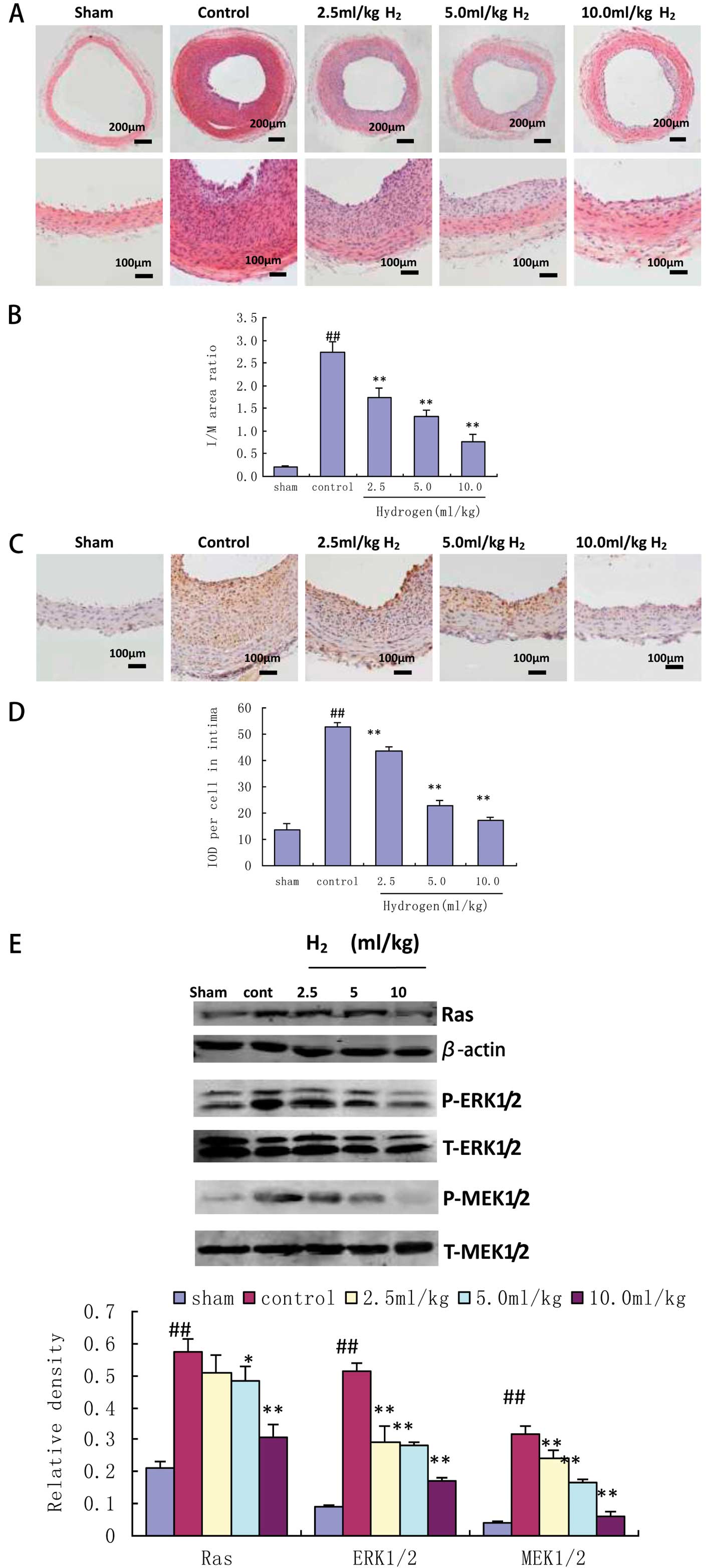 | Figure 1Hydrogen-rich saline inhibits
injury-induced neointimal hyperplasia in rat carotid arteries. (A)
Histological sections and (B) the intima to media area ratio of
carotid arteries 14 days after balloon injury.
##P<0.01, n=6, vs. sham group;
**P<0.01, n=6, vs. control group. (C) Immunostaining
and (D) integrated optical density (IOD) per cell of intima in
carotid artery section treated as in panel (A).
##P<0.01, n=6, vs. sham group;
**P<0.01, n=6, vs. control group. (E) Western blot
analysis of the active forms of Ras, ERK1/2 and MEK1/2 in carotid
arteries 14 days after balloon injury. ##P<0.01, n=4,
vs. sham group; *P<0.05; **P<0.01, n=4,
vs. control group. |
Western blot analysis revealed that the activation
of ERK1/2, MEK1/2 and Ras was strongly suppressed in a
dose-dependent manner in 14-day HRSS-treated samples compared with
control samples (P<0.01) (Fig.
1E).
PCNA-positive cells were abundant in the balloon
injury group. However, PCNA immunostaining was much less apparent
in cells treated with HRSS. Staining intensities were lower in
HRSS-treated rats (P<0.01) (Fig.
1C and D).
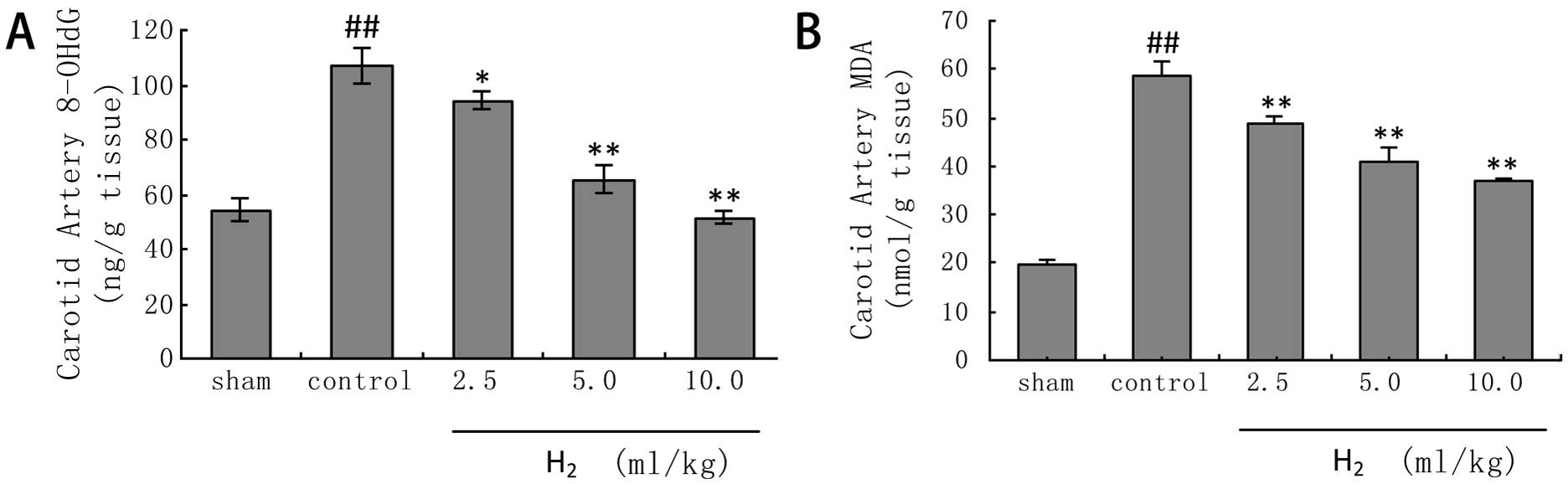
Treatment with HRSS significantly inhibited the
levels of carotid artery MDA and 8-OHdG in a dose-dependent manner
(Fig. 2).
