Introduction
Atherosclerosis is considered to be a chronic
inflammatory disease (1). It has
recently been suggested that atherosclerosis primarily proceeds
from disturbed blood flow regions, such as various branching points
and curvatures of the arteries (2). A previous study demonstrated that
blood flow with laminar shear stress (LSS) increases the expression
of specific genes and proteins and protects endothelial cells (ECs)
against atherosclerosis, whereas shear stress generated by
disturbed blood flow generally promotes atherogenesis. These data
suggest that shear stress generated by disturbed blood flow is a
main cause of atherosclerosis (3).
However, the exact regulatory mechanisms through
which fluid shear stress affects atherogenesis remain unclear.
Recently, Nam et al developed a mouse model of disturbed
blood flow by partial carotid artery ligation and showed that low
shear stress and oscillatory shear stress (OSS) cause endothelial
dysfunction and accelerate atherosclerosis. They suggested that
disturbed blood flow directly induces the development of
atherosclerosis (4).
Advanced glycation end products (AGEs) and their
cell surface receptors have been implicated in the pathogenesis of
diabetic complications. The receptor for advanced glycation end
products (RAGE) is a transmembrane receptor for AGEs and other
ligands, such as high mobility group box 1 (HMGB1). The binding of
RAGE and these ligands activate ECs and monocytes, leading to
inflammation (5). A number of
studies have demonstrated that the interaction between RAGE and its
ligands leads to the development and progression of atherosclerotic
disease. In animal studies, a correlation between RAGE and
atherosclerosis has been demonstrated (6). Apolipoprotein E (apoE) and RAGE
(apoE−/−/RAGE−/−) double knockout mice have
been shown to develop a significantly lower number of
atherosclerotic lesions compared with apoE−/− mice with
an intact expression of RAGE (7).
Although RAGE expression is increased in atherosclerotic plaque, it
is unknown whether arterial hemodynamics regulate RAGE expression
and activation (5).
Soluble RAGE (sRAGE) is the soluble receptor form of
RAGE lacking the intracellular domain through the proteolytic
cleavage of RAGE or alternative RNA splicing in humans (8). Typically, sRAGE functions as a
competitive inhibitor of RAGE by interacting with AGEs and other
ligands, such as HMGB1, resulting in the inhibition of RAGE-induced
cellular signaling, tissue damage and dysfunction (9). The administration of sRAGE to
diabetic ApoE−/− mice has been shown to repress
atherogenesis, as well as to stabilize established atherosclerosis
(6,10,11). In a model of carotid arterial
injury, the interruption of RAGE-ligand interaction by treatment
with sRAGE has been shown to result in the reduced proliferation of
smooth muscle cells and neointimal formation in both diabetic and
non-diabetic rats (12).
The regulatory mechanisms of sRAGE in the occurrence
of atherosclerotic plaque induced by disturbed blood flow remain
largely unknown. In this study, we demonstrate that RAGE expression
is directly modulated by fluid shear stress and that sRAGE inhibits
RAGE-induced pro-inflammatory responses induced by OSS. Therefore,
sRAGE, as a competent inhibitor of RAGE plays a protective role
during the development of atherosclerosis originating from
disturbed blood flow.
Materials and methods
Cell culture and blood flow
experiments
Human umbilical vein endothelial cells (HUVECs;
Gibco, Paisley, Scotland, UK) were grown in Medium 200 with 5%
fetal bovine serum and low-serum growth supplement (LSGS; Cascade
Biologics Inc., Portland, OR, USA) (13). U937 cells were purchased from the
Korean Cell Line Bank (KCLB). Cells were cultured in RPMI-1640
(Gibco, Scotland, UK) with 10% fetal bovine serum (Gibco) and 1%
penicillin-streptomycin (Corning Cellgro, Manassas, VA, USA) at
37°C and 5% CO2.
