Atrial fibrillation (AF), typically characteristic
of rapid and chaotic electrical activity in the atria with
subsequent desynchronized atrial contractions, constitutes the most
common type of cardiac arrhythmia in the setting of clinical
practice, accounting for approximately one-third of
hospitalizations for irregular heart rhythms (1). The prevalence of AF is approximately
1% in the general population, and increases markedly with advancing
age, from approximately 0.5% of individuals in their fifties to
nearly 10% of the octogenarian population (2). According to the Framingham Heart
Study, the lifetime risk for development of AF is projected to be
25% for persons over the age of 40 (3). Currently, 2.5 million Americans are
suffering from AF, but with the aging population and improved
cardiovascular survival, that number is expected to exceed 16
million by the year 2050 (4). AF
is responsible for substantially increased cardiovascular morbidity
and mortality. Patients with AF have a 5-fold increased risk of
stroke, and it is estimated that 15–20% of all strokes are
attributable to AF (5). The risk
of cerebrovascular thromboembolism ascribed to AF also increases
strikingly with advancing age, rising from 1.5% at 50–59 years of
age to 23.5% at 80–89 years of age (5). AF independently increases the risk
of congestive heart failure and increases the risk of mortality
2-fold (6). Additionally, AF may
contribute to complications such as adverse hemodynamics, reduced
exercise tolerance, degraded quality of life, impaired cognition or
dementia, and tachycardia-induced cardiomyopathy (7). Therefore, AF has become an immense
and growing public health burden. Only in the United States, the
costs attributable to the care of individuals with nonvalvular AF
are in excess of $6.4 billion/year (8). Despite the high prevalence and
clinical significance, the molecular mechanism underlying AF
remains poorly understood.
Traditionally, AF has been perceived as a condition
that occurs in the context of atrial electrical and structural
remodeling that can result from miscellaneous cardiac and systemic
disorders, including hypertension, coronary artery disease,
rheumatic heart disease, congenital heart disease, chronic
pulmonary heart disease, cardiomyopathy, cardiac surgery, diabetes
mellitus, hyperthyroidism and electrolyte imbalance (1). However, in 30–45% of AF patients, no
obvious risk factors can be identified by routine medical
examination, and such AF is termed ‘idiopathic’ or ‘lone’ (1).; up to 15% of these cases have a
positive family history, and are thus defined as familial AF
(9). Increasing evidence
demonstrates the familial aggregation of AF and an enhanced
vulnerability to AF in the close relatives of patients with AF,
suggesting that genetic defects may be involved in the pathogenesis
of AF in a subset of patients (10–16). Genome-wide genetic linkage
analysis with highly polymorphic microsatellite markers mapped
susceptibility loci for AF on human chromosomes 10q22, 6q14-16,
11p15.5, 5p13, 10p11-q21 and 5p15, of which AF-causing mutations in
two genes, KCNQ1 on chromosome 11p15.5 and NUP155 on
chromosome 5p13, were identified and functionally characterized
(17–23). In addition, genetic screening of
candidate genes revealed an increasing number of AF associated
mutations in genes encoding potassium channel subunits (KCNH2,
KCNA5, KCNJ2, KCNJ8 and KCNE1-5), sodium channel
subunits (SCN5A and SCN1B-3B), signaling peptide
(NPPA), gap junctions (GJA1 and GJA5), and others
(24–44). Nevertheless, these causative
mutations appear to be relatively rare causes of AF, and the
genetic determinants for AF in an overwhelming majority of patients
remain elusive.
Emerging evidence indicates that abnormal
embryological development of the cardiovascular system,
particularly the pulmonary venous myocardium, is a major anatomic
substrate for AF (45).
Developmental biology studies substantiate the key role for several
transcription factors, including NKX2.5, GATA4, GATA5 and GATA6, in
the normal cardiovascular morphogenesis (46–48), and multiple mutations in GATA4,
GATA5 and GATA6 have been causally associated with AF (49–57). NKX2.5 is a member of the
NK2-family of transcription factors and its expression and
functions overlap with those of the GATA family during
cardiovascular development, particularly in synergistic regulation
of target gene expressions cooperatively with GATA4 (58), which justifies NKX2.5 as a
prime candidate gene for AF.
A cohort of 110 unrelated index patients with
familial AF was recruited from the Han Chinese population in China.
The available relatives of the probands were enrolled. A total of
200 ethnically-matched unrelated healthy individuals were enlisted
as controls. Peripheral venous blood samples were prepared and
clinical data including medical records, electrocardiogram and
echocardiography reports were collected. The study subjects were
clinically classified using a consistently applied set of
definitions (9,57). Briefly, AF was diagnosed by a
standard 12-lead electrocardiogram demonstrating no P-waves and
irregular R-R intervals irrespective of clinical symptoms. Lone AF
was defined as AF occurring in individuals <60 years of age
without other cardiac or systemic diseases by physical examination,
electrocardiogram, transthoracic echocardiogram, and extensive
laboratory tests. Familial AF was referred to as the presence of
documented lone AF in additional two or more first- or
second-degree relatives. Relatives with AF occurring at any age in
the setting of structural heart disease (hypertensive, ischemic,
myocardial or valvular) were classified as ‘undetermined’ for
having an inherited form of AF. The ‘undetermined’ classification
was also used if documentation of AF on an electrocardiogram
tracing was lacking in relatives with symptoms consistent with AF
(palpitation, dyspnea and light-headedness), or if a screening
electrocardiogram and echocardiogram were not performed, regardless
of the symptoms. Relatives were classified as ‘unaffected’ if they
were asymptomatic and had a normal electrocardiogram. Paroxysmal AF
was defined as AF lasting >30 sec that terminated spontaneously.
Persistent AF was defined as AF lasting more than seven days and
requiring either pharmacologic therapy or electrical cardioversion
for termination. AF that was refractory to cardioversion or that
was allowed to continue was classified as permanent. The study
protocol was reviewed and approved by the local institutional
Ethics Committee and written informed consent was obtained from all
participants.
