Introduction
Bone metabolism is a highly coordinated process
performed mainly by two types of functional cells, osteoblasts and
osteoclasts. The former is responsible for bone formation and the
latter for bone resorption. At present, it is well recognized that
osteoblasts play a pivotal role in the regulation of bone
resorption through receptor activator of nuclear factor-κB ligand
(RANKL) expression which is responsive to numerous bone resorptive
stimuli (1). Osteoblasts, which
are differentiated from mesenchymal progenitors, express various
cell type-specific markers during the differentiation process.
Osteocalcin (OC) is a bone-specific protein that is modified
post-translationally by vitamin K-dependent γ-carboxylation, known
as bone Gla-protein (2). It is
well known that OC is synthesized specifically in osteoblasts;
therefore, OC is recognized as one of the markers of the mature
osteoblast phenotype. In contrast, OC-deficient mice reportedly
display an increase in bone formation without impairing bone
resorption, suggesting that OC is a determinant of moderate bone
formation (3). In addition, it
has recently been shown that un-carboxylated OC released from
osteoblasts regulates energy metabolism through acting on
pancreatic β-cells to increase insulin synthesis, adipocytes to
increase adiponectin and skeletal myocytes to glucose uptake
(4,5). These findings lead us to speculate
that bone, as an endocrine organ, plays a vital role also in energy
metabolism through the release of OC. However, the exact mechanism
underlying OC synthesis in osteoblasts remains to be clarified.
Thyroid hormone is an important regulator of
skeletal function as well as whole body metabolism. Thyroid hormone
excess, namely hyperthyroidism, is a major cause of secondary
osteoporosis (6). In the state of
hyperthyroidism, the serum levels of alkaline phosphatase and OC,
markers of the osteoblast phenotype, are elevated as well as the
excretion of pyridinoline and hydroxypyridinoline cross-link, which
reflects bone resorption (6). It
is known that an imbalance in bone remodeling causes the loss of
bone mass by hyperthyroidism (6).
The receptors for triiodothyronine (T3), a member of the
steroid hormone receptor superfamily, are expressed in osteoblasts
(6). It has been reported that
thyroid hormone stimulates alkaline phosphatase activity and
secretion of OC and insulin-like growth factors in osteoblasts and
that it modulates proliferation of osteoblasts (6–9).
In our previous studies (10,11), we reported that p38
mitogen-activated protein (MAP) kinase but not p44/p42 MAP kinase,
is involved in the T3-stimulated OC synthesis in
osteoblast-like MC3T3-E1 cells, and that the adenylyl cyclase-cAMP
system has an inhibitory role in OC synthesis via suppression of
p38 MAP kinase activation.
AMP-activated protein kinase (AMPK) is generally
known to regulate multiple metabolic pathways (12). AMPK has emerged over the last
decade as a key sensing mechanism in the regulation of cellular
energy homeostasis (13–15). The enzyme is activated by a
variety of physiological and pathological stresses which increase
the intracellular AMP:ATP ratio, either by increasing ATP
consumption or by decreasing ATP production in mammalian cells.
Activated AMPK acts to restore cellular energy balance by
ATP-generating pathways such as fatty acid oxidation, while
simultaneously inhibiting ATP utilizing pathways. Regarding bone
metabolism, metformin, which can activate AMPK (16), reportedly increases markers of
osteoblast differentiation including OC mRNA expression, and
enhances mineralization in osteoblastlike MC3T3-E1 cells (17). It has recently been reported that
AMPK activation regulates bone formation and bone mass (18). These previous findings led us to
speculate that AMPK influences bone metabolism through the
functional modulation of osteoblasts. We previously demonstrated
that AMPK plays a role in the synthesis of vascular endothelial
growth factor or interleukin-6 in osteoblasts (19,20). However, the exact role of AMPK in
bone metabolism, particularly in osteoblasts, has not yet been
fully elucidated.
In the present study, we investigated the mechanism
behind T3-stimulated OC synthesis in osteoblast-like
MC3T3-E1 cells and the involvement of AMPK in OC synthesis. We here
demonstrated that AMPK positively regulates
T3-stimulated OC synthesis in these cells.
Materials and methods
Materials
T3 was purchased from Sigma Chemical Co.
(St. Louis, MO, USA). Mouse OC enzyme-linked immunosorbent assay
(ELISA) kit was purchased from Biomedical Technologies, Inc.
(Stoughton, MA, USA). Compound C, a pyrrazolopyrimidine derivative
widely used as a specific and reversible AMPK inhibitor (16,21,22), was purchased from
Calbiochem-Novabiochem Corp. (La Jolla, CA, USA). Phospho-specific
AMPKα (Thr-172) antibodies, phosphospecific AMPKα (Ser-485)
antibodies, AMPKα antibodies, phospho-specific AMPKβ (Ser-108)
antibodies, phosphospecific AMPKβ (Ser-182) antibodies, AMPKβ
antibodies and phospho-specific acetyl-CoA carboxylase antibodies
were purchased from Cell Signaling Technology, Inc. (Beverly, MA,
USA). GAPDH antibodies were obtained from Santa Cruz Biotechnology,
Inc. (Santa Cruz, CA, USA). An ECL Western blotting detection
system was purchased from GE Healthcare, Ltd. (Buckinghamshire,
UK). Control short interfering RNA (siRNA; Silencer Negative
Control no. 1 siRNA) and AMPK-siRNA were purchased from Qiagen
(Hilden, Germany). siLentFect was purchased from Bio-Rad (Hercules,
CA, USA). TRIzol reagent and Omniscript Reverse Transcriptase kit
were purchased from Invitrogen (Carlsbad, CA, USA) and Qiagen,
respectively. Fast-start DNA Master SYBR-Green I was purchased from
Roche Diagnostics GmbH (Mannheim, Germany). Other materials and
chemicals were obtained from commercial sources. Compound C was
dissolved in dimethyl sulfoxide. The maximum concentration of
dimethyl sulfoxide was 0.1%, which did not affect the assay for OC
or detection of the protein level by western blot analysis.