Confluent HUVECs cultured in 60-mm dishes were
exposed to fluid shear stress. Cells were exposed to flow in a cone
and plate viscometer. We exposed a unidirectional steady flow
(shear stress of 15 dyne/cm2) for LSS and a
bidirectional disturbed flow (shear stress of ±5
dyne/cm2) for OSS as previously described (14).
Model of partial carotid artery
ligation
This animal study was performed in accordance with
the Guidelines for Animal Experiments of the Animal Experimentation
Ethics Committee of Ewha Womans University, Seoul, Korea. To
investigate the function of sRAGE in blood vessels under disturbed
flow conditions, we generated a mouse model of disturbed flow by
partial ligation of the carotid artery as previously described
(4). Male mice were ligated at 6
weeks of age. ApoE−/− and C57BL/6 mice were purchased
from the Central Animal Laboratory, Inc., Seoul, Korea. Partial
carotid artery ligated mice are viable and show acutely induced
disturbed blood flow with low shear stress and OSS, resulting in
endothelial dysfunction and atherosclerosis. All mice were fed a
chow diet and provided with water ad libitum until partial
ligation. Partial ligation of the left carotid artery (LCA) was
carried out as described in a previous study (4).
ApoE−/− mice were treated with sRAGE (80
μg/kg; A&RT Co., Korea) or PBS through intraperitoneal
injection daily for 2 weeks and subsequently sacrificed. C57BL/6
mice were sacrificed at 1, 3 and 7 days following partial carotid
artery ligation.
Immunofluorescence staining
The mice were sacrificed and perfused with saline
containing heparin (10 U/ml) through the left ventricle. The LCA
and right carotid artery (RCA) were collected en bloc with the
trachea and esophagus. For the cryosections, tissue was embedded in
Tissue-Tek optimum cutting temperature (OCT) medium, frozen on dry
ice and stored at −80°C until use (15). Cross cryosections (4 μm) were
air-dried for 1 h at room temperature. The tissue sections were
then placed in 10% formalin for 10 min at room temperature for
fixation. They sections were then washed in PBS for 5 min to remove
the OCT compound and then blocked with 10% normal donkey serum
(Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) in PBS for 2
h. The tissue sections were then stained with rabbit anti-RAGE
(1:50; Abcam, Cambridge, MA, USA) or rabbit anti-CD31 (1:50; Abcam)
overnight at 4°C. The samples were then incubated for 2 h in the
dark with anti-rabbit secondary antibody (1:200; Santa Cruz
Biotechnology, Inc.). Nuclei were counterstained with DAPI (100
ng/ml; Santa Cruz Biotechnology, Inc.) for 8 min in the dark. The
tissue sections were then routinely stained with hematoxylin and
eosin (H&E) (16). The slides
were viewed on an Olympus BX51 microscope (Olympus America Inc.,
Melville, NY, USA).
Immunohistochemical staining
Cryosections were fixed in 10% formalin for 10 min
and blocked with peroxide block (Cell Marque, Rocklin, CA, USA) for
10 min at room temperature. Samples were incubated with rabbit
anti-RAGE antibodies (1:50, 1:100; Abcam) or rabbit anti-CD31
(1:50; Abcam) for 1 h at room temperature. To visualize primary
antibodies, the Polink-2 Plus HRP Detection kit (Golden Bridge
International, Inc., Mukilteo, WA, USA) was used. Nuclei were
counterstained with Mayer’s hematoxylin (Merck KGaA, Darmstadt,
Germany) (6). Oil red O staining
was carried out with using frozen sections as previously described
(17).
Monocyte adhesion assay
HUVECs were treated with sRAGE (1 μg/ml) followed by
exposure to static, laminar (15 dyne/cm2) and
oscillatory flow (±5 dyne/cm2) for 24 h. For the
positive controls, cells were treated with 10 ng/ml tumor necrosis
factor-α (TNF-α, 10 ng/ml) for 6 h. The medium was then removed,
and U937 cells were added to the dishes and incubated for 30 min at
37°C. The unbound cells in the dishes were then removed by washing
3 times with serum-free medium. The adherent cells were counted in
5 randomly selected optical fields in each well. Phase-contrast
microphotographs of the cells in the plates were taken using an
Olympus CKX41 inverted microscope (Olympus America Inc.).