Genomic DNA from all participants was isolated from
blood lymphocytes with the Wizard Genomic DNA Extraction Kit
(Promega, Madison, WI, USA), according to the manufacturer’s
instructions. Initially, the whole coding region and splice
junctions of the NKX2.5 gene were sequenced in 110 unrelated
index patients with familial AF. Subsequently, genotyping
NKX2.5 in the available relatives of the mutation carrier
and 200 ethnically-matched unrelated healthy individuals who served
as controls, was performed. The referential genomic DNA sequence of
NKX2.5 was derived from GenBank (accession no. NT_023133), a
gene sequence database at the National Center for Biotechnical
Information (NCBI; http://www.ncbi.nlm.nih.gov/). Using the online Primer
3 software (http://frodo.wi.mit.edu/), the primer
pairs used to amplify the coding exons (exon 1–2) and intron-exon
boundaries of NKX2.5 by polymerase chain reaction (PCR) were
designed as follows: primer 1, forward, 5′-CAC GAT GCA GGG AAG
CTG-3′ and reverse, 5′-AGT TTC TTG GGG ACG AAA GC-3′ (the PCR
product was 477 base pairs in size); primer 2, forward, 5′-CCT CCA
CGA GGA TCC CTT AC-3′ and reverse, 5′-CGA GTC CCC TAG GCA TGG-3′
(the product was 463 base pairs); primer 3, forward, 5′-AGA ACC GGC
GCT ACA AGT G-3′ and reverse, 5′-GAG TCA GGG AGC TGT TGA GG-3′ (the
product was 473 base pairs). The PCR was performed using HotStar
TaqDNA Polymerase (Qiagen, Hilden, Germany) on a Veriti Thermal
Cycler (Applied Biosystems, Foster, CA, USA) with standard
conditions and concentrations of reagents. Amplified products were
purified with the QIAquick Gel Extraction Kit (Qiagen). Both
strands of each PCR product were sequenced with a
BigDye® Terminator v3.1 Cycle Sequencing Kit on an
ABI-PRISM 3130xl DNA Analyzer (both from Applied Biosystems). The
sequencing primers were the same as mentioned above for the
specific region amplifications. DNA sequences were analyzed with
the DNA Sequencing Analysis Software v5.1 (Applied Biosystems). The
variant was corroborated by re-sequencing of an independent
PCR-generated amplicon from the subject and met the quality control
threshold with a call rate exceeding 99%. For an identified
sequence variant, the Exome Variant Server (EVS; http://evs.gs.washington.edu/EVS) and NCBI’s
single nucleotide polymorphism (SNP; http://www.ncbi.nlm.nih.gov/SNP) online databases were
queried to confirm its novelty.
Conservation of the amino acid altered by missense
mutation was appraised by aligning human NKX2.5 to chimpanzee,
monkey, dog, cow, mouse, rat, chicken and zebrafish NKX2.5 using
the HomoloGene and Show Multiple Alignment links on the NCBI’s
website (http://www.ncbi.nlm.nih.gov/homologene).
The recombinant expression plasmid NKX2.5-pEFSA and
the atrial natriuretic factor (ANF)-luciferase reporter plasmid,
which contains the 2600-bp 5′-flanking region of the ANF
gene, i.e., ANF(−2600)-Luc, were kindly provided by Dr Ichiro
Shiojima, from the Department of Cardiovascular Science and
Medicine, Chiba University Graduate School of Medicine, Chuo-ku,
Chiba, Japan. The identified mutation was introduced into the
wild-type NKX2.5 using a QuickChange II XL Site-Directed
Mutagenesis Kit (Stratagene) with a complementary pair of primers.
The mutant was sequenced to confirm the desired mutation and to
exclude any other sequence variations.
COS-7 cells were cultured in Dulbecco’s modified
Eagle’s medium supplemented with 10% fetal calf serum. The
ANF(−2600)-Luc reporter construct and an internal control reporter
plasmid pGL4.75 (hRluc/CMV; Promega) were used in transient
transfection assays to examine the transcriptional activation
function of the NKX2.5 mutant. COS-7 cells were transfected
with 0.4 μg of wild-type or mutant NKX2.5-pEFSA expression vector,
1.0 μg of ANF(−2600)-Luc reporter construct, and 0.04 μg of pGL4.75
control reporter vector using PolyFect Transfection Reagent
(Qiagen). For co-transfection experiments, 0.2 μg of wild-type
NKX2.5-pEFSA, 0.2 μg of mutant NKX2.5-pEFSA or empty vector pEFSA,
1.0 μg of ANF(−2600)-Luc, and 0.04 μg of pGL4.75 were used. Firefly
luciferase and Renilla luciferase activities were measured with the
Dual-Glo Luciferase Assay System (Promega) 48 h after transfection.
The activity of the ANF promoter was presented as fold
activation of Firefly luciferase relative to Renilla luciferase.
Three independent experiments were performed at minimum for
wild-type and mutant NKX2.5.
Data are expressed as the means ± SD. Continuous
variables were tested for normality of distribution and student’s
unpaired t-test was used for comparison of numeric variables
between two groups. Comparison of the categorical variables between
two groups was performed using Pearson’s χ2 test or
Fisher’s exact test when appropriate. A two-tailed P-value of
<0.05 was considered to indicate a statistically significant
difference.
A cohort of 110 unrelated index patients with
familial AF and a total of 200 ethnically-matched unrelated healthy
individuals used as controls were enrolled and clinically
evaluated. None of the participants had apparent traditional risk
factors for AF. There was no significant difference between patient
and control groups in baseline characteristics including age,
gender, body mass index, blood pressure, fasting blood glucose,
serum lipid, left atrial dimension, left ventricular ejection
fraction, heart rate at rest, as well as life style (data not
shown). In the present study, four patients were also diagnosed
with hypertension in accordance with the criterion that the average
systolic or diastolic blood pressure (2 readings made after 5 min
of rest in the sitting position) is 140 or 90 mm Hg, respectively,
but at the time of initial diagnosis of AF, their blood pressures
were normal. The baseline clinical characteristics of the 110 index
patients with familial AF are summarized in Table I.