Cell culture
Cloned osteoblast-like MC3T3-E1 cells derived from
newborn mouse calvaria (23) were
maintained as previously described (24). Briefly, the cells were cultured in
α-minimum essential medium (α-MEM) containing 10% fetal calf serum
(FCS) at 37°C in a humidified atmosphere of 5% CO2/95%
air. The cells were seeded into 35-mm (5×104) or 90-mm
(2×105) diameter dishes in α-MEM containing 10% FCS.
After 5 days, the medium was exchanged for α-MEM containing 0.3%
FCS. The cells were used for experiments after 48 h.
OC assay
The cultured cells were stimulated by T3
in 1 ml of α-MEM containing 0.3% FCS for the indicated periods. The
conditioned medium was collected at the end of the incubation, and
the OC concentration was measured by an OC ELISA kit. When
indicated, the cells were pretreated with various doses of compound
C for 60 min.
Western blot analysis
Western blot analysis was performed as previously
described (25). The cultured
cells were stimulated by T3 or vehicle in α-MEM
containing 0.3% FCS for the indicated periods. When indicated, the
cells were pretreated with various doses of compound C for 60 min.
The cells were washed twice with phosphate-buffered saline and then
lysed, homogenized and sonicated in a lysis buffer containing 62.5
mM Tris/HCl; pH 6.8, 3% sodium dodecyl sulfate (SDS), 50 mM
dithiothreitol and 10% glycerol. SDS-polyacrylamide gel
electrophoresis (PAGE) was performed according to Laemmli (26) on 10% polyacrylamide gel. The
protein was fractionated and transferred onto Immun-Blot PVDF
membranes (Bio-Rad). Membranes were blocked with 5% fat-free dry
milk in Tris-buffered saline-Tween-20 (TBS-T; 20 mM Tris/HCl, pH
7.6, 137 mM NaCl, 0.1% Tween-20) for 2 h before incubation with the
primary antibodies. The following antibodies were used:
phospho-specific AMPKα (Thr-172) antibodies, phospho-specific AMPKα
(Ser-485) antibodies, AMPKα antibodies, phospho-specific AMPKβ
(Ser-108) antibodies, phospho-specific AMPKβ (Ser-182) antibodies,
AMPKβ antibodies, phospho-specific acetyl-CoA carboxylase
antibodies and GAPDH antibodies as primary antibodies.
Peroxidase-labeled antibodies raised in goat against rabbit IgG
[Kirkegaard & Perry Laboratories (KPL), Inc., Gaithersburg, MD,
USA] were used as secondary antibodies. The primary and secondary
antibodies were diluted at 1:1,000 with 5% fat-free dry milk in
TBS-T. Peroxidase activity on the membrane was visualized on X-ray
film by means of the ECL Western blotting detection system.
Real-time RT-PCR
The cultured cells were pretreated with compound C
for 60 min and stimulated by T3 for the indicated
periods. Total RNA was isolated and transcribed into cDNA using
TRIzol reagent and Omniscript Reverse Transcriptase kit. Real-time
RT-PCR was performed using a LightCycler system (Roche Diagnostics)
in capillaries and Fast-Start DNA Master SYBR-Green I provided with
the kit. Sense and antisense primers were synthesized based on the
report of Zhang et al (27) for mouse OC mRNA and GAPDH mRNA.
The amplified products were determined by melting curve analysis
and agarose electrophoresis. OC mRNA levels were normalized with
those of GAPDH mRNA.
siRNA transfection
To knockdown AMPK in MC3T3-E1 cells, the cells were
transfected with negative control siRNA or AMPK-siRNA utilizing
siLentFect according to the manufacturer’s protocol. In brief, the
cells (1×105 cells) were seeded into 35-mm diameter
dishes in α-MEM containing 10% FCS and subcultured for 48 h. The
cells were then incubated at 37°C with 50 nM siRNA-siLentFect
complexes. After 24 h, the medium was replaced with α-MEM
containing 0.3% FCS. The cells were then stimulated by
T3 in α-MEM containing 0.3% FCS for the indicated
periods.
Determination of the enzyme
activation
The absorbance of enzyme immunoassay samples was
measured at 450 nm using the EL340 Bio Kinetic Reader (Bio-Tek
Instruments, Inc., Winooski, VT, USA).
Statistical analysis
The data were analyzed by ANOVA followed by the
Bonferroni method for multiple comparisons between pairs, and a
P<0.05 was considered to indicate a statistically significant
result. All data are presented as the means ± SEM of triplicate
independent determinations.