RNA isolation, reverse-transcription
polymerase chain reaction (PCR)
Total RNA was isolated from the cultured HUVECs by
the use of a Total RNA Isolation kit (Qiagen Inc., Hilden, Germany)
(13). First-strand cDNA was
synthesized using the Super Script III first-strand synthesis
system (Invitrogen, Carlsbad, CA, USA). cDNA was amplified by PCR
for 30 cycles (Eppendorf AG, Hamburg, Germany). The following
oligonucleotide primers were used in this study: human RAGE sense,
5′-AGCGGCTGGAATGGAAACTGAACA-3′ and antisense,
5′-GAAGGGGCAAGGGCACACCATC-3′; human HMGB1 sense,
5′-GCGACTCTGTGCCTCGCTGA-3′ and antisense,
5′-ACGGGCCTTGTCCGCTTTTGC-3′; human VCAM-1 sense,
5′-GGCCTCAGTCAGTGTGA-3′ and antisense, 5′-AACCCCATTCAGCGTCA-3′;
human GAPDH sense, 5′-GAGTCAACGGATTTGGTCGT-3′ and antisense,
5′-TTGATTTTGGAGGGATCTCG-3′. The experiments were repeated 3
times.
Statistical analysis
All data are expressed as the means ± SEM from least
3 independent experiments with different sample sizes. A paired
t-test was used to assess the significance of the results between 2
groups. Differences between 3 or more groups were analyzed by
contrast analysis, using Super ANOVA. A p-value <0.05 was
considered to indicate a statistically significant difference.
Results
sRAGE represses RAGE-dependent
inflammatory effects under OSS conditions
To determine whether sRAGE is involved in the
RAGE-dependent pathway in ECs, we treated HUVECs with sRAGE (1
μg/ml of EC medium) and subsequently exposed the cells to LSS
