Introduction
Intracerebral hemorrhage (ICH) is a severe type of
stroke causing neurological dysfunction with a high mortality rate
(1). Current surgical therapies
for ICH are not effective and acceptable drugs have not yet been
developed for clinical trials. ICH-induced brain injury occurs
through multiple mechanisms, and is also mediated in part by an
apoptotic mechanism (2,3).
Apoptosis is a form of cell death that constitutes
part of a common mechanism in cell replacement, tissue remodeling,
and the removal of damaged cells. Apoptosis is triggered by a
variety of stimuli (4); however,
the inappropriate or excessive initiation of apoptosis has been
implicated in several types of neurodegenerative disorders,
including stroke (5). Apoptotic
cell death can be assessed using terminal deoxynucleotidyl
transferase-mediated dUTP nick end-labeling (TUNEL) staining, which
detects DNA fragmentation. The caspases, a family of 14 cysteine
proteases, are essential players in apoptotic cell death both as
initiators (caspase-2, -8, -9 and -10) and executioners (caspase-3,
-6 and -7) (6). Cell death in the
parenchyma occurs via apoptotic mechanisms during ICH, and
apoptotic cell death is associated with the induction of caspase-3
in cells adjacent to the hematoma (2,7).
Apart from the caspases, the Bcl-2 family proteins also play
important roles in the regulation of apoptosis. The Bcl-2 family
proteins are classified into anti-apoptotic proteins, including
Bcl-2 and Bcl-xL, and pro-apoptotic proteins, such as Bax and Bid.
The balance between pro-apoptotic and anti-apoptotic Bcl-2 family
members determines the mitochondrial response to apoptotic stimuli
(8).
Brain-derived neurotrophic factor (BDNF) is a small
dimeric protein, and functions through its receptor, tyrosine
kinase B (TrkB). BDNF modulates neuronal cell growth and survival,
and BDNF has been implicated in learning and memory processes;
therefore, dysfunction in BDNF is accompanied by cognitive deficits
(9). BDNF enhances
hippocampal-dependent memory and long-term potentiation, a form of
synaptic plasticity, via TrkB (10). A high level of BDNF is
concentrated in the hippocampus, and BDNF expression is selectively
increased following activity-dependent learning and memory tasks
(11). TrkB activation has been
shown to inhibit apoptosis following subarachnoid hemorrhage
(12).
Dexmedetomidine is a potent and highly selective
agonist for α2-adrenoreceptors with sedative,
anxiolytic, analgesic and anesthetic effects (13,14). The neuroprotective effects of
dexmedetomidine by stimulating α2-adrenoreceptors have
been reported (15–17). Dexmedetomidine has been shown to
inhibit apoptotic neuronal cell death in the hippocampus by
enhancing antioxidant activity following transient global cerebral
ischemia-reperfusion injury in rats (18). Dexmedetomidine has also been shown
to exert neuroprotective effects following subarachnoid
hemorrhage-induced hippocampal injury in rabbits (19) and to reduce oxidative stress
following subarachnoid hemorrhage in rats (20).
The neuroprotective effects of dexmedetomidine
against several brain injuries have been suggested; however, the
memory deteriorating effects of dexmedetomidine have also been
indicated. Dexmedetomidine infusion results in reversible sedation,
mild analgesia and memory impairment (21). Dexmedetomidine impairs long-term
potentiation in the mouse hippocampus via activation of
α2-adrenoreceptors (22). van Oostrom et al (23) reported that the suppressive effect
of dexmedetomidine on memory formation occurred at doses which
reduce central nervous system activity. In a study on humans,
dexmedetomidine-induced amnesia was caused by a failure of
information to be encoded into long-term memory (24).
In the present study, we investigated whether
dexmedetomidine ameliorates or exacerbates memory function under
ICH conditions. For this purpose, a step-down avoidance test for
short-term memory and a radial 8-arm maze test for spatial learning
memory were conducted using rats. The anti-apoptotic effect of
dexmedetomidine against ICH was also evaluated. Apoptosis in the
hippocampus was detected using TUNEL assay, immunohistochemistry
for caspase-3, and western blot analysis for Bcl-2, Bax, Bid and
caspase-3 expression in the hippocampus. Western blot analysis for
BDNF and TrkB was also performed for the detection of cell survival
in the hippocampus.
Materials and methods
Experimental animals and treatments
All experiments were performed in accordance with
the Animal Care Guidelines of the National Institutes of Health
(NIH) and the Korean Academy of Medical Sciences. The effects of
dexmedetomidine on ICH-induced brain injury in rats were evaluated.
Seven-week-old Sprague-Dawley rats (210±10 g) were randomly divided
into 5 groups (n=8 in each group): the sham-operated group, the
ICH-induced brain injury group, the ICH-induced brain injury and 1
μg/kg dexmedetomidine-treated group, the ICH-induced brain injury
and 5 μg/kg dexmedetomidine-treated group, and the ICH-induced
brain injury and 10 μg/kg dexmedetomidine-treated group. In
addition, the effects of dexmedetomidine on normal rats were also
evaluated.
Dexmedetomidine was procured from Hospira Inc.
(Rocky Mount, NC, USA). The animals in the dexmedetomidine-treated
groups received the dose of dexmedetomidine in 0.5 ml saline
intraperitoneally (i.p.) once a day for 14 consecutive days,
commencing 1 day after the induction of ICH. The animals in the
sham-operated group and the ICH-induced brain injury group received
an equivalent dose of saline i.p. once a day for the same
duration.
Induction of collagenase-induced ICH
To induce ICH, the rats were anesthetized with
Zoletil 50® (10 mg/kg, i.p.; Vibac Laboratories, Carros,
France) and placed in a stereotaxic frame. The needle of a 10-μl
Hamilton syringe (Micro 701; Hamilton Co., Reno, NV, USA) was
inserted through a burr hole into the right hippocampus to the
following coordinates: 2.2 mm anterior and 2.2 mm lateral to the
bregma, with a depth of 4.2 mm. Distilled water (1 μl) containing
0.2 U collagenase (Type 4; Sigma Chemical Co., St. Louis, MO, USA)
was infused over 1 min. The needle remained in place for an
additional 3 min following the infusion, and then was withdrawn
slowly.
Step-down avoidance test
The latency in the step-down avoidance test was
measured to evaluate short-term memory, as previously described
(25). The rats were trained in
the step-down avoidance test 14 days after the initiation of
dexmedetomidine treatment. The rat was placed on a 7×25 cm platform
that was 2.5 cm high. The platform faced a 45×25 cm grid of
parallel stainless steel bars, 0.1 cm in caliber, spaced 1 cm
apart. In the training session, the animal received a 0.2 mA
scrambled foot shock for 2 sec immediately upon stepping down. Two
hours after the training session, the latency (sec) in each group
was measured. The time the rat spent on the platform before
stepping down and placing all 4 paws on the grid was defined as the
latency period. Latency over 180 sec was counted as 180 sec.
Radial 8-arm maze test
Spatial learning memory was tested using a radial
8-arm maze test, as previously described (26). The radial-arm maze apparatus
consisted of a central octagonal plate (30 cm in diameter) and 8
radiating arms (50 cm in length and 10 cm in width). The apparatus
was placed 1 m above the floor. A small receptacle filled with
water (3 cm in diameter and 1 cm in depth) was located at the end
of the arms. The rat was trained 3 times before the spatial
learning test. During the training sessions, the rat was deprived
of water for 24 h and allowed to explore the water for 5 min after
finishing each session. The test was conducted on the 13th day
after the initiation of dexmedetomidine treatment. The time the rat
spent seeking water at the end of the arms was counted. The test
was terminated when a rat found the water in all 8 arms or when
>8 min elapsed. The number of correct choices made before the
first wrong choice was counted, and re-entry into previously
visited arms was counted as the number of wrong choices made.