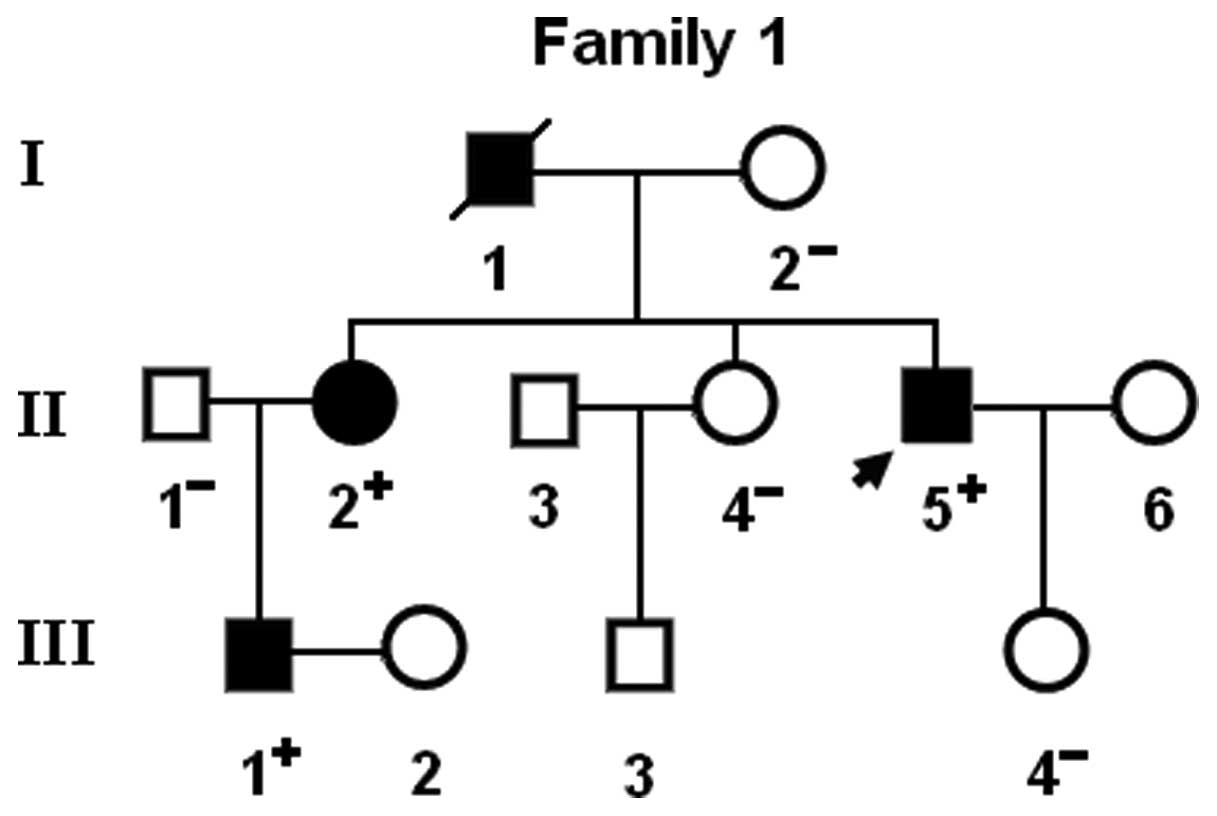
Notably, congenital atrial septal defect was
confirmed by the early echocardiogram in patient I-1 and patient
II-2 from family 1. In addition, two previously reported
NKX2.5 sequence polymorphisms, including c.63A>G and
c.606C>G, were observed in both AF patients and control
individuals. However, there was no significant difference in either
of the two allele frequencies between the AF patient and healthy
control groups. All the sequence variants and their allele
frequencies are listed in Table
III.
A cross-species alignment of NKX2.5 protein
sequences displayed that the altered amino acid was completely
conserved evolutionarily, underscoring its functional importance
(Fig. 4).
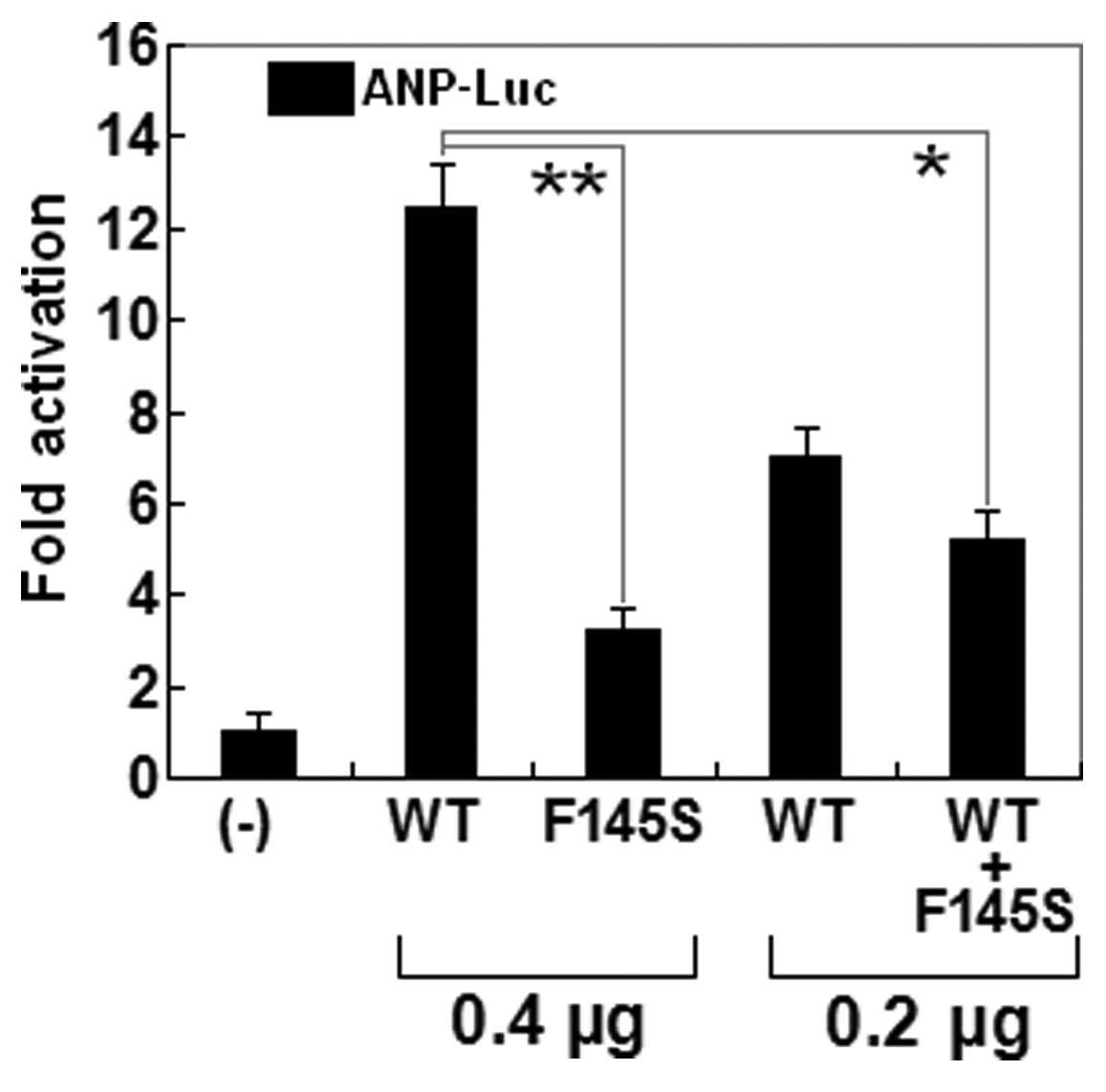
The transcriptional activation function of the
mutated NKX2.5 in COS-7 cells was characterized using a luciferase
reporter, which was driven by the promoter of ANP, one of
NKX2.5-directed cardiac target genes. The activity of the
ANP promoter was expressed as fold activation of Firefly
luciferase relative to Renilla luciferase. The same amounts of
wild-type (0.4 μg) and F145S-mutant NKX2.5 (0.4 μg)
activated the ANP promoter by ~12- and ~3-fold, respectively. When
the same amount of wild-type NKX2.5 (0.2 μg) was
cotransfected with F145S-mutant NKX2.5 (0.2 μg), the induced
activation of the ANP promoter was ~5-fold. These results suggest
that the NKX2.5 mutation results in a significantly reduced
transcriptional activation compared with its wild-type counterpart
(Fig. 5).