(fluid shear stress, 15 dyne/cm2) or OSS (shea stress,
±5 dyne/cm2) for 24 h. The cell lysates were collected,
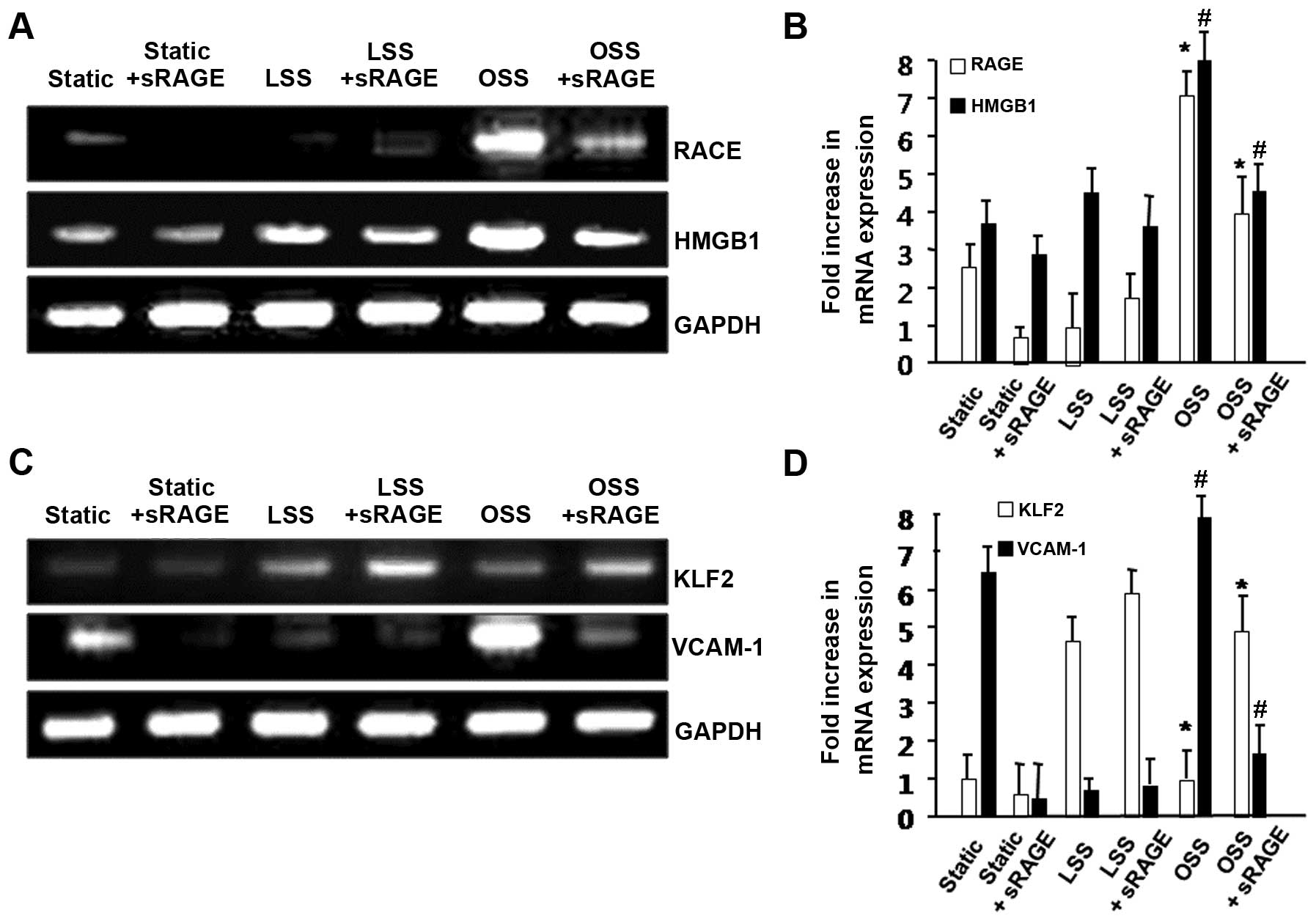
and the mRNA expression of RAGE was analyzed by RT-PCR. As shown in
Fig. 1A, sRAGE significantly
inhibited the OSS-induced expression of RAGE in the HUVECs. We also
observed that sRAGE reduced the mRNA expression level of HMGB1, a
ligand of RAGE. These results suggest that sRAGE plays an important
role in the RAGE-dependent pathway as a competent inhibitor of RAGE
under OSS conditions.
LSS plays a critical role in enhancing the survival
of ECs and regulating vascular tone; it also exerts
anti-inflammatory and anti-atheroscleroic effects (18–22). Previous studies have shown that
OSS generally upregulates genes with pro-inflammatory and
atherogenic properties (22–25). These effects occur through the
LSS-dependent induction of certain genes, such as Krüppel-like
factor -2 (KLF2) (26–29). KLF-2 is induced by LSS, which in
turn regulates a number of flow-responsive genes (26,27,30). Furthermore, KLF2 has been shown to
regulate leukocyte adhesion to the endothelium by downregulating
the expression of adhesion molecules that recruit leukocytes, such
as vascular cell adhesion molecule-1 (VCAM-1) (26,31,32). We used RT-PCR to analyze the
expression of KLF2 and VCAM-1. The mRNA expression level of KLF2
was increased by LSS; however, it was significantly reduced by OSS.