Preparation of tissues
The animals were sacrificed immediately after
determining the latency in the step-down avoidance test. The rats
were anesthetized using Zoletil 50® (10 mg/kg, i.p.;
Vibac Laboratories), transcardially perfused with 50 mM
phosphate-buffered saline (PBS), and fixed with a freshly prepared
solution consisting of 4% paraformaldehyde in 100 mM phosphate
buffer (PB, pH 7.4). The brains were dissected and stored overnight
in the same fixative solution. They were then transferred to a 30%
sucrose solution for cryoprotection. For immunohistochemistry, the
slices were coronal sectioned (40 μm thick) using a cryostat
(Leica, Nussloch, Germany). Ten slice sections on average in the
CA1 region were collected from each rat. The sections of 2.5 to 2.7
mm posterior to the bregma were used for TUNEL staining and
caspase-3 immunohistochemistry.
TUNEL staining
To visualize DNA fragmentation, a marker of
apoptosis, TUNEL staining was performed using an In Situ
Cell Death Detection kit® (Roche, Mannheim, Germany),
according to the manufacturer's instructions (25). The sections were post-fixed in
ethanol-acetic acid (2:1) and rinsed. The sections were then
incubated with proteinase K (100 μg/ml), rinsed and incubated in 3%
H2O2, permeabilized with 0.5% Triton X-100,
rinsed again, and incubated in the TUNEL reaction mixture. The
sections were rinsed and visualized using a converter-POD with
0.03% 3,3′-diaminobenzidine (DAB). Mayer's hematoxylin (Dako,
Glostrup, Denmark) was used as the counterstain, and the sections
were mounted onto gelatin-coated slides. The slides were air-dried
overnight at room temperature and dehydrated through a gradient of
ethanol and covered with coverslips using Permount®
(Fisher Scientific, New Jersey, NJ, USA).
Immunohistochemistry for caspase-3
To measure caspase-3 expression, caspase-3
immunohistochemistry was performed, as previously described
(25). The sections from each
brain were incubated overnight with mouse anti-caspase-3 antibody
(1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA), and then
with biotinylated mouse secondary antibody (1:200; Vector
Laboratories, Burlingame, CA, USA), and were then amplified for 1 h
using the Vector Elite ABC kit® (1:100; Vector
Laboratories). Antibody-biotin-avidin-peroxidase complexes were
visualized using 0.03% DAB, and the sections were mounted onto
gelatin-coated slides. The slides were air-dried overnight at room
temperature and dehydrated through a gradient of ethanol and
covered with coverslips using Permount® (Fisher
Scientific).
Western blot analysis
Western blot analysis was performed as previously