HRM inhibits FBS-induced VSMC
proliferation and migration
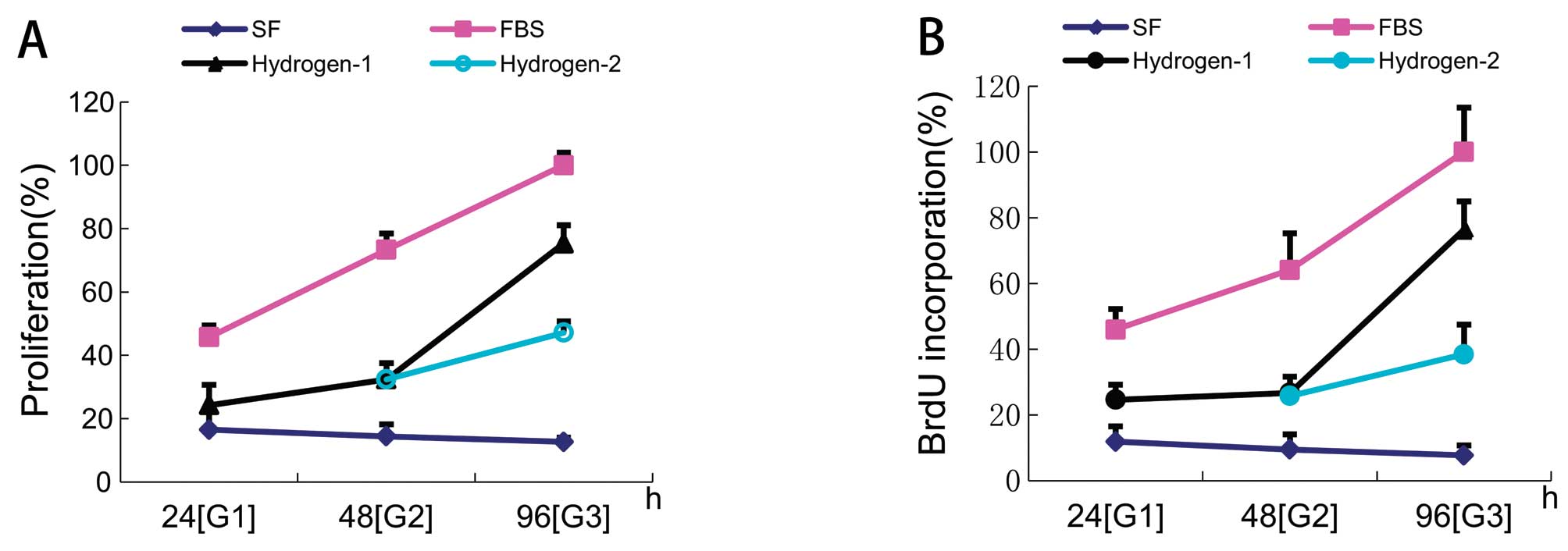
MTT assay showed that VSMC viability in the HRSS
group was significantly lower than in the control group (32.4±5.2
vs. 73.3±5.1%; P<0.01). Two VSMC cultures were treated with HRM
for 48 h; hydrogen was then ceased for one culture, while it was
continued in the other. Following the removal of hydrogen, cells
resumed viability (Fig. 3A).
Cells without hydrogen treatment exhibited a BrdU index of
64.1±11.1%, compared to 26.7±4.9% in the hydrogen group (Fig. 3B).
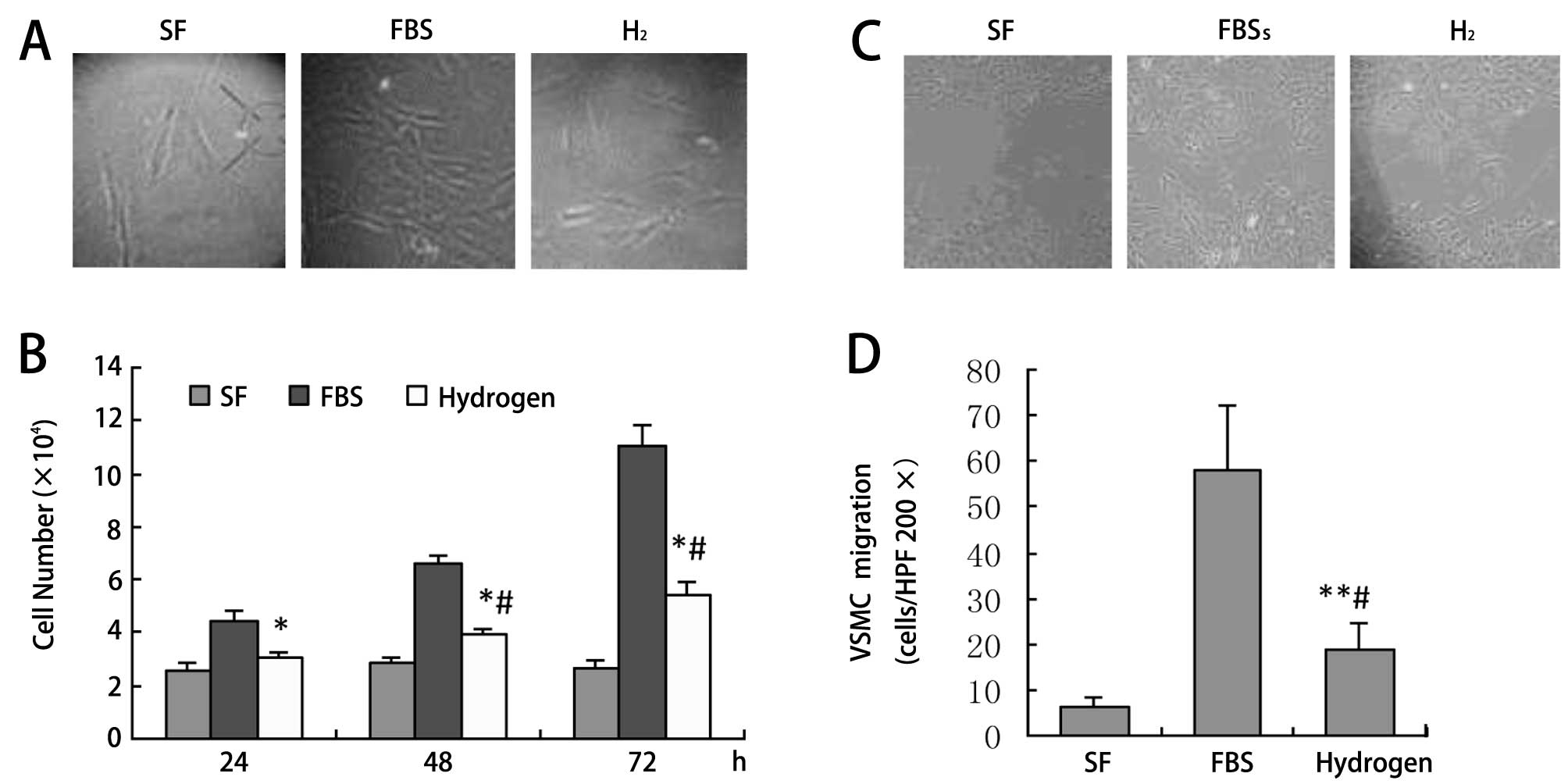
VSMC cell count was smaller in HRM-treated cells
after 48 and 72 h (40 and 51% reductions, respectively; P<0.01)
(Fig. 4A and B). VSMCs treated
with FBS for 24 h showed greater mobility (FBS 58.3±13.9 vs.