However, sRAGE significantly upregulated KLF2 expression in HUVECs
under OSS (Fig. 1B). By contrast,
when the cells were treated with sRAGE, the OSS-induced VCAM-1
expression was markedly attenuated (Fig. 1B). Takeb together, these data
demonstrate that sRAGE inhibits RAGE-dependent inflammation induced
by OSS.
sRAGE suppresses OSS-induced inflammation
in ECs
KLF2 exerts anti-inflammatory effects in ECs by
inhibiting the expression of VCAM-1, which regulates the
recruitment of monocytes to the ECs (26). To determine the potential role of
sRAGE in repressing the OSS-induced pro-inflammatory effects, we
examined monocyte adhesion to HUVECs under static, LSS and OSS
conditions with or without treatment with sRAGE (Fig. 2). ECs challenged with the
pro-inflammatory cytokine, TNF-α, were used as the positive
controls. As shown in Fig. 2, the
TNF-α and OSS-stimulated cells exhibited augmented monocyte
adhesion. By contrast, the sRAGE-treated ECs displayed
significantly less monocyte adhesion even under static conditions
with TNF-α treatment and OSS. Taken together, these results
indicate that sRAGE regulates monocyte-EC adhesion, stimulated by
OSS.
Partial carotid artery ligated mice have
enhanced RAGE expression in regions of disturbed blood flow
To directly examine variations in RAGE expression
under OSS conditions in vivo, we used a mouse model of
disturbed blood flow with low OSS induced by partial carotid artery
ligation (4).
As shown in Fig.
3, RAGE expression was markedly increased in the partially
ligated LCA, whereas there was a significantly lower expression
level of RAGE in the non-ligated RCA (Fig. 3A). To confirm whether shear stress
modulates the expression of RAGE in ECs, we performed
immunohistochemical staining on tissues from partially ligated
C57BL/6 mice (Fig. 3B). The
endothelial layer was stained with CD31 as an EC marker. The levels
of RAGE expression were significantly enhanced in the partially
ligated LCA compared with the non-ligated RCA. This suggests that
shear stress modulates the expression of RAGE in the vasculature.
These findings are consistent with those from previous studies,
showing that RAGE transcript levels are highly expressed at the
site of disturbed flow compared to the site of normal blood flow in
the arteries of swine (33). In
our study, the expression of RAGE was increased in the LCA even as