Results
Effects of T3 on the
phosphorylation of AMPK in MC3T3-E1 cells
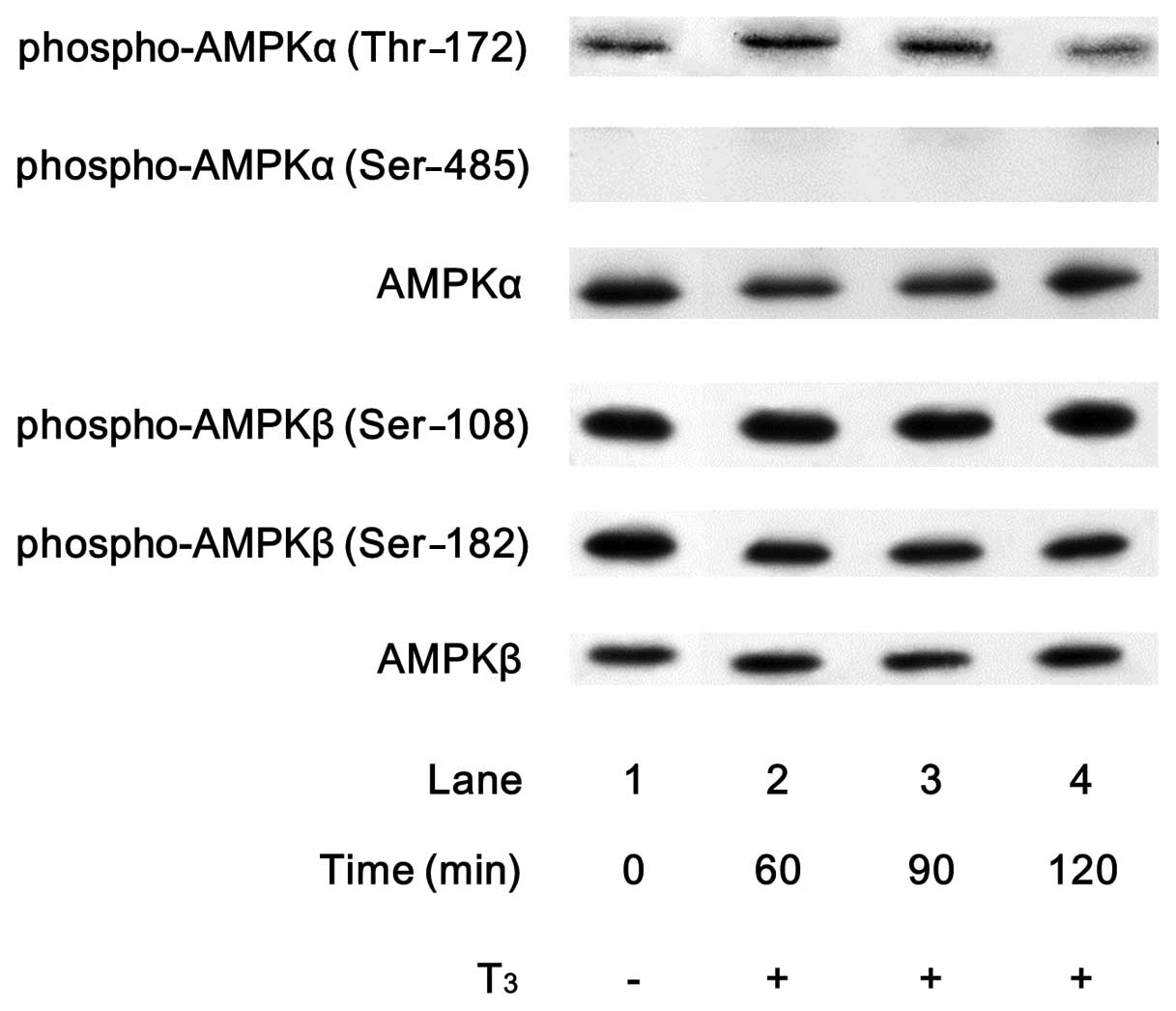
In order to investigate whether T3
activates AMPK in osteoblasts, we first examined the effects of
T3 on the phosphorylation of AMPK in osteoblast-like
MC3T3-E1 cells. T3 markedly induced the phosphorylation
of the AMPKα-subunit (Thr-172) (Fig.
1). The effect of T3 on the phosphorylation of the
AMPKα-subunit (Thr-172) reached its peak at 60–90 min and decreased
thereafter. However, T3 failed to affect the
phosphorylation levels of AMPKα-subunit (Ser-485), AMPKβ-subunit
(Ser-108) and AMPKβ-subunit (Ser-182) (Fig. 1).
Effect of compound C on the
phosphorylation of acetyl-CoA carboxylase in MC3T3-E1 cells
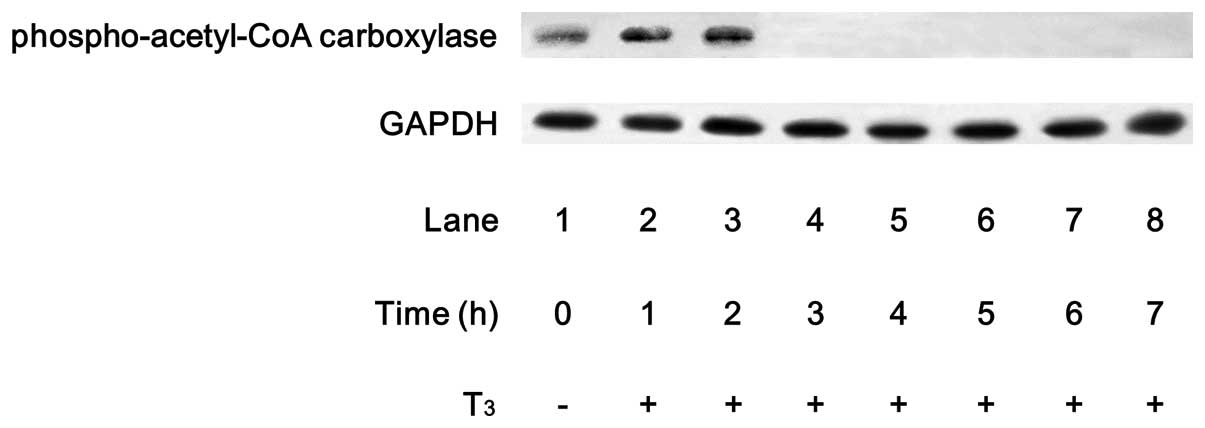
It is generally recognized that acetyl-CoA