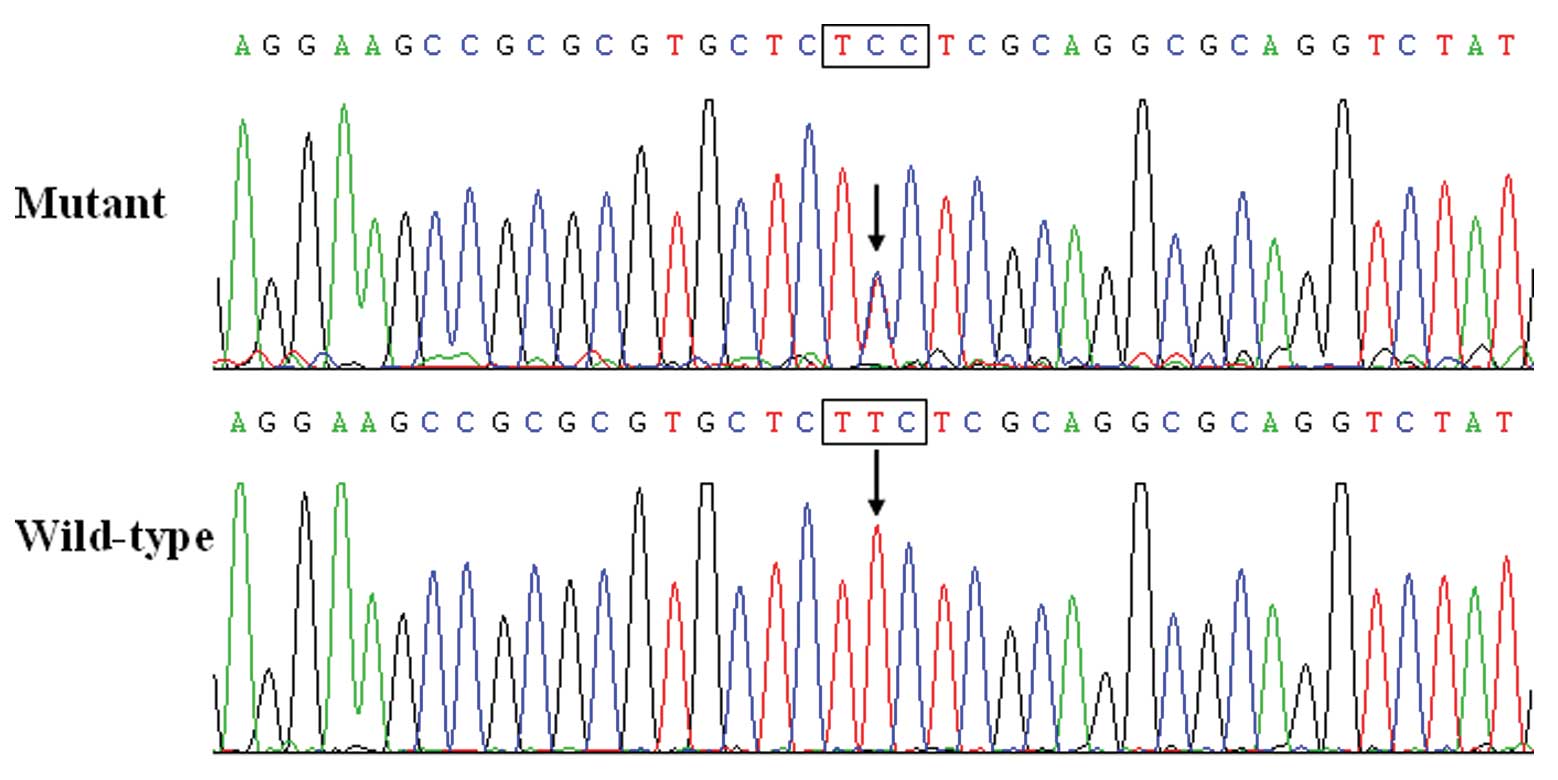
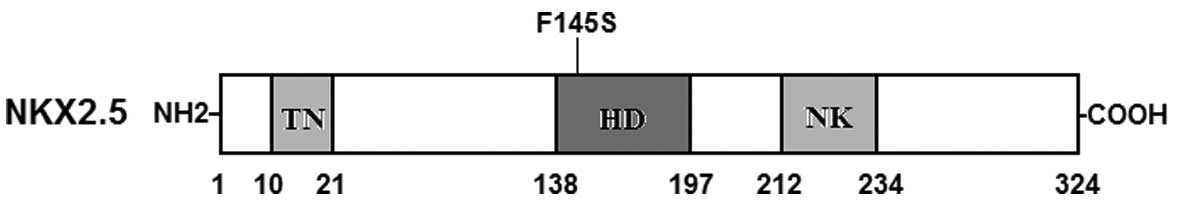
In the present study, a novel heterozygous NKX2.5
mutation, p.F145S, was identified in a family with familial AF.
This missense mutation was present in all the affected family
members examined but was absent in the unaffected family members
available and in the 400 normal chromosomes from a matched control
population. A cross-species alignment of multiple NKX2.5 protein
sequences exhibited that the altered amino acid was completely
conserved evolutionarily. Functional analysis demonstrated that the
mutant NKX2.5 was associated with a significantly decreased
transcriptional activity. Therefore, it is highly likely that
functionally impaired NKX2.5 is involved in the pathogenesis of AF
in this family. To our knowledge, this is the first report on the
relationship between NKX2.5 loss-of-function mutation and enhanced
susceptibility to AF.