described (26). The hippocampal
tissues were dissected and collected, and were then immediately
frozen at −70°C. The right hemisphere was homogenized on ice, and
lysed in lysis buffer containing 50 mM HEPES (pH 7.5), 150 mM NaCl,
10% glycerol, 1% Triton X-100, 1 mM PMSF, 1 mM EGTA, 1.5 mM
MgCl2·6H2O, 1 mM sodium orthovanadate and 100
mM sodium fluoride. Protein content was measured using a Bio-Rad
colorimetric protein assay kit (Hercules, CA, USA). Protein samples
(30 μg) were separated on a sodium dodecyl sulfate-polyacrylamide
gel and transferred onto nitrocellulose membranes.
The membranes were incubated with 5% skim milk in
Tris-buffered saline containing 0.1% Tween-20 and then incubated
overnight at 4°C with the following primary antibodies: mouse
anti-β-actin, anti-Bcl-2, anti-Bax, anti-caspase-3, and rabbit
anti-Bid, anti-BDNF and anti-TrkB (1:1,000; Santa Cruz
Biotechnology). Subsequently, the membranes were incubated for 1 h
with secondary antibodies (1:2,000; Vector Laboratories), and band
detection was performed using the enhanced chemiluminescence (ECL)
detection kit (Santa Cruz Biotechnology).
Data analysis
For the confirmation of the expression of apoptotic
proteins, the detected bands were calculated densitometrically
using Molecular Analyst™, version 1.4.1 (Bio-Rad). The numbers of
TUNEL-positive and caspase-3-positive cells in the hippocampal CA1
region were counted hemilaterally under a light microscope
(Olympus, Tokyo, Japan), and they were expressed as the numbers of
cells/mm2 of the CA1 area. The area of the CA1 region
was measured using the Image-Pro Plus image analysis system (Media
Cyberbetics Inc., Silver Spring, MD, USA).
Statistical analysis was performed using one-way
ANOVA followed by Duncan's post-hoc test, and the results are
expressed as the means ± standard error of the mean (SEM). A
P-value <0.05 was considered to indicate a statistically
signficant difference.
Results
Effect of dexmedetomidine on short-term
memory in the step-down avoidance test
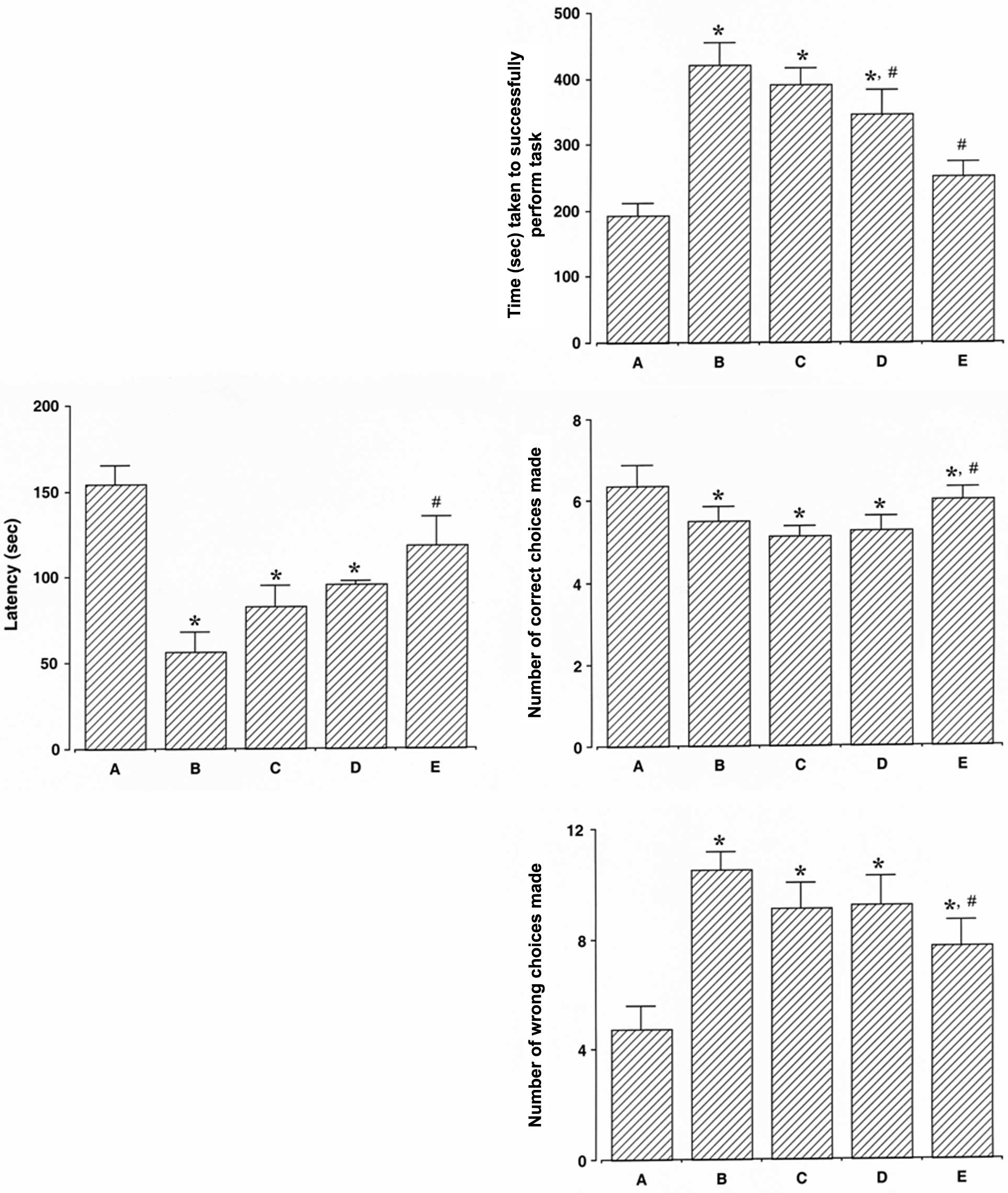
The latency of the reactions of the rats in the
step-down avoidance test is presented in Fig. 1 (left panel). The latency was
decreased following the induction of ICH (P<0.05) and treatment
with dexmedetomidine increased the latency in the rats with
ICH-induced brain injury (P<0.05). The present results revealed
that dexmedetomidine alleviated ICH-induced short-term memory
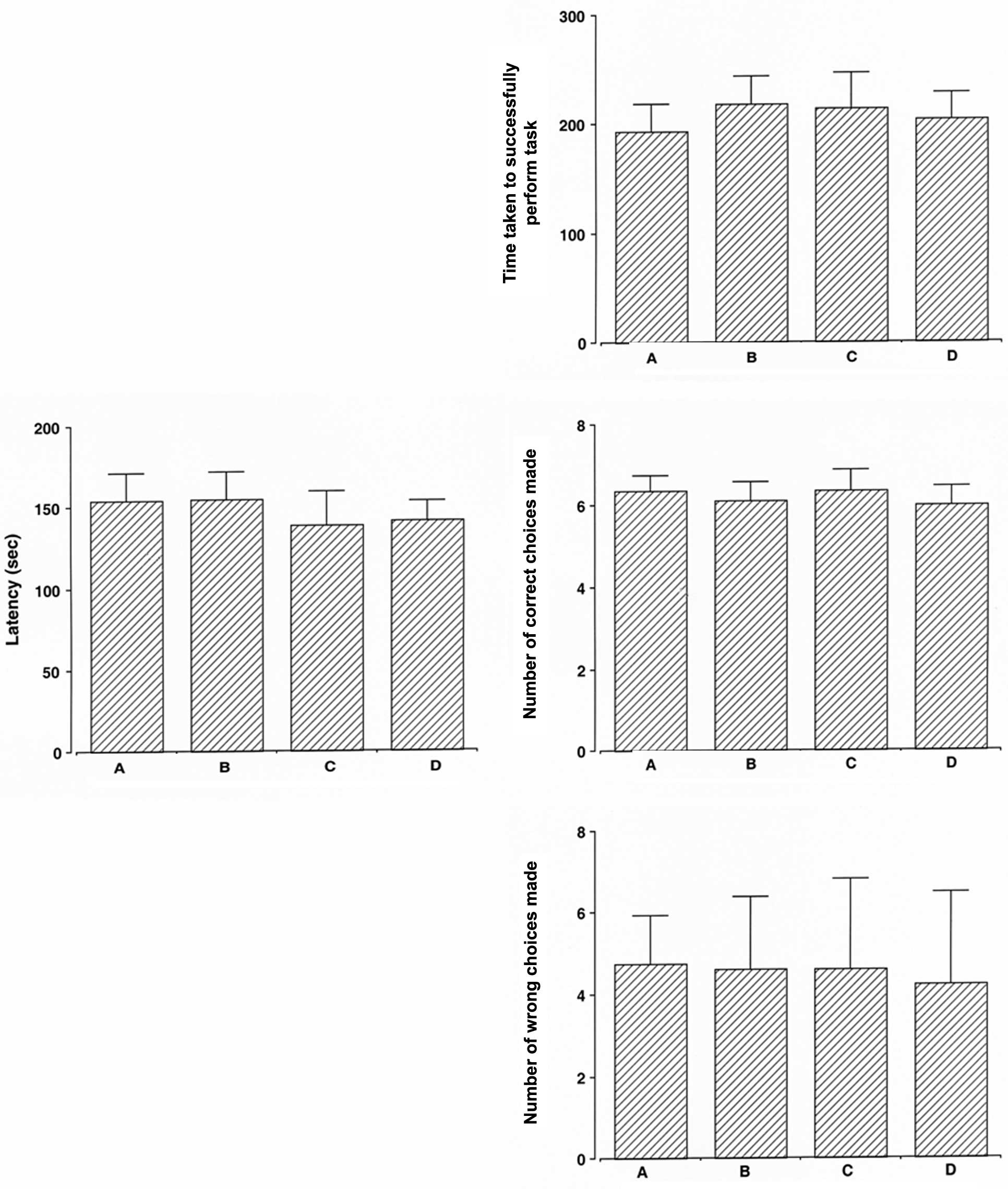
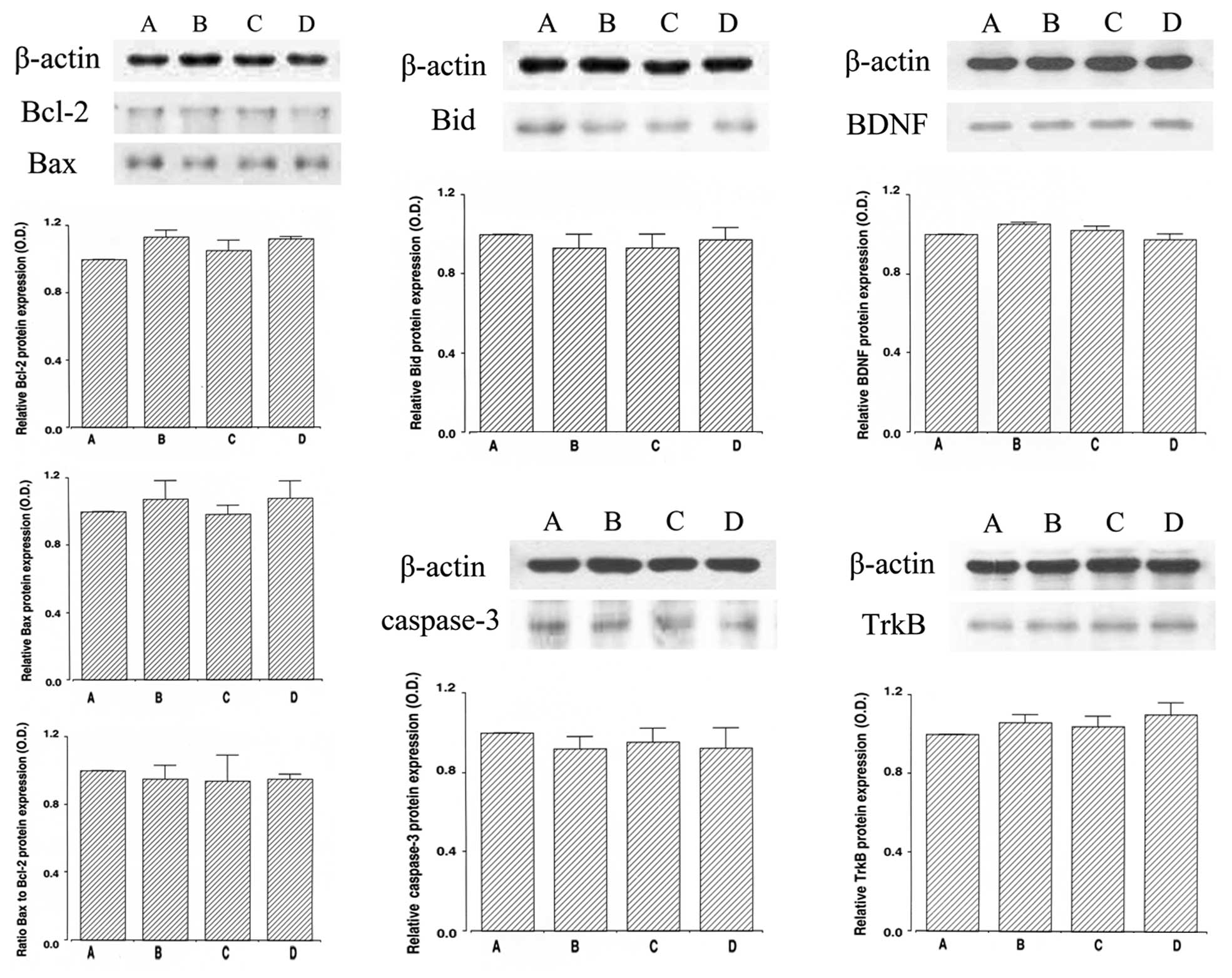
impairment. In the normal rats, dexmedetomidine exerted no
significant effect on latency (Fig.