carboxylase, which catalyzes an important step in lipid synthesis,
is a direct substrate of AMPK (15). We confirmed that T3
markedly stimulated the phosphorylation of acetyl-CoA carboxylase
in MC3T3-E1 cells (Fig. 2). The
effect of T3 on the acetyl-CoA carboxylase
phosphorylation reached its peak within 2 h and decreased
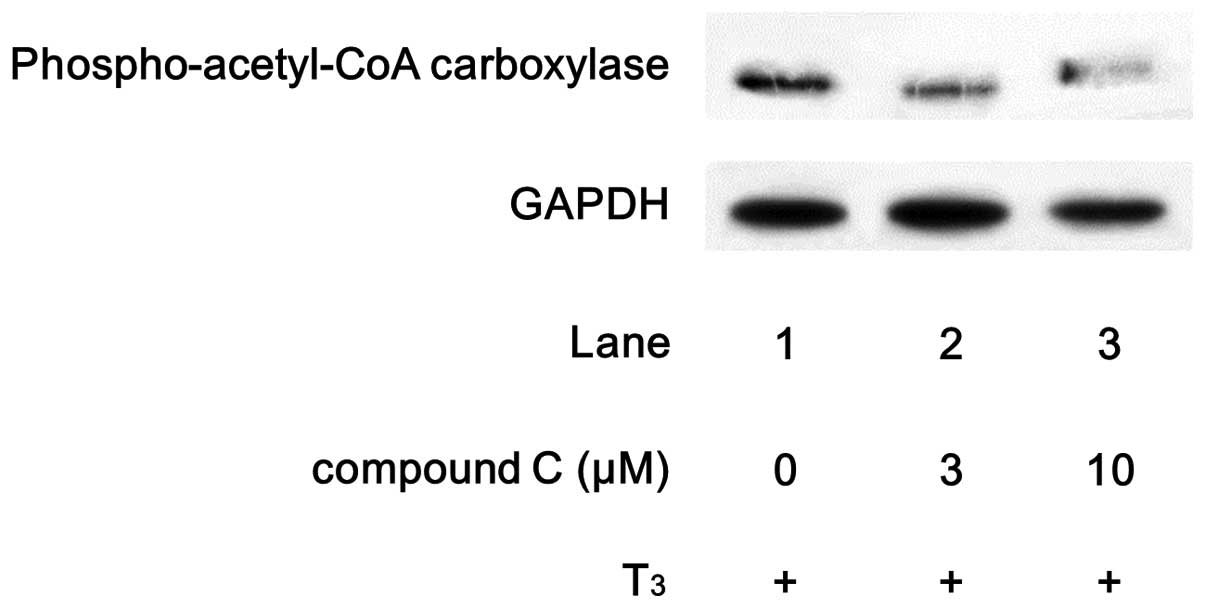
thereafter. We found that compound C, an inhibitor of AMPK
(16), markedly suppressed the
T3-stimulated phosphorylation of acetyl-CoA carboxylase
(Fig. 3).
Effect of compound C on the
T3-stimulated OC release in MC3T3-E1 cells
In order to clarify the involvement of AMPK in the
T3-stimulated OC release in osteoblasts, we next
examined the effect of compound C on the release of OC stimulated
by T3 in MC3T3-E1 cells. Compound C, which alone did not