Previous investigations revealed that NKX2.5 is an
upstream regulator of several genes expressed during embryogenesis
including the genes that encode ANF, brain natriuretic peptide and
α-cardiac actin (60). As a
well-known NKX2.5 downstream target molecule, ANF contains several
NKX2.5 binding sites in its proximal promoter region, including
−242 from the transcription start site, which has been confirmed as
an in vivo binding site of NKX2.5 (61). Therefore, the functional
characteristics of the NKX2.5 mutation can be explored by analysis
of the transcriptional activity of the ANF promoter in cells
transfected with NKX2.5 mutant in contrast to its wild-type
counterpart. In this study, the functional role of the novel
p.F145S mutation of NKX2.5 identified in familial AF patients was
analyzed by transcriptional activity assays and the results
demonstrated a significantly decreased transcriptional activity on
a downstream gene. These findings indicate that NKX2.5
haploinsufficiency caused by mutation is potentially an alternative
pathophysiological mechanism of AF.
The finding that functionally compromised NKX2.5
predisposes to AF may be partially due to the abnormally developed
pulmonary vein myocardium. The pulmonary venous vessel is
ensheathed by a layer of myocardium referred to as pulmonary
myocardial sleeve, which has been verified to be responsible for
the initiation and perpetuation of AF by several potential
arrhythmogenic mechanisms including enhanced intrinsic pacemaker
activity and liability to reentrance (62–64). Genetic-labeling lineage tracing
studies have validated that NKX2.5 is expressed in the atria as
well as in the pulmonary myocardium and is essential for the
localized formation of the sinoatrial node during embryogenesis.
NKX2.5 may functionally serve as a suppressor of the sinoatrial
node lineage gene program, which limits pacemaker activity to the
sinoatrial and atrioventricular nodes. When the level of NKX2.5
protein decreased in a hypomorphic model, the pulmonary
cardiomyocytes shifted to connexin40-negative, HCN4-positive cells,
a nodal-like phenotype with pacemaker activity (63). In NKX2.5-null mouse
embryos, HCN4 was activated along the entire embryonic heart
tube, whereas connexin40 expression was inhibited, ectopic
pacemaker cells were observed throughout the heart tube (64). Hence, NKX2.5 loss-of-function
mutation presumably contributes to formation of the pulmonary
myocardium sleeve and switch of the pulmonary myocardium to a
sinoatrial node-like phenotype, creating an atrial
electrophysiological matrix in favor of AF.
There are some downstream genes transactivated by
NKX2.5, and mutations in several target genes have been linked to
AF, including the genes encoding ANF and gap junction protein
connexin40 (38,39,41–43). Therefore, it is probable that
mutated NKX2.5 confers susceptibility to AF by decreasing
expressions of target genes.
It is of note that congenital atrial septal defect
was documented in 2 AF patients carrying p.F145S mutation of
NKX2.5. Similarly, congenital cardiovascular malformations were
previously confirmed in AF patients carrying GATA4, GATA5 or GATA6
mutations (49–57). Markedly, a long list of mutations
in these genes has been implicated in a wide variety of congenital
cardiovascular anomalies (65–75). These observational results
indicate that AF may share a common genetic origin with congenital
heart disease.
In conclusion, the findings of the present study
provide novel insights into the molecular mechanism of AF,
suggesting potential implications for early prophylaxis and
allele-specific therapy of this common arrhythmia.
The authors are grateful to the participants for
their dedication to the study. This study was supported by grants
from the National Natural Science Fund of China (81270161, 81070153
and 30570768), the National Basic Research Program of China
(2010CB912604), the Personnel Development Foundation of Shanghai,
China (2010019), and the Key Program of Basic Research of Shanghai,
China (10JC1414000, 10JC1414001 and 10JC1414002).
|
1
|
Fuster V, Rydén LE, Cannom DS, Crijns HJ,
Curtis AB, Ellenbogen KA, Halperin JL, Kay GN, Le Huezey JY, Lowe
JE, Olsson SB, Prystowsky EN, Tamargo JL, Wann LS, Smith SC Jr,
Priori SG, Estes NA III, Ezekowitz MD, Jackman WM, January CT, Lowe
JE, Page RL, Slotwiner DJ, Stevenson WG, Tracy CM, Jacobs AK,
Anderson JL, Albert N, Buller CE, Creager MA, Ettinger SM, Guyton
RA, Halperin JL, Hochman JS, Kushner FG, Ohman EM, Stevenson WG,
Tarkington LG and Yancy CW; American College of Cardiology
Foundation/American Heart Association Task Force. 2011 ACCF/AHA/HRS
focused updates incorporated into the ACC/AHA/ESC 2006 guidelines
for the management of patients with atrial fibrillation: a report
of the American College of Cardiology Foundation/American Heart
Association Task Force on practice guidelines. Circulation.
123:e269–e367. 2011.
|
|
2
|
Go AS, Hylek EM, Phillips KA, Chang Y,
Henault LE, Selby JV and Singer DE: Prevalence of diagnosed atrial
fibrillation in adults: national implications for rhythm management
and stroke prevention: the AnTicoagulation and Risk Factors in
Atrial Fibrillation (ATRIA) Study. JAMA. 285:2370–2375. 2001.
View Article : Google Scholar
|
|
3
|
Lloyd-Jones DM, Wang TJ, Leip EP, Larson
MG, Levy D, Vasan RS, D’Agostino RB, Massaro JM, Beiser A, Wolf PA
and Benjamin EJ: Lifetime risk for development of atrial
fibrillation: the Framingham Heart Study. Circulation.
110:1042–1046. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Miyasaka Y, Barnes ME, Gersh BJ, Cha SS,
Bailey KR, Abhayaratna WP, Seward JB and Tsang TS: Secular trends
in incidence of atrial fibrillation in Olmsted County, Minnesota,
1980 to 2000, and implications on the projections for future
prevalence. Circulation. 114:119–125. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wolf PA, Abbott RD and Kannel WB: Atrial
fibrillation as an independent risk factor for stroke: the
Framingham Study. Stroke. 22:983–988. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Benjamin EJ, Wolf PA, D’Agostino RB,
Silbershatz H, Kannel WB and Levy D: Impact of atrial fibrillation
on the risk of death: the Framingham Heart Study. Circulation.