2, left panel).
Effect of dexmedetomidine on spatial
learning memory in the radial 8-arm maze test
The time taken to successfully perform the task, the
number of correct choices made before the first wrong choice, and
the number of wrong choices made before the 8 successful
performances in the radial 8-arm maze test are presented in
Fig. 1 (right panel). The time
taken to successfully perform the task was longer, the number of
correct choices made was lower, and the number of wrong choices
made was higher in the rats with ICH-induced brain injury compared
to the control rats (P<0.05). Treatment with dexmedetomidine
reduced the time taken to successfully perform the task, increased
the number of correct choices made, and decreased the number of
wrong choices made in the rats with ICH-induced brain injury
(P<0.05). The present results revealed that treatment with
dexmedetomidine alleviated the ICH-induced spatial learning memory
impairment. In the normal rats, dexmedetomidine exerted no
significant effect on the time taken to successfully perform the
task, the number of correct choices made, and the number of wrong
choices made (Fig. 2, right
panel).
Effect of dexmedetomidine on the number
of TUNEL-positive cells in the hippocampal CA1 region
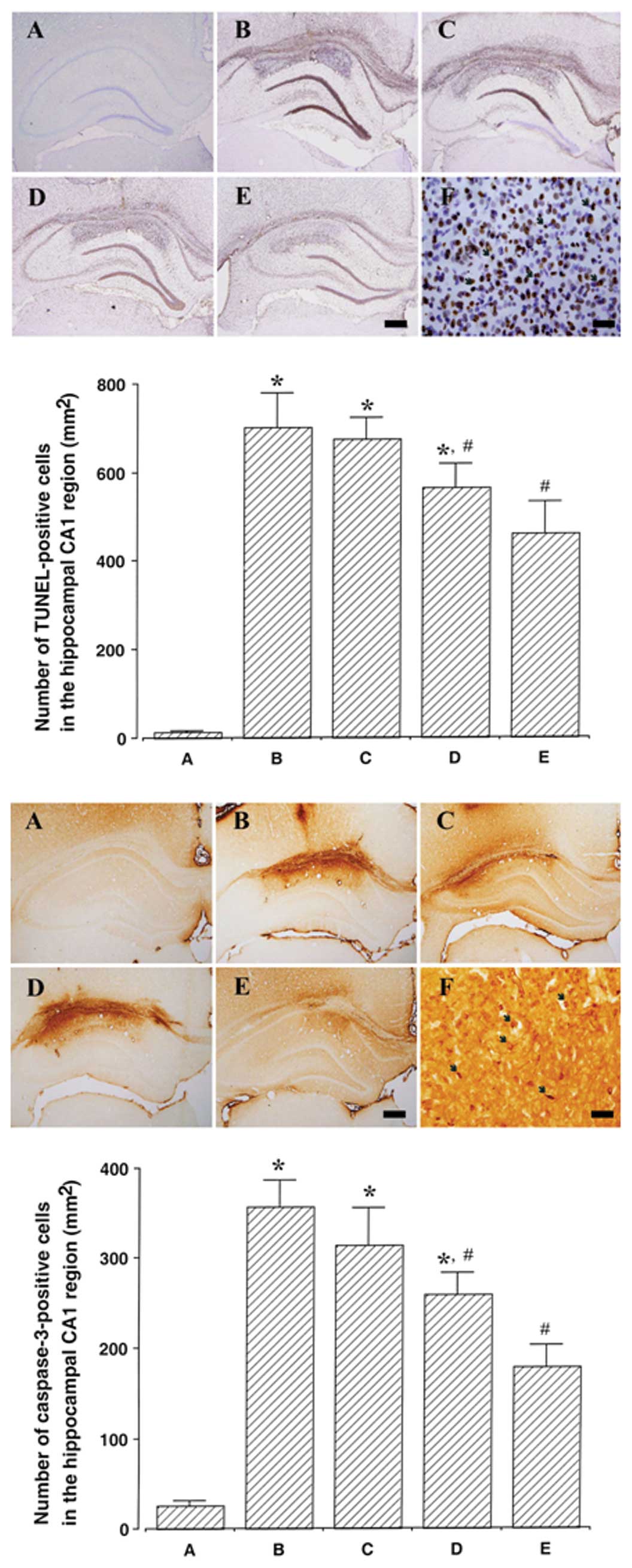
Photomicrographs of TUNEL-positive cells in the
hippocampal CA1 region are presented in Fig. 3 (upper panel). The induction of
ICH increased DNA fragmentation in the CA1 region (P<0.05) and
treatment with dexmedetomidine suppressed the ICH-induced DNA
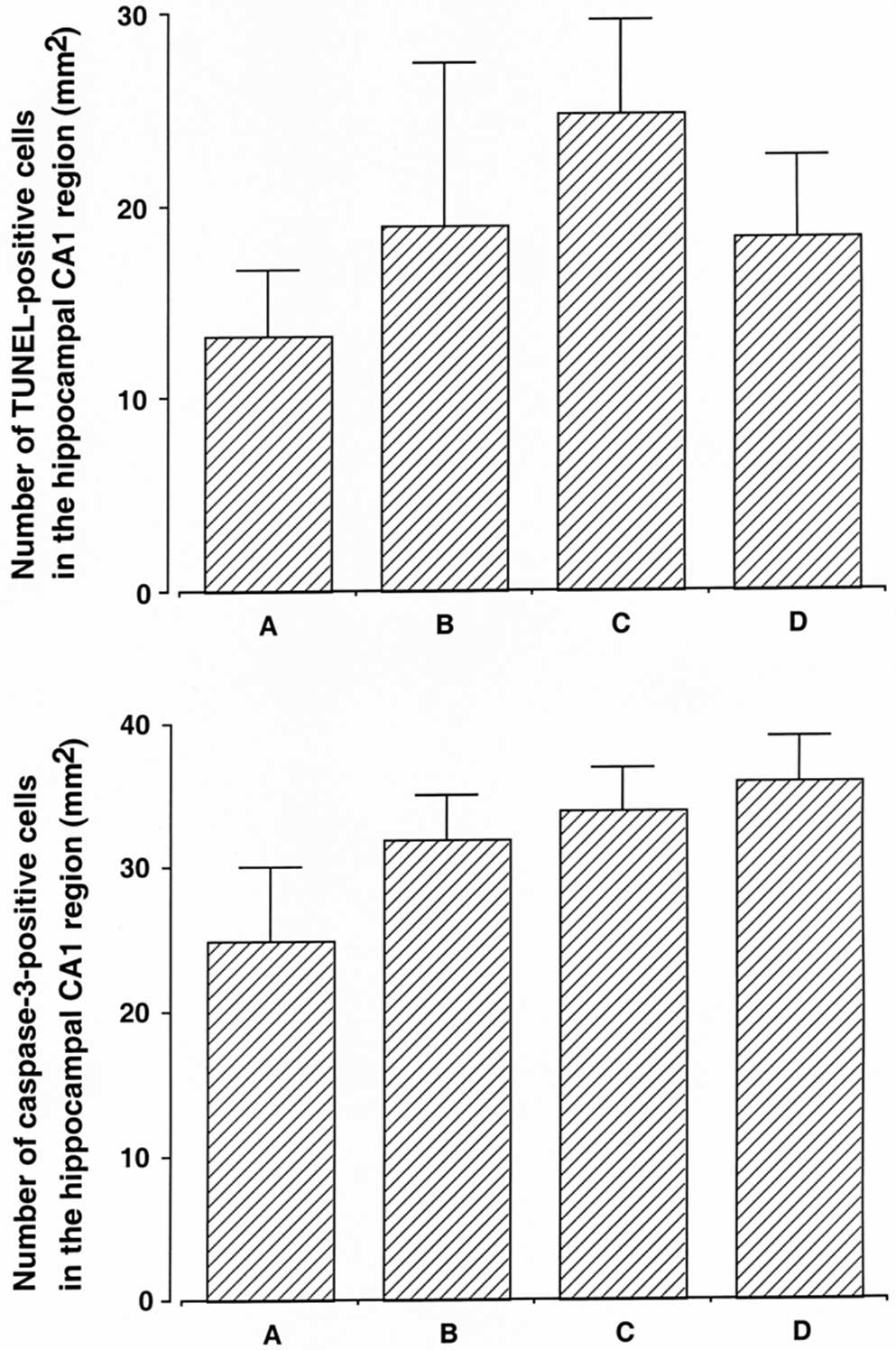
fragmentation (P<0.05). In the normal rats, dexmedetomidine
exerted no significant effect on DNA fragmentation (Fig. 4, upper panel).
Effect of dexmedetomidine on caspase-3
expression in the CA1 region
Photomicrographs of caspase-3-positive cells in the
hippocampal CA1 region are presented in Fig. 3 (lower panel). The induction of
ICH increased caspase-3 expression in the CA1 region (P<0.05)
and treatment with dexmedetomidine suppressed the ICH-induced
caspase-3 expression (P<0.05). In the normal rats,
dexmedetomidine exerted no significant effect on caspase-3
expression (Fig. 4, lower
panel).
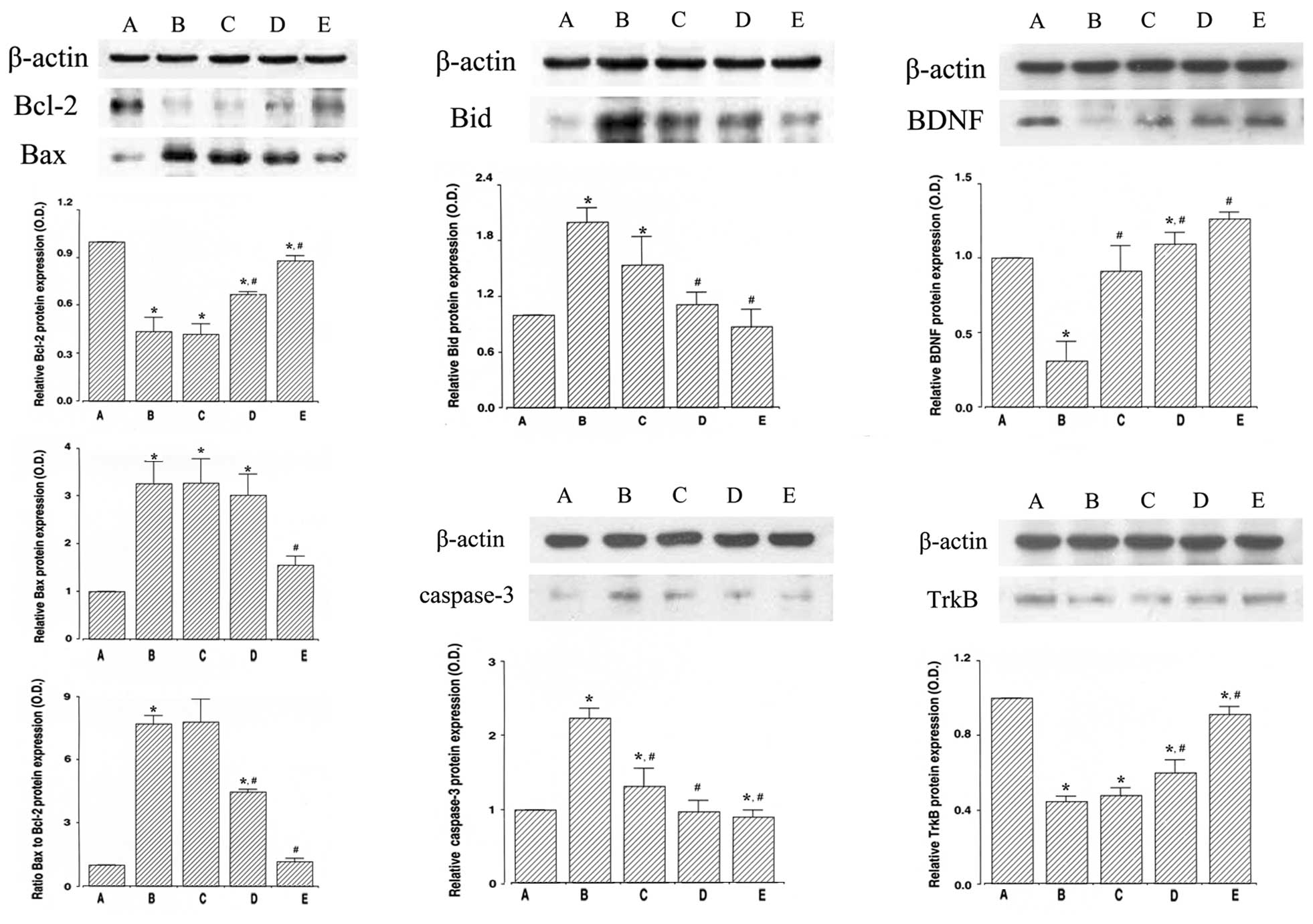
Effect of dexmedetomidine on the protein
levels of Bcl-2 and Bax in the hippocampus
To determine the expression of the anti-apoptotic
factor, Bcl-2, we evaluated the expression level of Bcl-2 (26–29
kDa) by western blot analysis (Fig.