hydrogen 18.6±5.8 cells; P<0.01) (Fig. 4C and D).
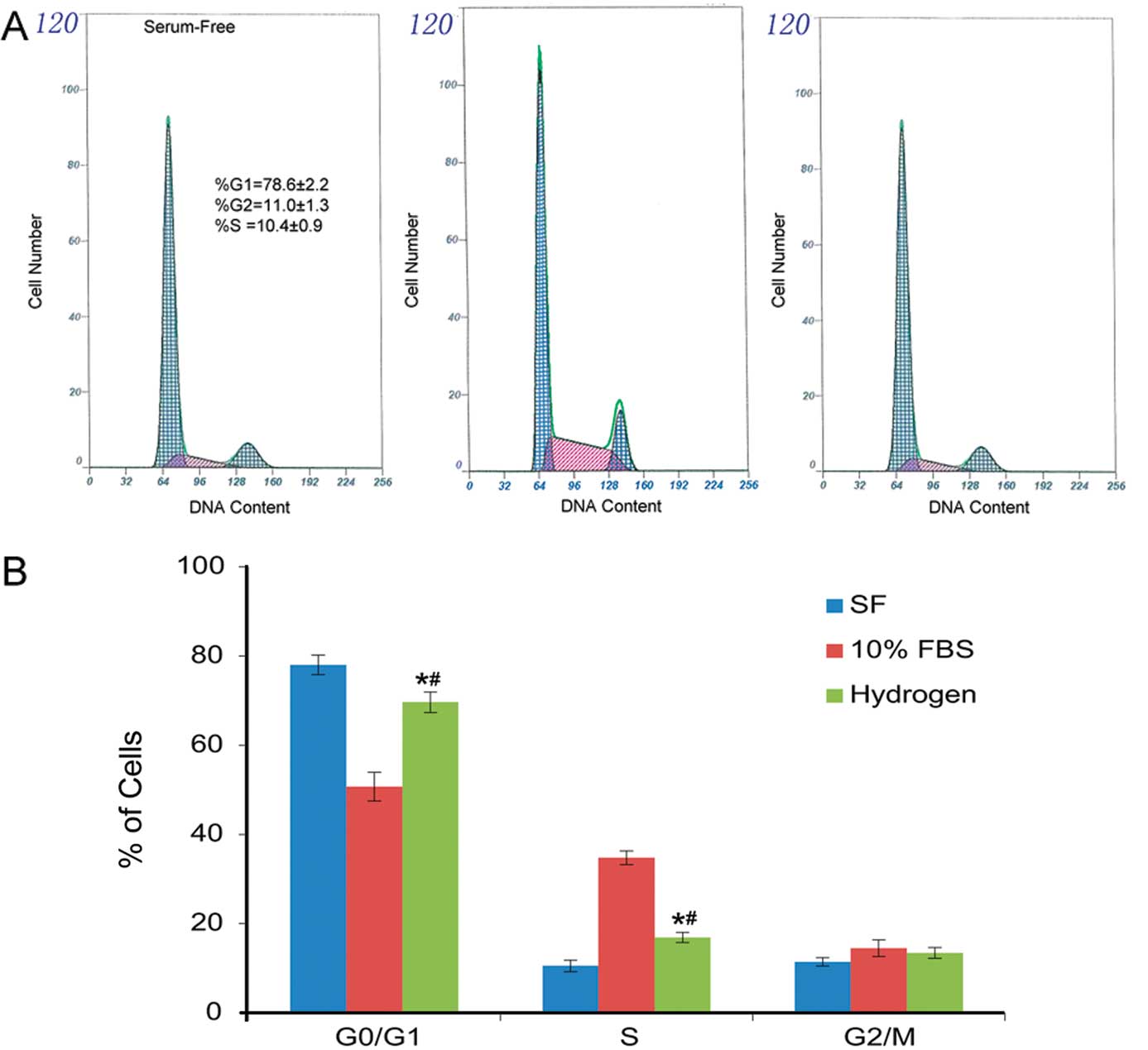
HRM prevents FBS-induced S-phase entry
in VSMCs
Treatment with HRM inhibited FBS-induced G1-S
progression, as demonstrated by the increase in G0/G1 cells
(68.8±2.2%) accompanied by concurrent decrease in S-phase cells
(16.7±2.0%) (Fig. 5A). These
results suggest that hydrogen may prevent FBS-induced S-phase entry
in VSMCs via a G0-G1 blocking mechanism.