98:946–952. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Magnani JW, Rienstra M, Lin H, Sinner MF,
Lubitz SA, McManus DD, Dupuis J, Ellinor PT and Benjamin EJ: Atrial
fibrillation: current knowledge and future directions in
epidemiology and genomics. Circulation. 124:1982–1993. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Coyne KS, Paramore C, Grandy S, Mercader
M, Reynolds M and Zimetbaum P: Assessing the direct costs of
treating nonvalvular atrial fibrillation in the United States.
Value Health. 9:348–356. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Darbar D, Herron KJ, Ballew JD, Jahangir
A, Gersh BJ, Shen WK, Hammill SC, Packer DL and Olson TM: Familial
atrial fibrillation is a genetically heterogeneous disorder. J Am
Coll Cardiol. 41:2185–2192. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ellinor PT, Yoerger DM, Ruskin JN and
MacRae CA: Familial aggregation in lone atrial fibrillation. Hum
Genet. 118:179–184. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Arnar DO, Thorvaldsson S, Manolio TA,
Thorgeirsson G, Kristjansson K, Hakonarson H and Stefansson K:
Familial aggregation of atrial fibrillation in Iceland. Eur Heart
J. 27:708–712. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Junttila MJ, Raatikainen MJ, Perkiömäki
JS, Hong K, Brugada R and Huikuri HV: Familial clustering of lone
atrial fibrillation in patients with saddleback-type ST-segment
elevation in right precordial leads. Eur Heart J. 28:463–468. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Christophersen IE, Ravn LS,
Budtz-Joergensen E, Skytthe A, Haunsoe S, Svendsen JH and
Christensen K: Familial aggregation of atrial fibrillation: a study
in Danish twins. Circ Arrhythm Electrophysiol. 2:378–383. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang YQ, Zhang XL, Wang XH, Tan HW, Shi
HF, Fang WY and Liu X: Familial aggregation of lone atrial
fibrillation in the Chinese population. Intern Med. 49:2385–2391.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lubitz SA, Yin X, Fontes JD, Magnani JW,
Rienstra M, Pai M, Villalon ML, Vasan RS, Pencina MJ, Levy D,
Larson MG, Ellinor PT and Benjamin EJ: Association between familial
atrial fibrillation and risk of new-onset atrial fibrillation.
JAMA. 304:2263–2269. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fox CS, Parise H, D’Agostino RB Sr,
Lloyd-Jones DM, Vasan RS, Wang TJ, Levy D, Wolf PA and Benjamin EJ:
Parental atrial fibrillation as a risk factor for atrial
fibrillation in offspring. JAMA. 291:2851–2855. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brugada R, Tapscott T, Czernuszewicz GZ,
Marian AJ, Iglesias A, Mont L, Brugada J, Girona J, Domingo A,
Bachinski LL and Roberts R: Identification of a genetic locus for
familial atrial fibrillation. N Engl J Med. 336:905–911. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ellinor PT, Shin JT, Moore RK, Yoerger DM
and MacRae CA: Locus for atrial fibrillation maps to chromosome
6q14-16. Circulation. 107:2880–2883. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang
Y, Xu WY, Jin HW, Sun H, Su XY, Zhuang QN, Yang YQ, Li YB, Liu Y,
Xu HJ, Li XF, Ma N, Mou CP, Chen Z, Barhanin J and Huang W: KCNQ1
gain-of-function mutation in familial atrial fibrillation. Science.
299:251–254. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Oberti C, Wang L, Li L, Dong J, Rao S, Du
W and Wang Q: Genome-wide linkage scan identifies a novel genetic
locus on chromosome 5p13 for neonatal atrial fibrillation
associated with sudden death and variable cardiomyopathy.
Circulation. 110:3753–3759. 2004. View Article : Google Scholar
|
|
21
|
Zhang X, Chen S, Yoo S, Chakrabarti S,
Zhang T, Ke T, Oberti C, Yong SL, Fang F, Li L, de la Fuente R,
Wang L, Chen Q and Wang QK: Mutation in nuclear pore component
NUP155 leads to atrial fibrillation and early sudden cardiac death.
Cell. 135:1017–1027. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Volders PG, Zhu Q, Timmermans C, Eurlings
PM, Su X, Arens YH, Li L, Jongbloed RJ, Xia M, Rodriguez LM and
Chen YH: Mapping a novel locus for familial atrial fibrillation on
chromosome 10p11-q21. Heart Rhythm. 4:469–475. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Darbar D, Hardy A, Haines JL and Roden DM:
Prolonged signal-averaged P-wave duration as an intermediate
phenotype for familial atrial fibrillation. J Am Coll Cardiol.
51:1083–1089. 2008. View Article : Google Scholar
|
|
24
|
Olesen MS, Bentzen BH, Nielsen JB,
Steffensen AB, David JP, Jabbari J, Jensen HK, Haunsø S, Svendsen
JH and Schmitt N: Mutations in the potassium channel subunit KCNE1
are associated with early-onset familial atrial fibrillation. BMC
Med Genet. 13:242012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang Y, Xia M, Jin Q, Bendahhou S, Shi J
and Chen Y, Liang B, Lin J, Liu Y, Liu B, Zhou Q, Zhang D, Wang R,
Ma N, Su X, Niu K, Pei Y, Xu W, Chen Z, Wan H, Cui J, Barhanin J
and Chen Y: Identification of a KCNE2 gain-of-function mutation in
patients with familial atrial fibrillation. Am J Hum Genet.
75:899–905. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lundby A, Ravn LS, Svendsen JH, Hauns S,
Olesen SP and Schmitt N: KCNE3 mutation V17M identified in a
patient with lone atrial fibrillation. Cell Physiol Biochem.
21:47–54. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zeng Z, Tan C, Teng S, Chen J, Su S, Zhou
X, Wang F, Zhang S, Gu D, Makielski JC and Pu J: The single
nucleotide polymorphisms of I(Ks) potassium channel genes and their
association with atrial fibrillation in a Chinese population.
Cardiology. 108:97–103. 2007. View Article : Google Scholar
|
|
28
|
Ravn LS, Aizawa Y, Pollevick GD,
Hofman-Bang J, Cordeiro JM, Dixen U, Jensen G, Wu Y, Burashnikov E,
Haunso S, Guerchicoff A, Hu D, Svendsen JH, Christiansen M and
Antzelevitch C: Gain of function in IKs secondary to a mutation in
KCNE5 associated with atrial fibrillation. Heart Rhythm. 5:427–435.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hong K, Bjerregaard P, Gussak I and
Brugada R: Short QT syndrome and atrial fibrillation caused by
mutation in KCNH2. J Cardiovasc Electrophysiol. 16:394–396. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xia M, Jin Q, Bendahhou S, He Y, Larroque
MM and Chen Y, Zhou Q, Yang Y, Liu Y, Liu B, Zhu Q, Zhou Y, Lin J,
Liang B, Li L, Dong X, Pan Z, Wang R, Wan H, Qiu W, Xu W, Eurlings
P, Barhanin J and Chen Y: A Kir2.1 gain-of-function mutation
underlies familial atrial fibrillation. Biochem Biophys Res Commun.