5, upper left panel). The induction of ICH suppressed Bcl-2
expression in the hippocampus (P<0.05) and treatment with
dexmedetomidine increased Bcl-2 expression in the rats with
ICH-induced brain injury (P<0.05). In the normal rats,
dexmedetomidine exerted no significant effect on Bcl-2 expression
(Fig. 6, upper left panel).
To determine the expression of the pro-apoptotic
factor, Bax, we evaluated the expression level of Bax (24 kDa) by
western blot analysis (Fig. 5,
middle left panel). The induction of ICH increased Bax expression
in the hippocampus (P<0.05) and treatment with dexmedetomidine
inhibited the ICH-induced Bax expression (P<0.05). In the normal
rats, dexmedetomidine exerted no significant effect on Bax
expression (Fig. 6, middle left
panel).
Furthermore, we analyzed the ratio of Bax to Bcl-2
(Fig. 5, lower left panel). The
induction of ICH enhanced the ratio of Bax to Bcl-2 in the
hippocampus (P<0.05) and treatment with dexmedetomidine
suppressed the ratio of Bax to Bcl-2 in the rats with ICH-induced
brain injury (P<0.05). In the normal rats, dexmedetomidine
exerted no significant effect on the Bax to Bcl-2 ratio (Fig. 6, lower left panel).
Effect of dexmedetomidine on the protein
expression level of Bid in the hippocampus
To determine the expression of another pro-apoptotic
factor, Bid, we examined the expression level of Bid (22 kDa) by
western blot analysis (Fig. 5,
upper middle panel). The induction of ICH increased Bid expression
in the hippocampus (P<0.05) and treatment with dexmedetomidine
suppressed the ICH-induced Bid expression in the rats with
ICH-induced brain injury (P<0.05). In the normal rats,
dexmedetomidine exerted no significant effect on Bid expression
(Fig. 6, upper middle panel).
Effect of dexmedetomidine on the protein
expression level of caspase-3 in the hippocampus
To determine the expression of the apoptosis
executioner, caspase-3, we evaluated the protein expression level
of caspase-3 (17 kDa) by western blot analysis (Fig. 5, lower middle panel). The
induction of ICH increased caspase-3 expression in the hippocampus
(P<0.05) and treatment with dexmedetomidine suppressed the
ICH-induced caspase 3 expression (P<0.05). In the normal rats,
dexmedetomidine exerted no significant effect on caspase 3
expression (Fig. 6, lower middle
panel).
Effect of dexmedetomidine on the protein
expression levels of BDNF and TrkB in the hippocampus
To determine the expression of BDNF and TrkB, we
examined the protein levels of BDNF (15 kDa) and TrkB (95–145 kDa)
by western blot analysis (Fig. 5,
right panel). The induction of ICH decreased BDNF and TrkB
expression in the hippocampus (P<0.05) and treatment with
dexmedetomidine enhanced BDNF and TrkB expression in the rats with
ICH-induced brain injury (P<0.05). In the normal rats,
dexmedetomidine exerted no significant effect on BDNF and TrkB
expression (Fig. 6, right
panel).
Discussion
The animal model of ICH, induced by an injection of
collagenase, has been used to study the mechanisms of brain
injuries. The intracerebral injection of collagenase into the
hippocampus induces a lesion with triggered apoptotic neuronal cell
death in the hippocampus (2,3).
Cognitive impairment is a common symptom following ICH, with
executive and perceptual disorders being the most frequent
(27). Widespread patterns of
cognitive deficits have been observed in ICH patients (28).
In the present study, a step-down avoidance test
showed decreased latency in the rats with ICH-induced brain injury,
indicating the ICH-induced deterioration of short-term memory.
Treatment with dexmedetomidine increased the latency in the rats
with ICH-induced brain injury, indicating that dexmedetomidine
alleviated ICH-induced short-term memory impairment (Fig. 1, left panel). A radial-arm maze
test also showed a longer time taken to successfully perform the
task, a lower number of correct choices made, and a higher number
of wrong choices made in the rats with ICH-induced brain injury,
indicating the ICH-induced deterioration of spatial learning
memory. Treatment with dexmedetomidine shortened the time taken to
successfully perform the task, increased the number of correct
choices made, and decreased the number of wrong choices made in the
rats with ICH-induced brain injury, demonstrating that
dexmedetomidine alleviated the ICH-induced impairment of spatial
learning memory (Fig. 1, right
panel).
Hypoxic ischemia injury has been shown to induce
short-term memory deterioration in a step-down avoidance test, as
well as the impairment of spatial learning memory in a radial 8-arm
maze test (29). In a previous
study, maternal separation induced a decrease in latency in a
step-down avoidance test, representing memory loss. By contrast,
the increase in latency (to what are considered normal levels)
indicated the alleviation of memory loss (30).
In this study, the numbers of TUNEL-positive and
caspase-3-positive cells were increased following the induction of
ICH. By contrast, treatment with dexmedetomidine decreased the
numbers of TUNEL-positive and caspase-3-positive cells in the rats
with ICH-induced brain injury (Fig.
3). In addition, the expression of Bid and caspase-3 increased
following the induction of ICH. By contrast, treatment with
dexmedetomidine suppressed the expression of Bid and caspase-3 in
the rats with ICH-induced brain injury (Fig. 5, middle panel). Our results
demonstrated that dexmedetomidine treatment alleviated ICH-induced
apoptosis in the hippocampus.
Apoptosis appears to play a key role in neuronal
cell death during stroke (31,32). The induction of ICH in rats has
been shown to induce neuronal cell death, and apoptosis has been
closely implicated in ICH-induced neuronal cell death (7,31).
The upregulation of caspase-3 is an important hallmark of apoptosis
following ischemic and hemorrhagic brain insults (3,33).
An increase in the numbers of TUNEL-positive and caspase-3-positive
cells in the hippocampus indicates an enhancement of apoptotic
neuronal cell death in the hippocampus (3,30).
The pro-apoptotic molecule, Bid, contributes to the demise of nerve
cells following cerebral ischemia by the release of cytochrome
c and activation of caspases (34). Bid expression in the hippocampus
has also been shown to be upregulated following transient global
cerebral ischemia in rats (35).
In our study, the expression of Bcl-2 was
downregulated and that of Bax was upregulated in the hippocampus
following the induction of ICH, resulting in an increase in the
ratio of Bax to Bcl-2. Treatment with dexmedetomidine enhanced
Bcl-2 expression and suppressed Bax expression in the rats with
ICH-induced brain injury, resulting in a decrease in the ratio of
Bax to Bcl-2 (Fig. 5, left
panel). Our results demonstrated that dexmedetomidine exerted its
anti-apoptotic effects through the upregulation of Bcl-2 and the
downregulation of Bax expression in the hippocampus of rats with
IHC-induced brain injury.
The Bcl-2 family proteins have one or more Bcl-2
homology domains and play a crucial role in intracellular apoptotic
signal transduction by regulating the permeability of the
mitochondrial membrane. Bax, Bcl-xL, Bak, Bid and Bad are
pro-apoptotic, and they eliminate the mitochondrial membrane
potential by affecting the permeability transition pore and
facilitating the release of cytochrome c. Conversely, Bcl-2
and Bcl-xL function to conserve the membrane potential and block
the release of cytochrome c (36). Bcl-2 and Bcl-xL form heterodimers
with the main pro-apoptotic member, Bax, and they are thus
incapacitated from their protective function (37). Thus, the balance of Bax/Bcl-2 is
one of the crucial factors determining whether the cells undergo
apoptosis (8). In the study of
Engelhard et al (38),
dexmedetomidine upregulated Bcl-2 expression, and they suggested
that the neuroprotective property of dexmedetomidine involves the
modulation of the balance between pro- and anti-apoptotic proteins.