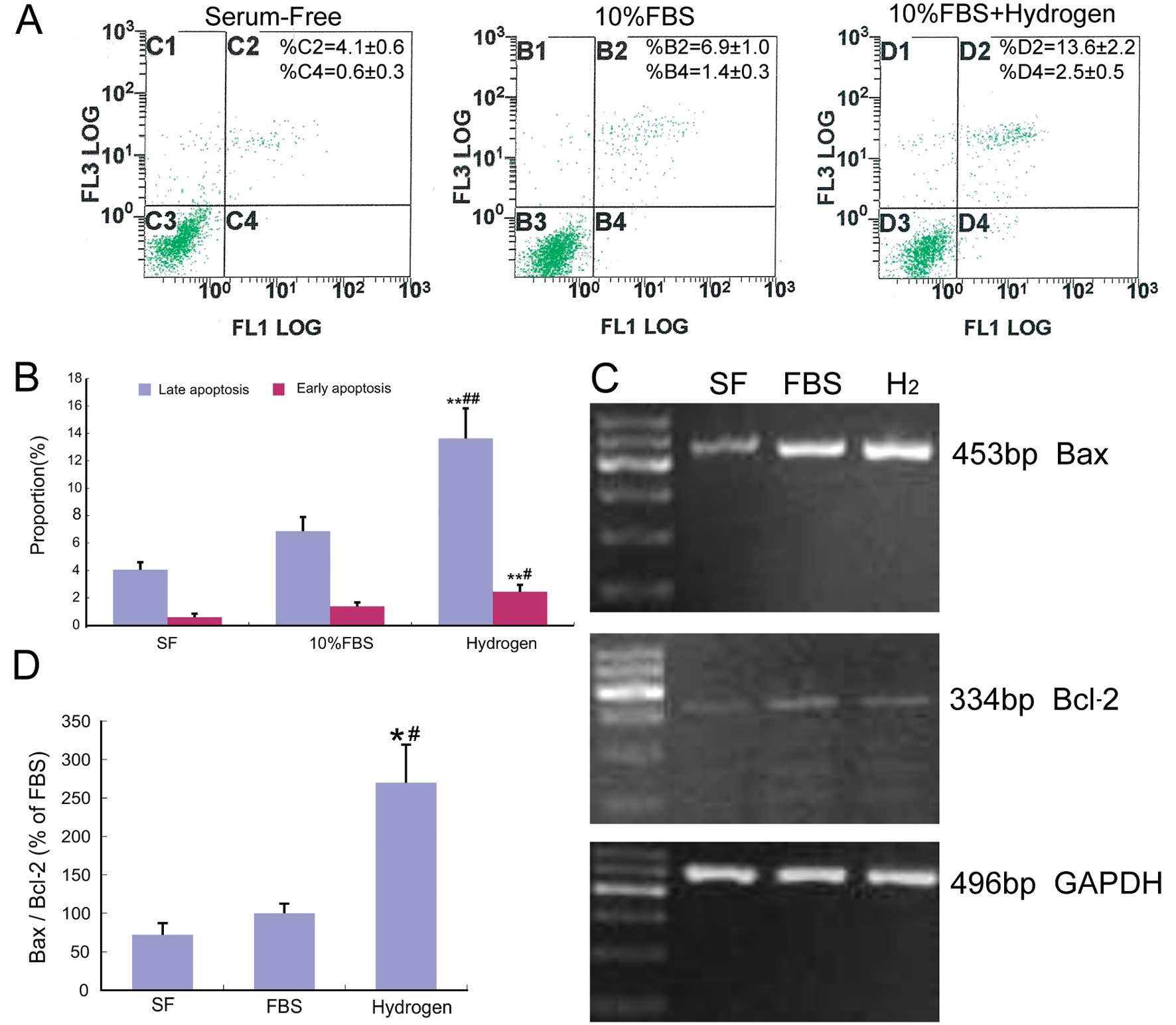
HRM increases the proportion of
apoptotic cells in VSMCs
Proportion of early apoptotic cells (Fig. 6A and B; lower right) increased
from 1.36% (FBS-treated) to 2.1% (FBS + hydrogen-treated)
(P<0.05). These results also showed that the proportion of late
apoptotic cells (Fig. 6A and B; upper
right) increased from 6.96 to 13.47% (P<0.01).
Following HRM treatment, a significant increase in
Bax/Bcl-2 ratio (P<0.01) indicated that VSMCs were, in fact,
progressing towards apoptosis (Fig.
6C and D).
ROS is involved in the effect of HRM
on FBS-stimulated VSMCs
FBS treatment significantly induced intracellular
peroxide production (Fig. 7A and
B). ROS generation in cells treated with HRM was 395.6±16.1,
compared with 483.0±15.1 for controls (P<0.01).
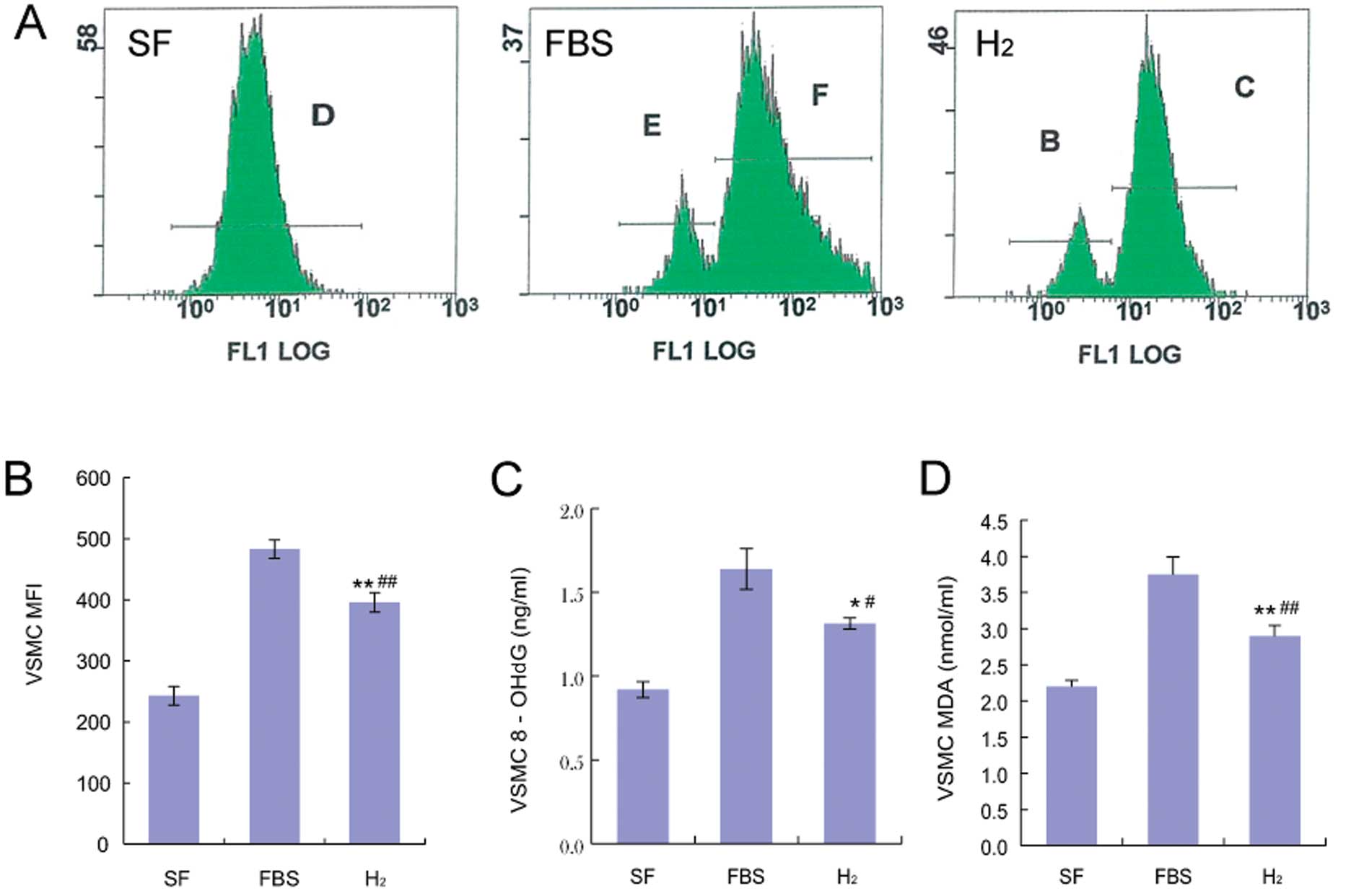 | Figure 7Hydrogen-rich medium inhibits
FBS-induced ROS production. (A) Levels of intracellular peroxide in
the presence of hydrogen-rich medium for 48 h. (B) Mean
fluorescence intensity (MFI) of DCF was expressed as means ± SEM.