332:1012–1019. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Delaney JT, Muhammad R, Blair MA, Kor K,
Fish FA, Roden DM and Darbar D: A KCNJ8 mutation associated with
early repolarization and atrial fibrillation. Europace.
14:1428–1432. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Olson TM, Alekseev AE, Liu XK, Park S,
Zingman LV, Bienengraeber M, Sattiraju S, Ballew JD, Jahangir A and
Terzic A: Kv1.5 channelopathy due to KCNA5 loss-of-function
mutation causes human atrial fibrillation. Hum Mol Genet.
15:2185–2191. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang Y, Li J, Lin X, Yang Y, Hong K, Wang
L, Liu J, Li L, Yan D, Liang D, Xiao J, Jin H, Wu J, Zhang Y and
Chen YH: Novel KCNA5 loss-of-function mutations responsible for
atrial fibrillation. J Hum Genet. 54:277–283. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Olson TM, Michels VV, Ballew JD, Reyna SP,
Karst ML, Herron KJ, Horton SC, Rodeheffer RJ and Anderson JL:
Sodium channel mutations and susceptibility to heart failure and
atrial fibrillation. JAMA. 293:447–454. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Watanabe H, Darbar D, Kaiser DW,
Jiramongkolchai K, Chopra S, Donahue BS, Kannankeril PJ and Roden
DM: Mutations in sodium channel beta1- and beta2-subunits
associated with atrial fibrillation. Circ Arrhythm Electrophysiol.
2:268–275. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang P, Yang Q, Wu X, Yang Y, Shi L, Wang
C, Wu G, Xia Y, Yang B, Zhang R, Xu C, Cheng X, Li S, Zhao Y, Fu F,
Liao Y, Fang F, Chen Q, Tu X and Wang QK: Functional
dominant-negative mutation of sodium channel subunit gene SCN3B
associated with atrial fibrillation in a Chinese GeneID population.
Biochem Biophys Res Commun. 398:98–104. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Olesen MS, Jespersen T, Nielsen JB, Liang
B, Møller DV, Hedley P, Christiansen M, Varró A, Olesen SP, Haunsø
S, Schmitt N and Svendsen JH: Mutations in sodium channel β-subunit
SCN3B are associated with early-onset lone atrial fibrillation.
Cardiovasc Res. 89:786–793. 2011.
|
|
38
|
Hodgson-Zingman DM, Karst ML, Zingman LV,
Heublein DM, Darbar D, Herron KJ, Ballew JD, de Andrade M, Burnett
JC Jr and Olson TM: Atrial natriuretic peptide frameshift mutation
in familial atrial fibrillation. N Engl J Med. 359:158–165. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ren X, Xu C, Zhan C, Yang Y, Shi L, Wang
F, Wang C, Xia Y, Yang B, Wu G, Wang P, Li X, Wang D, Xiong X, Liu
J, Liu Y, Liu M, Liu J, Tu X and Wang QK: Identification of NPPA
variants associated with atrial fibrillation in a Chinese GeneID
population. Clin Chim Acta. 411:481–485. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Thibodeau IL, Xu J, Li Q, Liu G, Lam K,
Veinot JP, Birnie DH, Jones DL, Krahn AD, Lemery R, Nicholson BJ
and Gollob MH: Paradigm of genetic mosaicism and lone atrial
fibrillation: physiological characterization of a connexin
43-deletion mutant identified from atrial tissue. Circulation.
122:236–244. 2010. View Article : Google Scholar
|
|
41
|
Gollob MH, Jones DL, Krahn AD, Danis L,
Gong XQ, Shao Q, Liu X, Veinot JP, Tang AS, Stewart AF, Tesson F,
Klein GJ, Yee R, Skanes AC, Guiraudon GM, Ebihara L and Bai D:
Somatic mutations in the connexin 40 gene (GJA5) in atrial
fibrillation. N Engl J Med. 354:2677–2688. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yang YQ, Zhang XL, Wang XH, Tan HW, Shi
HF, Jiang WF, Fang WY and Liu X: Connexin40 nonsense mutation in
familial atrial fibrillation. Int J Mol Med. 26:605–610.
2010.PubMed/NCBI
|
|
43
|
Yang YQ, Liu X, Zhang XL, Wang XH, Tan HW,
Shi HF, Jiang WF and Fang WY: Novel connexin40 missense mutations
in patients with familial atrial fibrillation. Europace.
12:1421–1427. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mahida S, Lubitz SA, Rienstra M, Milan DJ
and Ellinor PT: Monogenic atrial fibrillation as pathophysiological
paradigms. Cardiovasc Res. 89:692–700. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mommersteeg MT, Christoffels VM, Anderson
RH and Moorman AF: Atrial fibrillation: a developmental point of
view. Heart Rhythm. 6:1818–1824. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Akazawa H and Komuro I: Cardiac
transcription factor Csx/Nkx2-5: Its role in cardiac development
and diseases. Pharmacol Ther. 107:252–268. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pikkarainen S, Tokola H, Kerkelä R and
Ruskoaho H: GATA transcription factors in the developing and adult
heart. Cardiovasc Res. 63:196–207. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Peterkin T, Gibson A, Loose M and Patient
R: The roles of GATA-4, -5 and -6 in vertebrate heart development.
Semin Cell Dev Biol. 16:83–94. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Posch MG, Boldt LH, Polotzki M, Richter S,
Rolf S, Perrot A, Dietz R, Ozcelik C and Haverkamp W: Mutations in
the cardiac transcription factor GATA4 in patients with lone atrial
fibrillation. Eur J Med Genet. 53:201–203. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yang YQ, Wang MY, Zhang XL, Tan HW, Shi
HF, Jiang WF, Wang XH, Fang WY and Liu X: GATA4 loss-of-function
mutations in familial atrial fibrillation. Clin Chim Acta.