Blockade of α2-adrenoreceptors has been shown to
decrease the Bax mRNA level and increase the Bcl-xL mRNA level in
the cortex and hippocampus, indicating anti-apoptotic effects
(39). Dexmedetomidine has also
been shown to inhibit isoflurane-induced cortical apoptosis, and
this protective effect of dexmedetomidine was achieved by the
reversal of the isoflurane-induced decrease in Bcl-2 expression
(40).
In this study, the expression of BDNF and TrkB was
suppressed in the hippocampus following the inductionv of ICH. By
contrast, dexmedetomidine treatment increased BDNF and TrkB
expression in the rats with ICH-induced brain injury (Fig. 5, right panel). The results from
the present study indicate that the reduced BDNF and TrkB
expression in the hippocampus is involved in the short-term and
spatial learning memory impairment induced by ICH. The increase in
BDNF and TrkB expression following treatment with dexmedetomidine
ameliorated the ICH-induced memory deficits.
BDNF is involved in neuronal survival and
differentiation, and plays an important role in the learning
process due to its involvement in long-term potentiation in the
hippocampus (9,10,41). Plasminogen activator (tPA), by
activating the extracellular protease plasmin, converts the
precursor proBDNF to the mature BDNF and mature BDNF is a key
protein for long-term potentiation (42). BDNF expression in the hippocampus
has been shown to be suppressed following traumatic brain injury
and hemorrhage hypotension, suggesting that BDNF exerts
neuroprotective effects (30,43). The enhanced BDNF expression in the
hippocampus improves both short-term and long-term memory, and
contributes to neuronal survival and differentiation (44,45). The age-induced loss of short-term
and spatial working memory is accompanied with the suppression of
BDNF expression in the hippocampus, while the increased BDNF
expression contributes to memory enhancement (26). BDNF with fibrin-binding domain
significantly reduce the hematoma volume, alleviate tissue loss,
promote neuronal cell regeneration and improve behavioral
performance in rats with ICH-induced brain injury (46).
As mentioned above, the anti-apoptotic and
neuroprotective effects of dexmedetomidine against various brain
insults have been well documented. However, sedative-hypnotic drugs
are known to impair memory function; however, the details regarding
the nature of these effects are unknown. The memory-deteriorating
effects of dexmedetomidine have also been reported (22–24).
In the present study, we focused on the
memory-enhancing effects of dexmedetomidine under ICH conditions.
The present results showed that dexmedetomidine ameliorated
ICH-induced memory impairment. Dexmedetomidine also exerted
anti-apoptotic effects and increased BDNF expression in the
hippocampus of rats with ICH-induced brain injury. In the normal
rats, dexmedetomidine exerted no significant effects on apoptosis
and memory function, indicating that dexmedetomidine exerts no
detrimental effects on normal rats (Figs. 2, 4 and 6). Based on the present results,
dexmedetomidine may be used as a therapeutic agent for the
conservation of memory function in stroke patients.
Acknowledgements
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
funded by the Ministry of Education, Science and Technology of
Korea (2011-0013878).
References
|
1
|
Ferro JM: Update on intracerebral
haemorrhage. J Neurol. 253:985–999. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee HH, Kim H, Lee MH, Chang HK, Lee TH,
Jang MH, Shin MC, Lim BV, Shin MS, Kim YP, Yoon JH, Jeong IG and
Kim CJ: Treadmill exercise decreases intrastriatal
hemorrhage-induced neuronal cell death via suppression on caspase-3
expression in rats. Neurosci Lett. 352:33–36. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Suh HJ, So SM, Na YG, Ko IG, Kim SE, Sung
YS, Shin MS, Kim CJ, Cho YS and Kim KH: Neuroprotective effects of
tamsulosin on intracerebral hemorrhage. Neural Regen Res.
6:2505–2510. 2011.
|
|
4
|
Thompson CB: Apoptosis in the pathogenesis
and treatment of disease. Science. 267:1456–1462. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Johnson EM Jr, Greenlund LJ, Atkins PT and
Hsu CY: Neuronal apoptosis: current understanding of molecular
mechanisms and potential role in ischemic brain injury. J
Neurotrauma. 12:843–852. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Reed CJ: Apoptosis and cancer: strategies
for integrating programmed cell death. Semin Hematol. 37(Suppl 7):
9–16. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gong C, Boulis N, Qian J, Turner DE, Hoff
JT and Keep RF: Intracerebral hemorrhage-induced neuronal death.
Neurosurgery. 48:875–882. 2001.PubMed/NCBI
|
|
8
|
Upadhyay D, Panduri V, Ghio A and Kamp DW:
Particulate matter induces alveolar epithelial cell DNA damage and
apoptosis: role of free radicals and the mitochondria. Am J Respir
Cell Mol Biol. 29:180–187. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gomez-Pinilla F and Vaynman S: A
‘deficient environment’ in prenatal life may compromise systems
important for cognitive function by affecting BDNF in the
hippocampus. Exp Neurol. 192:235–243. 2005.
|
|
10
|
Minichiello L: TrkB signalling pathways in
LTP and learning. Nat Rev Neurosci. 10:850–860. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zimmerberg B, Foote HE and Van Kempen TA:
Olfactory association learning and brain-derived neurotrophic
factor in an animal model of early deprivation. Dev Psychobiol.
51:333–344. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hasegawa Y, Suzuki H, Altay O and Zhang
JH: Preservation of tropomyosin-related kinase B (TrkB) signaling
by sodium orthovanadate attenuates early brain injury after
subarachnoid hemorrhage in rats. Stroke. 42:477–483. 2011.
View Article : Google Scholar
|
|
13
|
Ard J, Doyle W and Bekker A: Awake
craniotomy with dexmedetomidine in pediatric patients. J Neurosurg
Anesthesiol. 15:263–266. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ramsay MA and Luterman DL: Dexmedetomidine
as a total intravenous anesthetic agent. Anesthesiology.
101:787–790. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dahmani S, Rouelle D, Gressens P and Mantz
J: Effects of dexmedetomidine on hippocampal focal adhesion kinase
tyrosine phosphorylation in physiologic and ischemic conditions.
Anesthesiology. 103:969–977. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Laudenbach V, Mantz J, Lagercrantz H,
Desmonts JM, Evrard P and Gressens P: Effects of
α2-adrenoceptor agonists on perinatal excitotoxic brain
injury: comparison of clonidine and dexmedetomidine.
Anesthesiology. 96:134–141. 2002.
|
|
17
|
Ma D, Hossain M, Rajakumaraswamy N, Arshad
M, Sanders RD, Franks NP and Maze M: Dexmedetomidine produces its
neuroprotective effect via the α2A-adrenoceptor subtype.
Eur J Pharmacol. 502:87–97. 2004.PubMed/NCBI
|
|
18
|
Eser O, Fidan H, Sahin O, Cosar M, Yaman
M, Mollaoglu H, Songur A and Buyukbas S: The influence of
dexmedetomidine on ischemic rat hippocampus. Brain Res.