affect the OC levels, significantly suppressed the
T3-stimulated OC release in a time-dependent manner
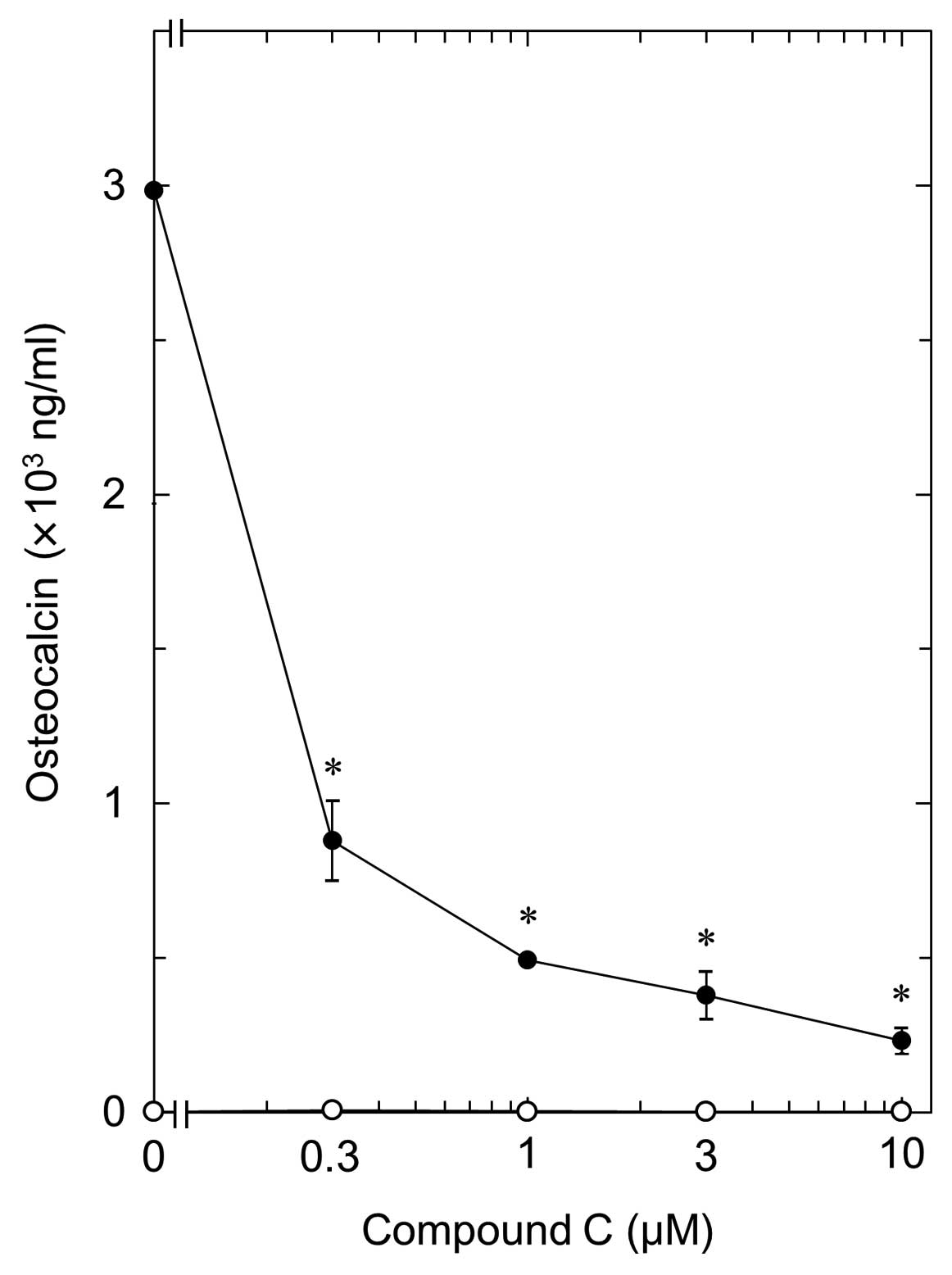
(Fig. 4). The inhibitory effect
of compound C was dose-dependent in the range between 0.3 and 10
μM (Fig. 5). The maximum
effect of compound C was observed at 10 μM, which resulted
in ∼90% inhibition compared to the OC level with T3
alone.
Effect of compound C on the
T3-induced OC mRNA expression in MC3T3-E1 cells
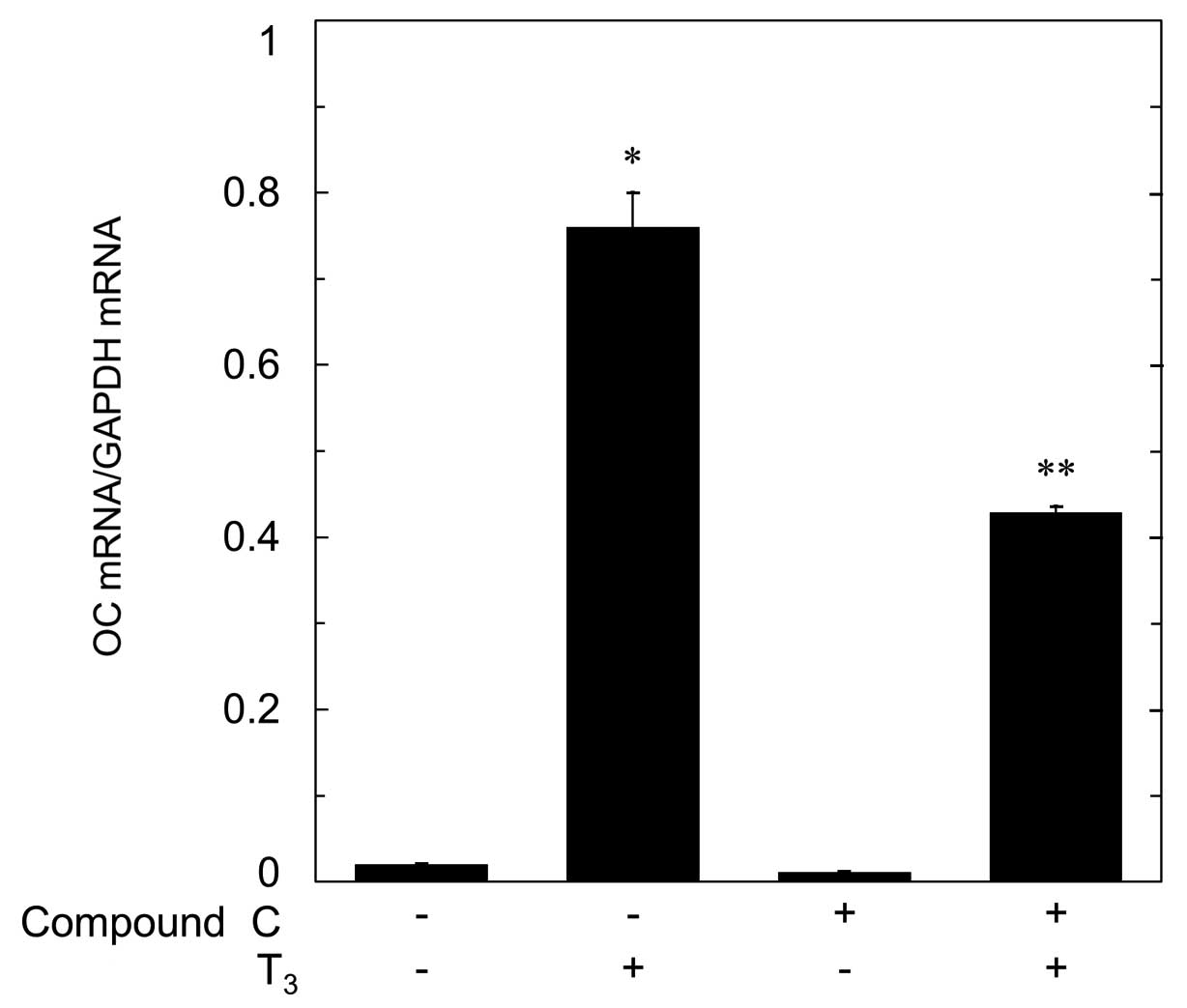
It has been shown that T3 induces the
mRNA expression of OC in osteoblasts (9). In order to investigate whether the
suppression of T3-stimulated OC synthesis by compound C
is mediated through transcriptional events, we next examined the
effect of compound C on T3-induced OC mRNA expression.