early as 1 day following ligation (Fig. 3C). On day 7, RAGE expression was
markedly upregulated in the LCA compared with the RCA.
sRAGE reduces atherosclerotic plaque
formation in vivo
To explore the role of sRAGE in disturbed
flow-induced atherosclerotic plaque formation in vivo, we
used a mouse model of partial carotid artery ligation using
ApoE−/− mice. The mice were fed a high-fat diet for 2
weeks following surgery. sRAGE (80 μg/kg) or PBS was injected into
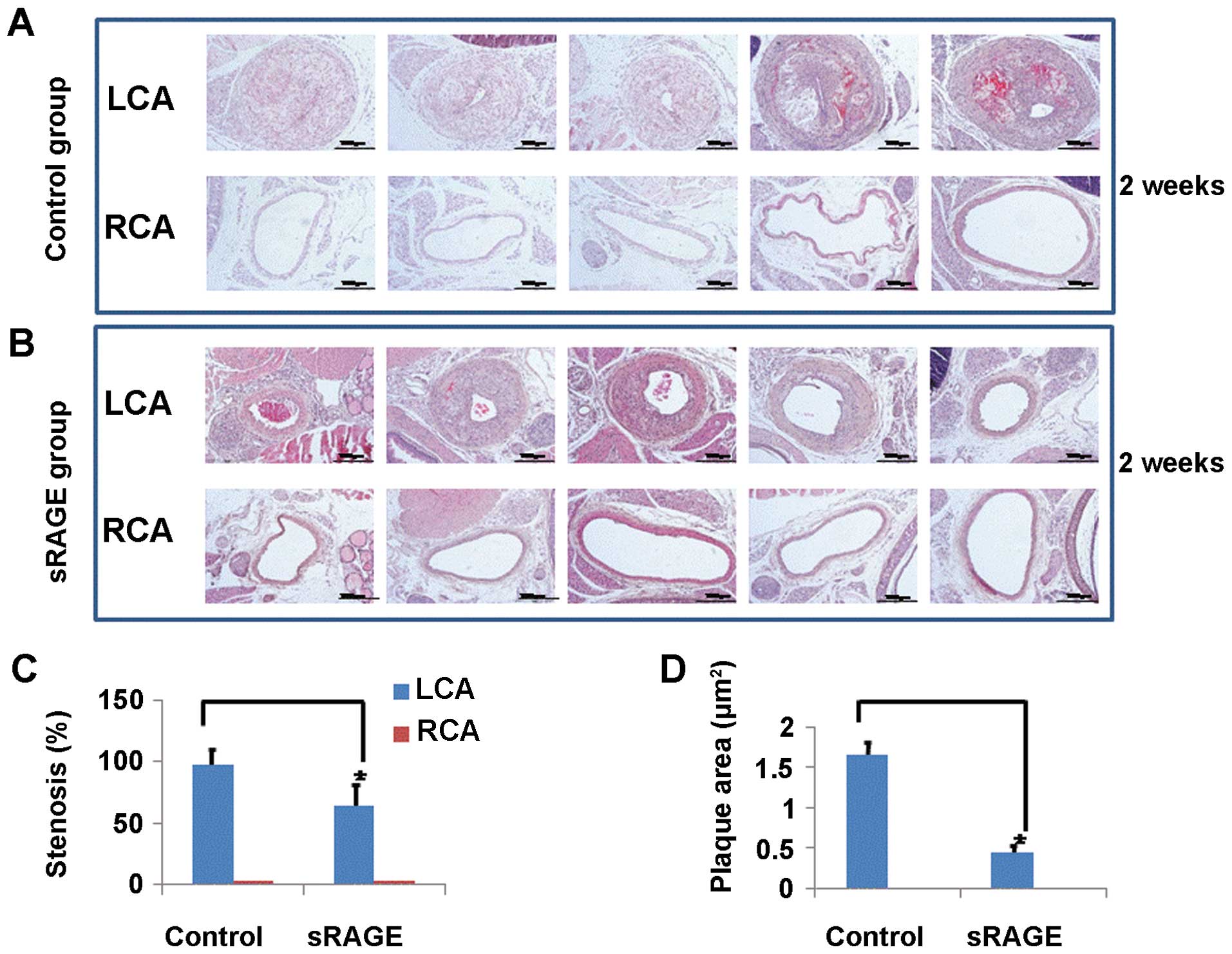
the intraperitoneal space, daily for 2 weeks. As shown in Fig. 4, the LCA of the control group
showed severe atherosclerotic plaque formation as determined by Oil
red O staining (Fig. 4A).
However, the LCA of the sRAGE-treated group showed significantly
reduced plaque formation (Fig.
4B). To better understand the effect of sRAGE in disturbed
flow-induced atherosclerotic plaque formation, the severity of
stenosis (Fig. 4C) and plaque
area (Fig. 4D) were compared
between the control and sRAGE-treated group. The non-ligated RCA in
both groups did not show any evidence of luminal stenosis and
atherosclerotic plaque formation. In contrast to the RCA, the
ligated LCA of the control group showed severe stenosis and a
significantly increased plaque area, while the sRAGE-treated group
showed significantly reduced luminal stenosis and plaque area.
These results suggest that sRAGE inhibits the development of
atherosclerotic plaque induced by disturbed blood flow.
Discussion
The novel finding of the present study is that sRAGE
plays a critical role as an inhibitor of atheromatous plaque
formation induced by disturbed blood flow. First, sRAGE repressed
the pro-inflammatory response induced by OSS through the
downregulation of RAGE, HMGB1 and VCAM-1 expression. According to a
previous study, HMGB1, the specific ligand for RAGE, is upregulated
at sites of activated ECs during vascular inflammation (9). In the current study, the expression
of HMGB1 under OSS conditions was significantly inhibited following
treatment with sRAGE. Monocyte binding was also inhibited following
treatment with sRAGE through the decreased expression of VCAM-1.
RAGE expression itself was decreased by sRAGE under OSS conditions.
Therefore, these findings suggest that sRAGE may be an important
inhibitor of vascular inflammatory responses generated by disturbed
blood flow.