1218:250–256. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cosar M, Eser O, Fidan H, Sahin O,
Buyukbas S, Ela Y, Yagmurca M and Ozen OA: The neuroprotective
effect of dexmedetomidine in the hippocampus of rabbits after
subarachnoid hemorrhage. Surg Neurol. 71:54–59. 2009. View Article : Google Scholar
|
|
20
|
Ayoglu H, Gul S, Hanci V, Bahadir B,
Bektas S, Mungan AG, Turan IO and Acikgoz B: The effects of
dexmedetomidine dosage on cerebral vasospasm in a rat subarachnoid
haemorrhage model. J Clin Neurosci. 17:770–773. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hall JE, Uhrich TD, Barney JA, Arain SR
and Ebert TJ: Sedative, amnestic, and analgesic properties of
small-dose dexmedetomidine infusions. Anesth Analg. 90:699–705.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Takamatsu I, Iwase A, Ozaki M, Kazama T,
Wada K and Sekiguchi M: Dexmedetomidine reduces long-term
potentiation in mouse hippocampus. Anesthesiology. 108:94–102.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
van Oostrom H, Stienen PJ, Doornenbal A
and Hellebrekers LJ: The α2-adrenoceptor agonist
dexmedetomidine suppresses memory formation only at doses
attenuating the perception of sensory input. Eur J Pharmacol.
629:58–62. 2010.
|
|
24
|
Hayama HR, Drumheller KM, Mastromonaco M,
Reist C, Cahill LF and Alkire MT: Event-related functional magnetic
resonance imaging of a low dose of dexmedetomidine that impairs
long-term memory. Anesthesiology. 117:981–995. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ko IG, Shin MS, Kim BK, Kim SE, Sung YH,
Kim TS, Shin MC, Cho HJ, Kim SC, Kim SH, Kim KH, Shin DH and Kim
CJ: Tadalafil improves short-term memory by suppressing
ischemia-induced apoptosis of hippocampal neuronal cells in
gerbils. Pharmacol Biochem Behav. 91:629–635. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim SE, Ko IG, Kim BK, Shin MS, Cho S, Kim
CJ, Kim SH, Baek SS, Lee EK and Jee YS: Treadmill exercise prevents
aging-induced failure of memory through an increase in neurogenesis
and suppression of apoptosis in rat hippocampus. Exp Gerontol.
45:357–365. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nys GM, van Zandvoort MJ, de Kort PL,
Jansen BP, de Haan EH and Kappelle LJ: Cognitive disorders in acute
stroke: prevalence and clinical determinants. Cerebrovasc Dis.
23:408–416. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Su CY, Chen HM, Kwan AL, Lin YH and Guo
NW: Neuro-psychological impairment after hemorrhagic stroke in
basal ganglia. Arch Clin Neuropsychol. 22:465–474. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ko IG, Cho H, Kim SE, Kim JE, Sung YH, Kim
BK, Shin MS, Cho S, Pak YK and Kim CJ: Hypothermia alleviates
hypoxic ischemia-induced dopamine dysfunction and memory impairment
in rats. Anim Cells Syst. 15:279–286. 2011. View Article : Google Scholar
|
|
30
|
Baek SS, Jun TW, Kim KJ, Shin MS, Kang SY
and Kim CJ: Effects of postnatal treadmill exercise on apoptotic
neuronal cell death and cell proliferation of maternal-separated
rat pups. Brain Dev. 34:45–56. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Matsushita K, Meng W, Wang X, Asahi M,
Asahi K, Moskowitz MA and Lo EH: Evidence for apoptosis after
intercerebral hemorrhage in rat striatum. J Cereb Blood Flow Metab.
20:396–404. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Qureshi AI: Nonsteroidal anti-inflammatory
drugs and the risk of intracerebral hemorrhage. Stroke. 34:379–386.
2003.PubMed/NCBI
|
|
33
|
Benchoua A, Braudeau J, Reis A, Couriaud C
and Onténiente B: Activation of proinflammatory caspases by
cathepsin B in focal cerebral ischemia. J Cereb Blood Flow Metab.
24:1272–1279. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Plesnila N, Zinkel S, Amin-Hanjani S, Qiu
J, Korsmeyer SJ and Moskowitz MA: Function of BID - a molecule of
the bcl-2 family - in ischemic cell death in the brain. Eur Surg
Res. 34:37–41. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Niizuma K, Endo H, Nito C, Myer DJ, Kim GS
and Chan PH: The PIDDosome mediates delayed death of hippocampal
CA1 neurons after transient global cerebral ischemia in rats. Proc
Natl Acad Sci USA. 105:16368–16373. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sugawara T, Fujimura M, Noshita N, Kim GW,
Saito A, Hayashi T, Narasimhan P, Maier CM and Chan PH: Neuronal
death/survival signaling pathways in cerebral ischemia. NeuroRx.
1:17–25. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kuwana T and Newmeyer DD: Bcl-2-family
proteins and the role of mitochondria in apoptosis. Curr Opin Cell
Biol. 15:691–699. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Engelhard K, Werner C, Eberspächer E,
Bachl M, Blobner M, Hildt E, Hutzler P and Kochs E: The effect of
the α2-agonist dexmedetomidine and the
N-methyl-D-aspartate antagonist S(+)-ketamine on the expression of
apoptosis-regulating proteins after incomplete cerebral ischemia
and reperfusion in rats. Anesth Analg. 96:524–531. 2003.
|
|
39
|
Il'inykh FA, Bannova AV, Kalinina TS and
Dygalo NN: Effects of ligands of α2-adrenoceptors on
mRNA level of apoptotic proteins in the developing rat brain. Izv
Akad Nauk Ser Biol. 1:104–109. 2008.(In Russian).
|
|
40
|
Sanders RD, Sun P, Patel S, Li M, Maze M
and Ma D: Dexmedetomidine provides cortical neuroprotection: impact
on anaesthetic-induced neuroapoptosis in the rat developing brain.
Acta Anaesthesiol Scand. 54:710–716. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xu B, Gottschalk W, Chow A, Wilson RI,
Schnell E, Zang K, Wang D, Nicoll RA, Lu B and Reichardt LF: The
role of brain-derived neurotrophic factor receptors in the mature
hippocampus: modulation of long-term potentiation through a
presynaptic mechanism involving TrkB. J Neurosci. 20:6888–6897.
2000.
|
|
42
|
Pang PT, Teng HK, Zaitsev E, Woo NT,
Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL and Lu B: Cleavage
of proBDNF by tPA/plasmin is essential for long-term hippocampal
plasticity. Science. 306:487–491. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hellmich HL, Garcia JM, Shimamura M, Shah
SA, Avila MA, Uchida T, Parsley MA, Capra BA, Eidson KA, Kennedy
DR, Winston JH, DeWitt DS and Prough DS: Traumatic brain injury and
hemorrhagic hypotension suppress neuroprotective gene expression in
injured hippocampal neurons. Anesthesiology. 102:806–814. 2005.
View Article : Google Scholar
|
|
44
|
Sairanen M, Lucas G, Ernfors P, Castrén M
and Castrén E: Brain-derived neurotrophic factor and antidepressant
drugs have different but coordinated effects on neuronal turnover,
proliferation, and survival in the adult dentate gyrus. J Neurosci.
25:1089–1094. 2005. View Article : Google Scholar
|
|
45
|
Suzuki A, Fukushima H, Mukawa T, Toyoda H,
Wu LJ, Zhao MG, Xu H, Shang Y, Endoh K, Iwamoto T, Mamiya N, Okano
E, Hasegawa S, Mercaldo V, Zhang Y, Maeda R, Ohta M, Josselyn SA,
Zhuo M and Kida S: Upregulation of CREB-mediated transcription
enhances both short- and long-term memory. J Neurosci.
31:8786–8802. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Han QQ, Jin W, Xiao ZF, Huang JC, Ni HB,
Kong J, Wu J, Chen B, Liang WB and Dai JW: The promotion of
neurological recovery in an intracerebral hemorrhage model using
fibrin-binding brain derived neurotrophic factor. Biomaterials.
32:3244–3252. 2011. View Article : Google Scholar
|