Compound C, which by itself had little effect on the basal level of
OC mRNA, significantly reduced the T3-induced level of
OC mRNA (Fig. 6).
Effect of AMPK-siRNA on
T3-stimulated OC release in MC3T3-E1 cells
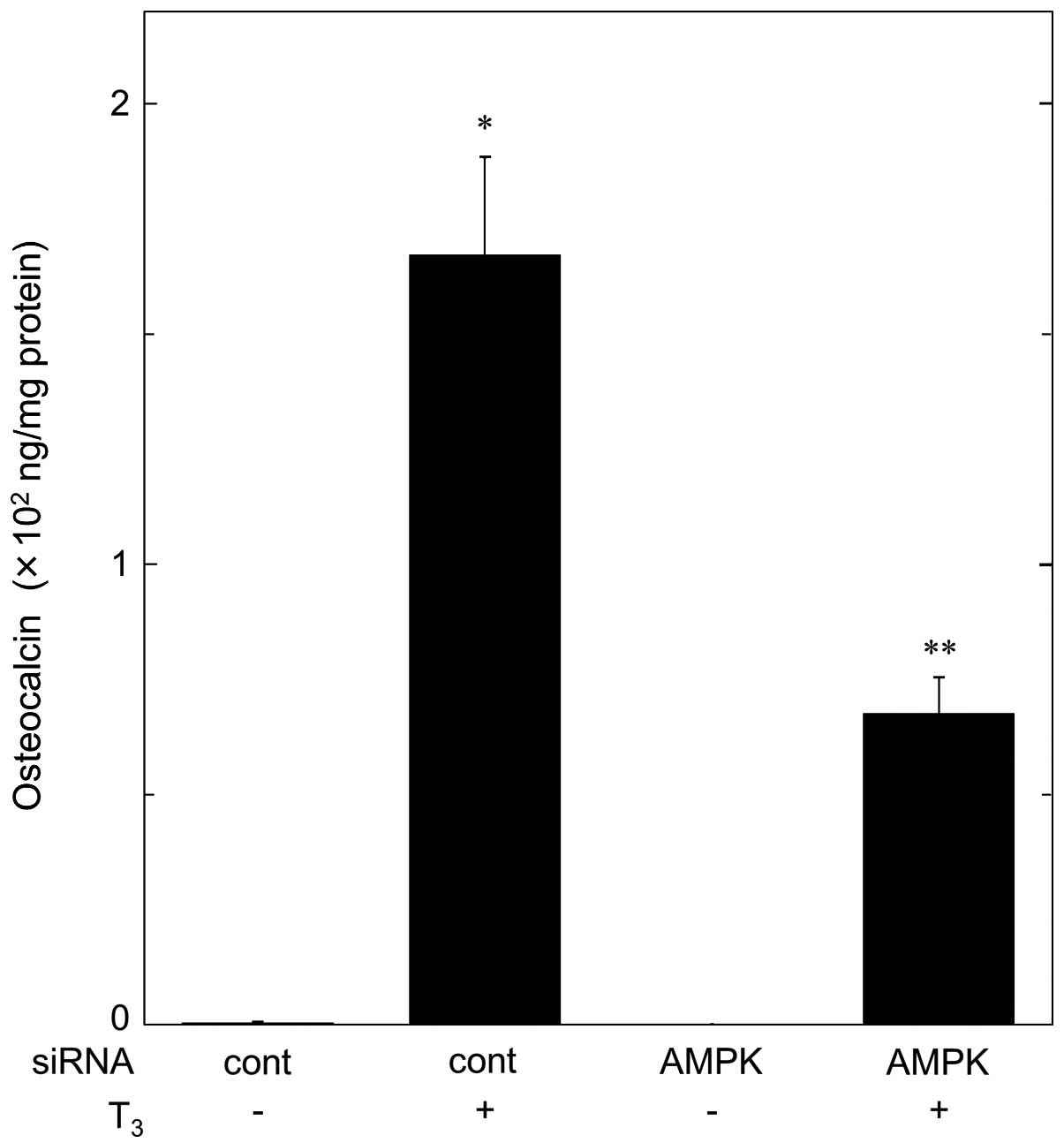
We further investigated the effect of AMPK knockdown
on the OC release stimulated by T3 in MC3T3-E1 cells. In
comparison with the control cells, the T3-stimulated OC
release was significantly diminished in AMPK-knockdown cells by
AMPK-siRNA (Fig. 7).
Approximately 60% suppression of the T3-stimulated OC
release resulted following the treatment with AMPK-siRNA.
Discussion
In the present study, we showed that T3
induced the phosphorylation of AMPK in osteoblast-like MC3T3-E1
cells, using phospho-specific AMPKα-subunit (Thr-172) antibodies.
It is generally recognized that the phosphorylation of Thr-172 in
the AMPKα-subunit is necessary for AMPK activity (12,28). Therefore, it is likely that
T3 stimulates the activation of AMPK in osteoblast-like
MC3T3-E1 cells. To the best of our knowledge, this is probably the
first report showing the involvement of AMPK in the intracellular
signaling of T3 in osteoblasts. On the other hand,
T3 failed to induce the phosphorylation of AMPKα-subunit
(Ser-485). It seems unlikely that the phosphorylation of Ser-485 in
the α-subunit is involved in T3-induced AMPK activation
in MC3T3-E1 cells. In addition, we found that AMPKβ-subunits
(Ser-108 and Ser-182), which were phosphorylated without
stimulation, were barely affected by T3. It has been
shown that phosphorylation at Ser-108 of the AMPKβ-subunit is
required for the activation of AMPK, while phosphorylation of
Ser-182 affects AMPK localization (29). Moreover, we showed here that the
phosphorylation of acetyl-CoA carboxylase, known as a direct
substrate of AMPK (30), was
significantly induced by T3. The time course of the
phosphorylation of AMPKα-subunit (Thr-172) stimulated by
T3 appears to be more rapid than that of acetyl-CoA
carboxylase. Furthermore, we found that compound C, an AMPK
inhibitor (16), reduced the
phosphorylation levels of acetyl-CoA carboxylase by T3.