Furthermore, the expression of KLF2, an
anti-inflammatory factor, was induced following treatment with
sRAGE under OSS conditions. This result indicates that sRAGE
induces the expression of atheroprotective factors which protect
cells in a pro-inflammatory environment, such as during the early
atherosclerotic process.
Second, we demonstrated that sRAGE suppresses
OSS-induced inflammatory responses, such as monocyte-EC adhesion in
in vitro experiments, as well as the development of
atherosclerotic plaque in partially ligated carotid arteries of
ApoE−/− mice in vivo. We showed that RAGE
expression was enhanced in the LCA, particularly in the endothelium
of the lesion of disturbed blood flow, and that the administration
of sRAGE suppressed atheromatous plaque formation in the partially
ligated carotid arteries of ApoE−/− mice. These findings
are consistent with those from previous studies, showing that the
treatment of ApoE−/− mice with sRAGE reduced the
development and progression of atherosclerosis in a dose-dependent
manner and that the progression of atherosclerosis was halted by
the administration of sRAGE in diabetic ApoE−/− mice
(5,6). However, these previous studies did
not determine the correlation between disturbed blood flow, RAGE
expression, sRAGE and atheromatous plaque formation. By comparison,
in our study, RAGE expression was increased by disturbed blood flow
and it was suppressed following treatment with sRAGE. Furthermore,
we showed that sRAGE suppressed atheromatous plaque formation
induced by disturbed blood flow. Blood vessels, particularly ECs
are consistently exposed to fluid shear stress, the dragging force
generated by blood flow. Certain studies have suggested that fluid
shear stress modulates vascular homeostasis and the focal
distribution of atherosclerosis by regulating endothelial gene
expression (19,34). Investigating the regulation of
blood flow-mediated genes is therefore mandatory in order to gain a
better understanding of atheroprotection.
To our knowledge, this is the first study
demonstrating that sRAGE regulates the process of atherogenesis
induced by disturbed blood flow. Previous studies using
static-cultured ECs have demonstrated that inflammatory signaling
is downregulated by sRAGE through the blockade of RAGE (35,36). However, in this study, we
demonstrate that fluid shear stress itself regulates the expression
of RAGE and its ligand, HMGB1. Another major finding of this study
was that sRAGE plays a crucial role in the regulation of the
RAGE-dependent pathway involving the expression of pro-inflammatory
and anti-inflammatory genes (VCAM-1 and KLF2) even in the absence
of additional RAGE ligands, such as HMGB1 and certain S100 family
members. This study unveils a novel role of sRAGE in regulating
atherogenesis in response to hemodynamic forces, and thus provides
the foundation for novel approaches for the treatment of
atherosclerosis.
Acknowledgements
The present study was supported by the Bio and
Medical Technology Development Program of the National Research
Foundation (NRF) funded by the Korean government (MEST) (no.
2011-0019695).