**P<0.01, n=3, vs. the FBS group;
##P<0.01, n=3, vs. the serum-free (SF) group. (C)
Effect of hydrogen on 8-OHdG. *P<0.05, n=3, vs. FBS
group; #P<0.05, n=3, vs. SF. (D) Effect of hydrogen
on MDA concentration. *P<0.05, n=3, vs. FBS group;
#P<0.05, n=3, vs. SF group. |
The 8-OHdG levels in cells treated with HRM were
1.3±0.0 ng/ml, compared to 1.6±0.1 ng/ml in controls (P<0.01)
(Fig. 7C). Fig. 7D shows that treatment with HRM
significantly inhibited MDA generation (FBS 3.7±0.2 vs. hydrogen
2.9±0.1; P<0.01).
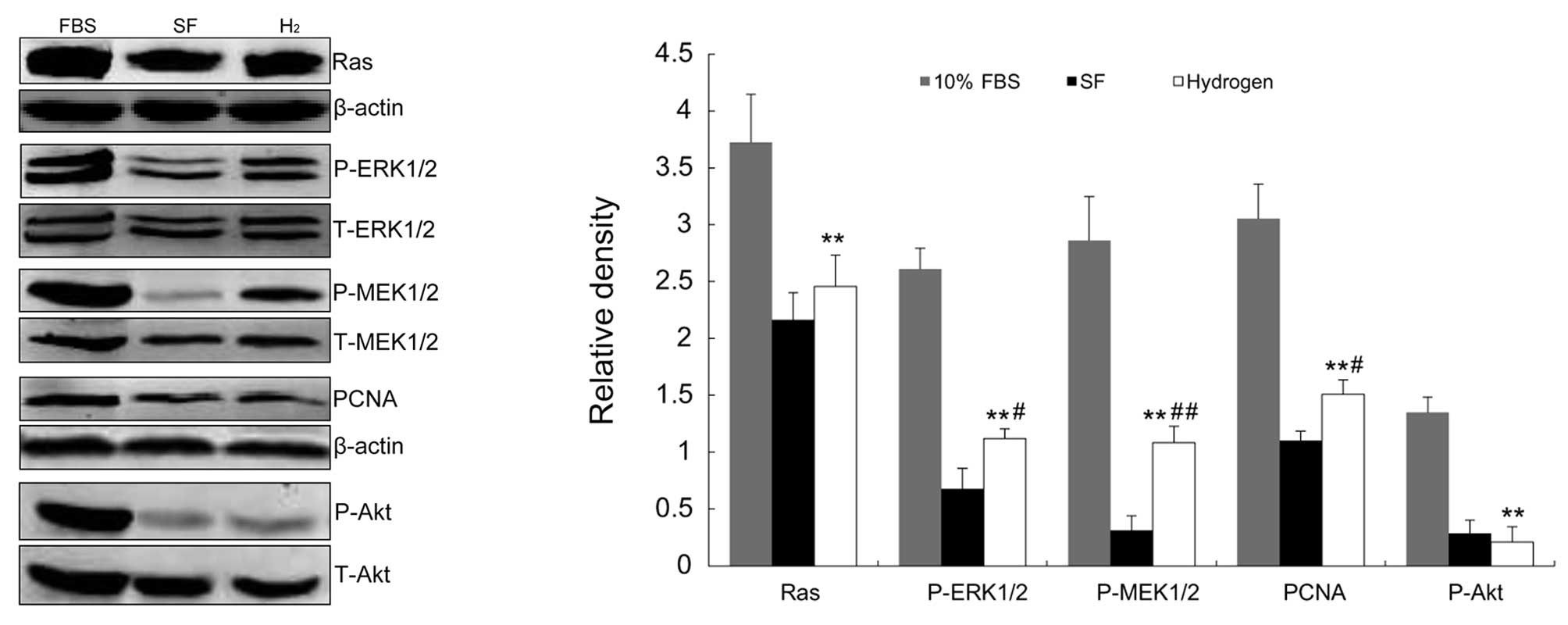
Ras-MEK1/2-ERK1/2 pathway and Akt
phosphorylation are involved in the effect of HRM on FBS-stimulated
VSMCs
Stimulation of cells with 10% FBS induced Ras
activation. Cells treated with HRM exhibited a slight inhibition of
FBS-induced Ras activation (Fig.
8A). Treatment of FBS-stimulated VSMCs with HRM markedly
decreased phosphorylated MEK1/2 levels (Fig. 8C). Data showed that FBS also
induced a profound increase in ERK1/2 activation. Treatment with
HRM significantly inhibited FBS-stimulated phosphorylation of
ERK1/2 (Fig. 8B). By contrast,
total ERK1/2 protein levels were not altered by treatment with
hydrogen. FBS induced a profound increase in Akt activation. The
level of Akt phosphorylation following FBS stimulation was also
significantly inhibited by treatment with HRM (Fig. 5E). As shown in Fig. 8D, FBS-induced PCNA expression was
significantly inhibited by HRM by 50% (P<0.01).