It is probable that the inhibitory effect of compound C is exerted
through the suppression of AMPK. Based on our findings, it is most
likely that T3 positively regulates AMPK activity via
the phosphorylation of the AMPKα-subunit (Thr-172) in
osteoblast-like MC3T3-E1 cells.
We previously demonstrated that T3
induces OC synthesis which is a marker of osteoblast
differentiation in osteoblast-like MC3T3-E1 cells (10,11). Thus, we next investigated whether
or not AMPK is involved in the T3-stimulated OC
synthesis in MC3T3-E1 cells. We showed that the
T3-stimulated OC release was significantly suppressed by
compound C, an inhibitor of AMPK (16), suggesting that AMPK is involved in
the OC release induced by T3 in these cells.
Additionally, compound C markedly reduced the OC mRNA expression
induced by T3, suggesting that AMPK regulates OC
synthesis at a point upstream of the transcriptional process. We
further examined the effect of AMPK knockdown by siRNA on
T3-stimulated OC release and demonstrated the
concomitant reduction in T3-stimulated induction of OC
by AMPK knockdown in the osteoblast-like MC3T3-E1 cells. Taking our
findings into account, it is most likely that AMPK functions as a
positive regulator in T3-stimulated OC synthesis in
osteoblasts.
Thyroid hormone excess, or hyperthyroidism, is a
major cause of secondary osteoporosis. Both bone formation and bone
resorption are accelerated in the state of hyperthyroidism, and the
predominance of bone resorption rather than bone formation results
in the loss of bone mass (6). On
the other hand, deficiency of the thyroid hormone causes severe
skeletal growth retardation in infants. These clinical states
indicate the importance of the thyroid hormone in bone metabolism.
Thus, it is essential to clarify the precise mechanism of
T3 action, an active form of thyroid hormone,
particularly in osteoblasts which promote bone formation and
regulate bone resorption. OC, a γ-carboxylated calcium-binding
protein, is generally recognized to be produced by mature
osteoblasts (2). It is well known
that the serum level of OC is elevated in patients with
hyperthyroidism (6). OC-deficient
mice reportedly develop hyperostosis (3), suggesting its crucial role in the
bone remodeling process. Thus, our present findings demonstrating
the AMPK-dependent OC release induced by T3 provide a
new concept of bone metabolism. In addition, it has recently been
shown that un-carboxylated OC released from osteoblasts as a
bone-derived hormone regulates energy metabolism by acting on
pancreatic β-cells to increase insulin synthesis, on adipocytes to
increase adiponectin and on skeletal myocytes for glucose uptake in
rodent (4,5). This significant effect of
un-carboxylated OC has not yet been clarified in human; however,
osteoblast-releasing OC may participate in whole body energy
regulation. In addition, metformin, an AMPKactivating substance,
has already been widely used as an agent for diabetes. AMPK plays a
vital role as an energy sensor promoting the development of tissue
derived from mesenchymal cells such as muscle and bone. Taking our
present findings into account as a whole, AMPK regulation of OC
release by osteoblasts may be a therapeutic target for metabolic
disorders. Further investigation is required to clarify the details
of AMPK in bone metabolism. In conclusion, our results strongly
suggest that AMPK positively regulates T3-stimulated OC
synthesis in osteoblasts.
Acknowledgements
We are very grateful to Emiko Fuseya
and Yumiko Kurokawa for their skillful technical assistance. This
investigation was supported in part by a Grant-in-Aid for
Scientific Research (16590873 and 16591482) from the Ministry of
Education, Science, Sports and Culture of Japan and the Research
Funding for Longevity Sciences (21A-4 and 22A-22) from the National
Center for Geriatrics and Gerontology (NCGG), Japan.
References
|
1
|
Boyce BF and Xing L: Functions of
RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem
Biophys. 473:139–146. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hauschka PV, Lian JB, Cole DE and Gundberg
CM: Osteocalcin and matrix Gla protein: vitamin K-dependent
proteins in bone. Physiol Rev. 69:990–1047. 1989.PubMed/NCBI
|
|
3
|
Ducy P, Desbois C, Boyce B, Pinero G,
Story B, Dunstan C, Smith E, Bonadio J, Goldstein S, Gundberg C,
Bradley A and Karsenty G: Increased bone formation in
osteocalcin-deficient mice. Nature. 382:448–452. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee NK and Karsenty G: Reciprocal
regulation of bone and energy metabolism. Trends Endocrinol Metab.
19:161–166. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rosen CJ: Bone remodeling, energy
metabolism, and the molecular clock. Cell Metab. 7:7–10. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gogakos AI, Duncan Bassett JH and Williams
GR: Thyroid and bone. Arch Biochem Biophys. 503:129–136. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rizzoli R, Poser J and Burgi U: Nuclear
thyroid hormone receptors in cultured bone cells. Metabolism.