412:1825–1830. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Jiang JQ, Shen FF, Fang WY, Liu X and Yang
YQ: Novel GATA4 mutations in lone atrial fibrillation. Int J Mol
Med. 28:1025–1032. 2011.PubMed/NCBI
|
|
52
|
Wang J, Sun YM and Yang YQ: Mutation
spectrum of the GATA4 gene in patients with idiopathic atrial
fibrillation. Mol Biol Rep. 39:8127–8135. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yang YQ, Wang J, Wang XH, Wang Q, Tan HW,
Zhang M, Shen FF, Jiang JQ, Fang WY and Liu X: Mutational spectrum
of the GATA5 gene associated with familial atrial fibrillation. Int
J Cardiol. 157:305–307. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wang XH, Huang CX, Wang Q, Li RG, Xu YJ,
Liu X, Fang WY and Yang YQ: A novel GATA5 loss-of-function mutation
underlies lone atrial fibrillation. Int J Mol Med. 31:43–50.
2013.PubMed/NCBI
|
|
55
|
Yang YQ, Wang XH, Tan HW, Jiang WF, Fang
WY and Liu X: Prevalence and spectrum of GATA6 mutations associated
with familial atrial fibrillation. Int J Cardiol. 155:494–496.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yang YQ, Li L, Wang J, Zhang XL, Li RG, Xu
YJ, Tan HW, Wang XH, Jiang JQ, Fang WY and Liu X: GATA6
loss-of-function mutation in atrial fibrillation. Eur J Med Genet.
55:520–526. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Li J, Liu WD, Yang ZL and Yang YQ: Novel
GATA6 loss-of-function mutation responsible for familial atrial
fibrillation. Int J Mol Med. 30:783–790. 2012.PubMed/NCBI
|
|
58
|
Zhang Y, Rath N, Hannenhalli S, Wang Z,
Cappola T, Kimura S, Atochina-Vasserman E, Lu MM, Beers MF and
Morrisey EE: GATA and Nkx factors synergistically regulate
tissue-specific gene expression and development in vivo.
Development. 134:189–198. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Pradhan L, Genis C, Scone P, Weinberg EO,
Kasahara H and Nam HJ: Crystal structure of the human NKX2.5
homeodomain in complex with DNA target. Biochemistry. 51:6312–6319.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Tanaka M, Chen Z, Bartunkova S, Yamasaki N
and Izumo S: The cardiac homeobox gene Csx/Nkx2.5 lies genetically
upstream of multiple genes essential for heart development.
Development. 126:1269–1280. 1999.PubMed/NCBI
|
|
61
|
Warren SA, Terada R, Briggs LE,
Cole-Jeffrey CT, Chien WM, Seki T, Weinberg EO, Yang TP, Chin MT,
Bungert J and Kasahara H: Differential role of Nkx2-5 in activation
of the atrial natriuretic factor gene in the developing versus
failing heart. Mol Cell Biol. 31:4633–4645. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Haïssaguerre M, Jaïs P, Shah DC, Takahashi
A, Hocini M, Quiniou G, Garrigue S, Le Mouroux A, Le Métayer P and
Clémenty J: Spontaneous initiation of atrial fibrillation by
ectopic beats originating in the pulmonary veins. N Engl J Med.
339:659–666. 1998.PubMed/NCBI
|
|
63
|
Mommersteeg MT, Brown NA, Prall OW, de
Gier-de Vries C, Harvey RP, Moorman AF and Christoffels VM: Pitx2c
and Nkx2-5 are required for the formation and identity of the
pulmonary myocardium. Circ Res. 101:902–909. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Mommersteeg MT, Hoogaars WM, Prall OW, de
Gier-de Vries C, Wiese C, Clout DE, Papaioannou VE, Brown NA,
Harvey RP, Moorman AF and Christoffels VM: Molecular pathway for
the localized formation of the sinoatrial node. Circ Res.
100:354–362. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Schott JJ, Benson DW, Basson CT, Pease W,
Silberbach GM, Moak JP, Maron BJ, Seidman CE and Seidman JG:
Congenital heart disease caused by mutations in the transcription
factor NKX2-5. Science. 281:108–111. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wang J, Xin YF, Liu XY, Liu ZM, Wang XZ
and Yang YQ: A novel NKX2-5 mutation in familial ventricular septal
defect. Int J Mol Med. 27:369–375. 2011.PubMed/NCBI
|
|
67
|
Liu XY, Wang J, Yang YQ, Zhang YY, Chen
XZ, Zhang W, Wang XZ, Zheng JH and Chen YH: Novel NKX2-5 mutations
in patients with familial atrial septal defects. Pediatr Cardiol.
32:193–201. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Wang J, Liu XY and Yang YQ: Novel NKX2-5
mutations responsible for congenital heart disease. Genet Mol Res.
10:2905–2915. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Garg V, Kathiriya IS, Barnes R,
Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS,
Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC and Srivastava D:
GATA4 mutations cause human congenital heart defects and reveal an
interaction with TBX5. Nature. 424:443–447. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Wang J, Fang M, Liu XY, Xin YF, Liu ZM,
Chen XZ, Wang XZ, Fang WY, Liu X and Yang YQ: A novel GATA4
mutation responsible for congenital ventricular septal defects. Int
J Mol Med. 28:557–564. 2011.PubMed/NCBI
|
|
71
|
Liu XY, Wang J, Zheng JH, Bai K, Liu ZM,
Wang XZ, Liu X, Fang WY and Yang YQ: Involvement of a novel GATA4
mutation in atrial septal defects. Int J Mol Med. 28:17–23.
2011.PubMed/NCBI
|
|
72
|
Wang J, Luo XJ, Xin YF, Liu Y, Liu ZM,
Wang Q, Li RG, Fang WY, Wang XZ and Yang YQ: Novel GATA6 mutations
associated with congenital ventricular septal defect or tetralogy
of fallot. DNA Cell Biol. 31:1610–1617. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Yang YQ, Li L, Wang J, Liu XY, Chen XZ,
Zhang W, Wang XZ, Jiang JQ, Liu X and Fang WY: A novel GATA4
loss-of-function mutation associated with congenital ventricular
septal defect. Pediatr Cardiol. 33:539–546. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zheng GF, Wei D, Zhao H, Zhou N, Yang YQ
and Liu XY: A novel GATA6 mutation associated with congenital
ventricular septal defect. Int J Mol Med. 29:1065–1071.
2012.PubMed/NCBI
|
|
75
|
McCulley DJ and Black BL: Transcription
factor pathways and congenital heart disease. Curr Top Dev Biol.
100:253–277. 2012. View Article : Google Scholar : PubMed/NCBI
|