Discussion
In the process of opening the artery, PTCA may
injure the vascular wall (31).
VSMC proliferation in the arterial wall is crucial in the
development of post-angioplasty restenosis and atherosclerosis
(32,33). Currently, the most common approach
to reduce neointimal formation and its consequences is the use of
DES (34). However, DES presents
a number of issues, such as incomplete stent coverage, inadequate
stent placement or failure of the eluting drug to inhibit VSMC
proliferation (35–37). The current study demonstrates that
hydrogen inhibits VSMC proliferation and migration and provides
important mechanistic information; this inhibitory effect involves
the blockage of the G1-S-phase cell cycle progression, thus
increasing apoptosis and inhibition of Ras-MEK1/2-ERK1/2 and Akt
signaling pathways. HRSS treatment also inhibits neointimal
hyperplasia induced by balloon injury via suppression of the
Ras-MEK1/2-ERK1/2 signaling pathway.
Oxidative stress is involved in vascular diseases
(38,39). Initial observations focused on ROS
derived from invading macrophages, indicating possible involvement
in oxidative lipid modifications of vessel walls. ROS can attack
circulating lipoproteins, contributing to atherosclerotic plaque
development (40), and can attack
vessel walls, maintaining a pro-inflammatory state (41). Ohsawa et al (18) used an apolipoprotein E knockout
mouse model to demonstrate the role of hydrogen as an antioxidant.
In their study, oral ingestion of HRSS for 6 months prevented
atherosclerosis development by decreasing oxidative stress in the
blood vessels. Moreover, controlled ROS production occurs in all
vascular cells, where these compounds act as secondary messengers
in the regulation of varied cellular functions. Notably, VSMC
proliferation and stenosis following vascular injury was shown to
coincide with elevated ROS levels in experimental animals (42,43).
The O2 and H2O2 ROS
are primarily detoxified by antioxidant defense enzymes, unlike
radical •OH and ONOO− species.
Hydrogen gas has been demonstrated to selectively reduce these 2
ROS species (6). Thus, 8-OHdG
formed by deoxyguanosine in DNA in the presence of hydroxyl free
radicals may be a useful biomarker for intracellular oxidative
stress (44). MDA is also a
non-specific marker of lipid peroxidation. Assessment of 8-OHdG
levels demonstrated that increasing ROS levels were inhibited by
HRSS in vitro in FBS-treated VSMCs. Furthermore, increases
in ROS levels in neointimal tissues in vivo were inhibited
by HRSS. These findings are consistent with previous reports,
including the report by Qin et al (14) that demonstrates hydrogen ability
to reduce injury-induced excess superoxide anions and inflammation.
Results from the present study also show that ROS reduction
coincides with a decrease in VSMC proliferation and proliferative
pathways. This moderate antioxidant ability indicates that hydrogen
may cause only minimal disturbance to normal physiological
functions involving ROS.
Upon exposure to growth factors or serum,
protein-tyrosine kinase (PTK) receptors are activated, resulting in
the activation of Ras/Raf/MAPK/MEK and downstream proteins. ERK
also plays a central role in this pathway (45). It has also been suggested that ERK
may play a pivotal role in VSMC proliferation control (46,47). Specifically, it may inhibit MAPK
phosphorylation, thus markedly inhibiting VSMC growth in
vitro (48). The activation
of ERK1/2 can increase the expression of downstream transcription
genes, such as cyclin D1, resulting in protein synthesis and cell
proliferation (49). Therefore,
confirmation that HRSS inhibits the ERK pathway partly explains the
antiproliferative mechanisms of such treatments.
Furthermore, Ras, a major upstream signaling protein
of Raf-MEK1/2-ERK1/2, is primarily stimulated by mitogenic factors
(50). It plays a pivotal role in
G1 progression and in the G1-S transition. Hydrogen treatment
significantly and efficiently reduces phosphorylated ERK (active
form) and phosphorylated MEK, resulting in a slight decrease in Ras
expression, suggesting that HRSS is capable of effectively
inhibiting the ERK pathway. This effect likely contributes to the
observed anti-proliferative activity of hydrogen in VSMCs. HRSS
effects on the ERK signaling cascade activated by balloon injury
may also be associated with Ras-MEK1/2-ERK1/2 signaling pathway
involvement in suppression of neointimal hyperplasia.
Dose-dependent inhibition of ROS, VSMC proliferation and of ERK1/2,
MEK1/2, and Ras activations were observed in cases of balloon
injury treated with lercanidipine (51). Our results indicate that the same
outcomes are achieved simply using hydrogen and that hydrogen acts
through similar mechanisms. These results further support the
ability of HRSS to inhibit the FBS- or injury-induced ERK1/2
signaling cascade, thus affecting VSMC proliferation both in
vitro and in vivo, which has not previously been
reported as a major hydrogen effect in a review on hydrogen’s
medical properties (7). HRSS was
also demonstrated to inhibit the PI3K pathway in VSMCs. PI3K
activates Akt (serine/threonine kinase). In its active state, Akt
promotes cell survival and growth (52); however, it remains unclear whether
the PI3K pathway is affected by HRM treatment. If HRM can inhibit
the PI3K pathway in VSMCs, this effect would also contribute to the
anti-proliferative effect of HRM on VSMCs.
Suppressed PCNA levels in the G0/G1-phase were
associated with hydrogen treatment, demonstrating the effect of
hydrogen treatment on the regulation of VSMCs at the nuclear level.
PCNA is synthesized in the early G1- and S-phase, making it a
useful marker for proliferation (50,53). The anti-proliferative effect of
HRM may result from its ability to block the entry of cells into
the S-phase due to interference in the early G0/G1 transition
phase. Preventing cell cycle transition between the G1- and S-phase
in VSMCs may be beneficial in reducing cell proliferation and
migration, as well as restenosis (54,55). The current evaluation of VSMC cell
cycle alteration in response to treatment with HRM showed a
considerable increase in cells in the G0/G1-phase and a concomitant
decrease in S-phase cells, which may result from reduced
proliferation, increased apoptosis, or a combination of the two.
Additionally, HRM treatment revealed increased apoptosis rates.
Cellular apoptotic events are known to be governed by levels of
anti-apoptotic (Bcl-xL family) and pro-apoptotic proteins (Bax,
Bak) (56), indicating that the
significant reduction in Bcl-2/Bax ratio observed in VSMCs treated
with HRM supports the involvement of hydrogen in the apoptotic
pathway.