35:71–74. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kasono K, Sato K, Han DC, Fujii Y,
Tsushima T and Shizume K: Stimulation of alkaline phosphatase
activity by thyroid hormone in mouse osteoblast-like cells
(MC3T3-E1): a possible mechanism of hyperalkaline phosphatasia in
hyperthyroidism. Bone Miner. 4:355–363. 1988.
|
|
9
|
Kassem M, Mosekilde L and Eriksen EF:
Effects of triiodothyronine on DNA synthesis and differentiation
markers of normal human osteoblast-like cells in vitro. Biochem Mol
Biol Int. 30:779–788. 1993.PubMed/NCBI
|
|
10
|
Ishisaki A, Tokuda H, Yoshida M, Hirade K,
Kunieda K, Hatakeyama D, Shibata T and Kozawa O: Activation of p38
mitogen-activated protein kinase mediates thyroid
hormone-stimulated osteocalcin synthesis in osteoblasts. Mol Cell
Endocrinol. 214:189–195. 2004. View Article : Google Scholar
|
|
11
|
Kanno Y, Ishisaki A, Yoshida M, Nakajima
K, Tokuda H, Numata O and Kozawa O: Adenylyl cyclase-cAMP system
inhibits thyroid hormone-stimulated osteocalcin synthesis in
osteoblasts. Mol Cell Endocrinol. 229:75–82. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fogarty S and Hardie DG: Development of
protein kinase activators: AMPK as a target in metabolic disorders
and cancer. Biochim Biophys Acta. 1804:581–591. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hardie DG, Hawley SA and Scott JW:
AMP-activated protein kinase - development of the energy sensor
concept. J Physiol. 574:7–15. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lage R, Dieguez C, Vidal-Puig A and Lopez
M: AMPK: a metabolic gauge regulating whole-body energy
homeostasis. Trends Mol Med. 14:539–549. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Steinberg GR and Kemp BE: AMPK in health
and disease. Physiol Rev. 89:1025–1078. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou G, Myers R, Li Y, Chen Y, Shen X,
Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N,
Hirshman MF, Goodyear LJ and Moller DE: Role of AMP-activated
protein kinase in mechanism of metformin action. J Clin Invest.
108:1167–1174. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kanazawa I, Yamaguchi T, Yano S, Yamauchi
M and Sugimoto T: Metformin enhances the differentiation and
mineralization of osteoblastic MC3T3-E1 cells via AMP kinase
activation as well as eNOS and BMP-2 expression. Biochem Biophys
Res Commun. 375:414–419. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shah M, Kola B, Bataveljic A, Arnett TR,
Viollet B, Saxon L, Korbonits M and Chenu C: AMP-activated protein
kinase (AMPK) activation regulates in vitro bone formation and bone
mass. Bone. 47:309–319. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kato K, Tokuda H, Adachi S,
Matsushima-Nishiwaki R, Natsume H, Yamakawa K, Gu Y, Otsuka T and
Kozawa O: AMP-activated protein kinase positively regulates FGF-2-
stimulated VEGF synthesis in osteoblasts. Biochem Biophys Res
Commun. 400:123–127. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kato K, Otsuka T, Kondo A,
Matsushima-Nishiwaki R, Natsume H, Kozawa O and Tokuda H:
AMP-activated protein kinase regulates PDGF-BB-stimulated
interleukin-6 synthesis in osteoblasts: involvement of
mitogen-activated protein kinases. Life Sci. 90:71–76. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim EK, Miller I, Aja S, Landree LE, Pinn
M, McFadden J, Kuhajda FP, Moran TH and Ronnett GV: C75, a fatty
acid synthase inhibitor, reduces food intake via hypothalamic
AMP-activated protein kinase. J Biol Chem. 279:19970–19976. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nam M, Lee WH, Bae EJ and Kim SG: Compound
C inhibits clonal expansion of preadipocytes by increasing p21
level irrespectively of AMPK inhibition. Arch Biochem Biophys.
479:74–81. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sudo H, Kodama HA, Amagai Y, Yamamoto S
and Kasai S: In vitro differentiation and calcification in a new
clonal osteogenic cell line derived from newborn mouse calvaria. J
Cell Biol. 96:191–198. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kozawa O, Tokuda H, Miwa M, Kotoyori J and
Oiso Y: Cross-talk regulation between cyclic AMP production and
phosphoinositide hydrolysis induced by prostaglandin E2 in
osteoblast-like cells. Exp Cell Res. 198:130–134. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kato K, Ito H, Hasegawa K, Inaguma Y,
Kozawa O and Asano T: Modulation of the stress-induced synthesis of
hsp27 and alpha B-crystallin by cyclic AMP in C6 rat glioma cells.
J Neurochem. 66:946–950. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Laemmli UK: Cleavage of structural
proteins during the assembly of the head of bacteriophage T4.
Nature. 227:680–685. 1970. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang W, Yang N and Shi XM: Regulation of
mesenchymal stem cell osteogenic differentiation by
glucocorticoid-induced leucine zipper (GILZ). J Biol Chem.
283:4723–4729. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hawley SA, Davison M, Woods A, Davies SP,
Beri RK, Carling D and Hardie DG: Characterization of the
AMP-activated protein kinase kinase from rat liver and
identification of threonine 172 as the major site at which it
phosphorylates AMP-activated protein kinase. J Biol Chem.
271:27879–27887. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Warden SM, Richardson C, O’Donnell J Jr,
Stapleton D, Kemp BE and Witters LA: Post-translational
modifications of the beta-1 subunit of AMP-activated protein kinase
affect enzyme activity and cellular localization. Biochem J.
354:275–283. 2001. View Article : Google Scholar
|
|
30
|
Park SH, Gammon SR, Knippers JD, Paulsen
SR, Rubink DS and Winder WW: Phosphorylation-activity relationships
of AMPK and acetyl-CoA carboxylase in muscle. J Appl Physiol.
92:2475–2482. 2002. View Article : Google Scholar : PubMed/NCBI
|