The use of FBS to induce VSMC growth in vitro
could be questioned. However, a previous study also using rat VSMCs
showed that 10% FBS was as effective as 20 ng/ml of
plateled-derived growth factor (PDGF) to promote VSMC proliferation
(51). Furthermore, lercanidipine
had the same efficacy in FBS- and PDGF-induced VSMCs, and inhibited
Ras-ERK1/2 signaling in the same way, suggesting that FBS and PDGF
promote VSMC proliferation using the same pathways.
Significant evidence shows that enhanced VSMC
proliferation is a fundamental feature of atherosclerosis
pathogenesis. In conclusion, data presented in this report
demonstrate that treatment with HRM can effectively inhibit VSMC
proliferation and migration in vitro and protect neointimal
formation in vivo following vascular injury by balloon
angioplasty. These results demonstrate that HRM suppresses the
proliferation of VSMCs by inhibiting the ROS, Ras-MEK1/2-ERK1/2 and
P13K/Akt pathways. Thus, hydrogen treatment promotes cell cycle
arrest in VSMCs at the G0/G1-phase and increases apoptosis rates.
HRSS is therefore a potentially useful antioxidant that may prove
clinically useful in patients undergoing coronary artery
angioplasty, stenting or coronary artery bypass to prevent
restenosis.
References
|
1
|
Landau C, Lange RA and Hillis LD:
Percutaneous transluminal coronary angioplasty. N Engl J Med.
330:981–993. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
van Domburg RT, Foley DP, Breeman A, van
Herwerden LA and Serruys PW: Coronary artery bypass graft surgery
and percutaneous transluminal coronary angioplasty. Twenty-year
clinical outcome. Eur Heart J. 23:543–549. 2002.PubMed/NCBI
|
|
3
|
Sturek M and Reddy HK: New tools for
prevention of restenosis could decrease the ‘oculo-stento’ reflex.
Cardiovasc Res. 53:292–293. 2002.PubMed/NCBI
|
|
4
|
Sasaki T, Maruyama H, Kase Y, Takeda S and
Aburada M: Antianginal effects of lercanidipine on the vasopressin
or methacholine induced anginal model in rats. Biol Pharm Bull.
28:811–816. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Park J, Ha H, Seo J, et al: Mycophenolic
acid inhibits platelet-derived growth factor-induced reactive
oxygen species and mitogen-activated protein kinase activation in
rat vascular smooth muscle cells. Am J Transplant. 4:1982–1990.
2004. View Article : Google Scholar
|
|
6
|
Ohsawa I, Ishikawa M, Takahashi K, et al:
Hydrogen acts as a therapeutic antioxidant by selectively reducing
cytotoxic oxygen radicals. Nat Med. 13:688–694. 2007. View Article : Google Scholar
|
|
7
|
Hong Y, Chen S and Zhang JM: Hydrogen as a
selective antioxidant: a review of clinical and experimental
studies. J Int Med Res. 38:1893–1903. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang C, Li J, Liu Q, et al: Hydrogen-rich
saline reduces oxidative stress and inflammation by inhibit of JNK
and NF-kappaB activation in a rat model of amyloid-beta-induced
Alzheimer’s disease. Neurosci Lett. 491:127–132. 2011.PubMed/NCBI
|
|
9
|
Sun H, Chen L, Zhou W, et al: The
protective role of hydrogen-rich saline in experimental liver
injury in mice. J Hepatol. 54:471–480. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sun Q, Kang Z, Cai J, et al: Hydrogen-rich
saline protects myocardium against ischemia/reperfusion injury in
rats. Exp Biol Med. 234:1212–1219. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kajiya M, Silva MJ, Sato K, Ouhara K and
Kawai T: Hydrogen mediates suppression of colon inflammation
induced by dextran sodium sulfate. Biochem Biophys Res Commun.
386:11–15. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nakao A, Kaczorowski DJ, Wang Y, et al:
Amelioration of rat cardiac cold ischemia/reperfusion injury with
inhaled hydrogen or carbon monoxide, or both. J Heart Lung
Transplant. 29:544–553. 2010. View Article : Google Scholar
|
|
13
|
Hayashida K, Sano M, Ohsawa I, et al:
Inhalation of hydrogen gas reduces infarct size in the rat model of
myocardial ischemiareperfusion injury. Biochem Biophys Res Commun.
373:30–35. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qin ZX, Yu P, Qian DH, et al:
Hydrogen-rich saline prevents neointima formation after carotid
balloon injury by suppressing ROS and the TNF-alpha/NF-kappaB
pathway. Atherosclerosis. 220:343–350. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang F, Yu G, Liu SY, et al: Hydrogen-rich
saline protects against renal ischemia/reperfusion injury in rats.
J Surg Res. 167:e339–e344. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shingu C, Koga H, Hagiwara S, et al:
Hydrogen-rich saline solution attenuates renal ischemia-reperfusion
injury. J Anesth. 24:569–574. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun Q, Cai J, Zhou J, et al: Hydrogen-rich
saline reduces delayed neurologic sequelae in experimental carbon
monoxide toxicity. Crit Care Med. 39:765–769. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ohsawa I, Nishimaki K, Yamagata K,
Ishikawa M and Ohta S: Consumption of hydrogen water prevents
atherosclerosis in apolipoprotein E knockout mice. Biochem Biophys
Res Commun. 377:1195–1198. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Itoh T, Fujita Y, Ito M, et al: Molecular
hydrogen suppresses FcepsilonRI-mediated signal transduction and
prevents degranulation of mast cells. Biochem Biophys Res Commun.
389:651–656. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zheng X, Mao Y, Cai J, et al:
Hydrogen-rich saline protects against intestinal
ischemia/reperfusion injury in rats. Free Radic Res. 43:478–484.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Clowes AW, Reidy MA and Clowes MM:
Kinetics of cellular proliferation after arterial injury. I Smooth
muscle growth in the absence of endothelium. Lab Invest.
49:327–333. 1983.PubMed/NCBI
|
|
22
|
Wei GL, Krasinski K, Kearney M, Isner JM,
Walsh K and Andres V: Temporally and spatially coordinated
expression of cell cycle regulatory factors after angioplasty. Circ
Res. 80:418–426. 1997.PubMed/NCBI
|
|
23
|
Yeh JL, Liou SF, Chang YP, et al:
Isoeugenodilol inhibits smooth muscle cell proliferation and
neointimal thickening after balloon injury via inactivation of
ERK1/2 pathway. J Biomed Sci. 15:375–389. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liou SF, Yeh JL, Liang JC, Chiu CC, Lin YT
and Chen IJ: Inhibition of mitogen-mediated proliferation of rat
vascular smooth muscle cells by labedipinedilol-A through PKC and
ERK 1/2 pathway. J Cardiovasc Pharmacol. 44:539–551. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Parenti A, Bellik L, Brogelli L, Filippi S
and Ledda F: Endogenous VEGF-A is responsible for mitogenic effects
of MCP-1 on vascular smooth muscle cells. Am J Physiol Heart Circ
Physiol. 286:H1978–H1984. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Berridge MV, Herst PM and Tan AS:
Tetrazolium dyes as tools in cell biology: new insights into their
cellular reduction. Biotechnol Annu Rev. 11:127–152. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Muir D, Varon S and Manthorpe M: An
enzyme-linked immunosorbent assay for bromodeoxyuridine
incorporation using fixed microcultures. Anal Biochem. 185:377–382.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li JJ, Han M, Wen JK and Li AY:
Osteopontin stimulates vascular smooth muscle cell migration by
inducing FAK phosphorylation and ILK dephosphorylation. Biochem
Biophys Res Commun. 356:13–19. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fishbein MC, Wang T, Matijasevic M, Hong L
and Apple FS: Myocardial tissue troponins T and I. An
immunohistochemical study in experimental models of myocardial
ischemia. Cardiovasc Pathol. 12:65–71. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Salvemini D, Riley DP and Cuzzocrea S: SOD
mimetics are coming of age. Nat Rev Drug Discov. 1:367–374. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schwartz RS and Henry TD: Pathophysiology
of coronary artery restenosis. Rev Cardiovasc Med. 3(Suppl 5):
S4–S9. 2002.
|
|
32
|
Braun-Dullaeus RC, Mann MJ and Dzau VJ:
Cell cycle progression: new therapeutic target for vascular
proliferative disease. Circulation. 98:82–89. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen Z, Lee FY, Bhalla KN and Wu J: Potent
inhibition of platelet-derived growth factor-induced responses in
vascular smooth muscle cells by BMS-354825 (dasatinib). Mol
Pharmacol. 69:1527–1533. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stettler C, Wandel S, Allemann S, et al:
Outcomes associated with drug-eluting and bare-metal stents: a
collaborative network meta-analysis. Lancet. 370:937–948. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lemos PA, Saia F, Ligthart JM, et al:
Coronary restenosis after sirolimus-eluting stent implantation:
morphological description and mechanistic analysis from a
consecutive series of cases. Circulation. 108:257–260. 2003.
View Article : Google Scholar
|
|
36
|
Fujii K, Mintz GS, Kobayashi Y, et al:
Contribution of stent underexpansion to recurrence after
sirolimus-eluting stent implantation for in-stent restenosis.
Circulation. 109:1085–1088. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Takebayashi H, Kobayashi Y, Mintz GS, et
al: Intravascular ultrasound assessment of lesions with target
vessel failure after sirolimus-eluting stent implantation. Am J
Cardiol. 95:498–502. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Griendling KK and Ushio-Fukai M: Redox
control of vascular smooth muscle proliferation. J Lab Clin Med.
132:9–15. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kunsch C and Medford RM: Oxidative stress
as a regulator of gene expression in the vasculature. Circ Res.
85:753–766. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Boullier A, Bird DA, Chang MK, et al:
Scavenger receptors, oxidized LDL, and atherosclerosis. Ann NY Acad
Sci. 947:214–223. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mizuno Y, Jacob RF and Mason RP:
Inflammation and the development of atherosclerosis. J Atheroscler
Thromb. 18:351–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Szocs K, Lassegue B, Sorescu D, et al:
Upregulation of Nox-based NAD(P)H oxidases in restenosis after
carotid injury. Arterioscler Thromb Vasc Biol. 22:21–27. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shi Y, Niculescu R, Wang D, Patel S,
Davenpeck KL and Zalewski A: Increased NAD(P)H oxidase and reactive
oxygen species in coronary arteries after balloon injury.
Arterioscler Thromb Vasc Biol. 21:739–745. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kasai H: Analysis of a form of oxidative
DNA damage, 8-hydroxy-2′-deoxyguanosine, as a marker of cellular
oxidative stress during carcinogenesis. Mutat Res. 387:147–163.
1997.
|
|
45
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lewis TS, Shapiro PS and Ahn NG: Signal
transduction through MAP kinase cascades. Adv Cancer Res.
74:49–139. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lee JH, Johnson PR, Roth M, Hunt NH and
Black JL: ERK activation and mitogenesis in human airway smooth
muscle cells. Am J Physiol Lung Cell Mol Physiol. 280:L1019–L1029.
2001.PubMed/NCBI
|
|
48
|
Koyama H, Olson NE, Dastvan FF and Reidy
MA: Cell replication in the arterial wall: activation of signaling
pathway following in vivo injury. Circ Res. 82:713–721. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Roovers K, Davey G, Zhu X, Bottazzi ME and
Assoian RK: Alpha5beta1 integrin controls cyclin D1 expression by
sustaining mitogen-activated protein kinase activity in growth
factor-treated cells. Mol Biol Cell. 10:3197–3204. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hirata A, Igarashi M, Yamaguchi H, et al:
Nifedipine suppresses neointimal thickening by its inhibitory
effect on vascular smooth muscle cell growth via a MEK-ERK pathway
coupling with Pyk2. Br J Pharmacol. 131:1521–1530. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wu JR, Liou SF, Lin SW, et al:
Lercanidipine inhibits vascular smooth muscle cell proliferation
and neointimal formation via reducing intracellular reactive oxygen
species and inactivating Ras-ERK1/2 signaling. Pharmacol Res.
59:48–56. 2009. View Article : Google Scholar
|
|
52
|
Lawlor MA and Alessi DR: PKB/Akt: a key
mediator of cell proliferation, survival and insulin responses? J
Cell Sci. 114:2903–2910. 2001.PubMed/NCBI
|
|
53
|
Ranganna K, Yatsu FM, Hayes BE, Milton SG
and Jayakumar A: Butyrate inhibits proliferation-induced
proliferating cell nuclear antigen expression (PCNA) in rat
vascular smooth muscle cells. Mol Cell Biochem. 205:149–161. 2000.
View Article : Google Scholar
|
|
54
|
Wu CH, Lin CS, Hung JS, et al: Inhibition
of neointimal formation in porcine coronary artery by a Ras mutant.
J Surg Res. 99:100–106. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Grube E, Gerckens U, Muller R and
Bullesfeld L: Drug eluting stents: initial experiences. Z Kardiol.
91(Suppl 3): S44–S48. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Donovan M and Cotter TG: Control of
mitochondrial integrity by Bcl-2 family members and
caspase-independent cell death. Biochim Biophys Acta. 1644:133–147.
2004. View Article : Google Scholar : PubMed/NCBI
|