References
|
1
|
Ross R: Atherosclerosis - an inflammatory
disease. N Engl J Med. 340:115–126. 1999. View Article : Google Scholar
|
|
2
|
Chiu JJ, Usami S and Chien S: Vascular
endothelial responses to altered shear stress: pathologic
implications for atherosclerosis. Ann Med. 41:19–28. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chiu JJ and Chien S: Effects of disturbed
flow on vascular endothelium: pathophysiological basis and clinical
perspectives. Physiol Rev. 91:327–387. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nam D, Ni CW, Rezvan A, Suo J, Budzyn K,
Lianos A, Harrison D, Giddens D and Jo H: Partial carotid ligation
is a model of acutely induced disturbed flow, leading to rapid
endothelial dysfunction and atherosclerosis. Am J Physiol Heart
Circ Physiol. 297:H1535–H1543. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
DeVerse JS, Bailey KA, Jackson KN and
Passerini AG: Shear stress modulates RAGE-mediated inflammation in
a model of diabetes-induced metabolic stress. Am J Physiol Heart
Circ Physiol. 302:H2498–H2508. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lindsey JB, Cipollone F, Abdullah SM and
McGuire DK: Receptor for advanced glycation end-products (RAGE) and
soluble RAGE (sRAGE): cardiovascular implications. Diab Vasc Dis
Res. 6:7–14. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Harja E, Bu DX, Hudson BI, Chang JS, Shen
X, Hallam K, Kalea AZ, Lu Y, Rosario RH, Oruganti S, Nikolla Z,
Belov D, Lalla E, Ramasamy R, Yan SF and Schmidt AM: Vascular and
inflammatory stresses mediate atherosclerosis via RAGE and its
ligands in apoE−/− mice. J Clin Invest. 118:183–194.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schlueter C, Hauke S, Flohr AM, Rogalla P
and Bullerdiek J: Tissue-specific expression patterns of the RAGE
receptor and its soluble forms - a result of regulated alternative
splicing? Biochim Biophys Acta. 1630:1–6. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Herold K, Moser B, Chen Y, Zeng S, Yan SF,
Ramasamy R, Emond J, Clynes R and Schmidt AM: Receptor for advanced
glycation end products (RAGE) in a dash to the rescue: inflammatory
signals gone awry in the primal response to stress. J Leukoc Biol.
82:204–212. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ
Jr, Chow WS, Stern D and Schmidt AM: Suppression of accelerated
diabetic atherosclerosis by the soluble receptor for advanced
glycation endproducts. Nat Med. 4:1025–1031. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bucciarelli LG, Wendt T, Qu W, Lu Y, Lalla
E, Rong LL, Goova MT, Moser B, Kislinger T, Lee DC, Kashyap Y,
Stern DM and Schmidt AM: RAGE blockade stabilizes established
atherosclerosis in diabetic apolipoprotein E-null mice.
Circulation. 106:2827–2835. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou Z, Wang K, Penn MS, Marso SP, Lauer
MA, Forudi F, Zhou X, Qu W, Lu Y, Stern DM, Schmidt AM, Lincoff AM
and Topol EJ: Receptor for AGE (RAGE) mediates neointimal formation
in response to arterial injury. Circulation. 107:2238–2243. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ha CH, Wang W, Jhun BS, Wong C, Hausser A,
Pfizenmaier K, McKinsey TA, Olson EN and Jin ZG: Protein kinase
D-dependent phosphorylation and nuclear export of histone
deacetylase 5 mediates vascular endothelial growth factor-induced
gene expression and angiogenesis. J Biol Chem. 283:14590–14599.
2008. View Article : Google Scholar
|
|
14
|
Jin ZG, Ueba H, Tanimoto T, Lungu AO,
Frame MD and Berk BC: Ligand-independent activation of vascular
endothelial growth factor receptor 2 by fluid shear stress
regulates activation of endothelial nitric oxide synthase. Circ
Res. 93:354–363. 2003. View Article : Google Scholar
|
|
15
|
Dave SH, Tilstra JS, Matsuoka K, Li F,
DeMarco RA, Beer-Stolz D, Sepulveda AR, Fink MP, Lotze MT and Plevy
SE: Ethyl pyruvate decreases HMGB1 release and ameliorates murine
colitis. J Leukoc Biol. 86:633–643. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sasaki T, Kuzuya M, Cheng XW, Nakamura K,
Tamaya-Mori N, Maeda K, Kanda S, Koike T, Sato K and Iguchi A: A
novel model of occlusive thrombus formation in mice. Lab Invest.
84:1526–1532. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou J, Lhotak S, Hilditch BA and Austin
RC: Activation of the unfolded protein response occurs at all
stages of atherosclerotic lesion development in apolipoprotein
E-deficient mice. Circulation. 111:1814–1821. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Berk BC: Atheroprotective signaling
mechanisms activated by steady laminar flow in endothelial cells.
Circulation. 117:1082–1089. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Davies PF: Flow-mediated endothelial
mechanotransduction. Physiol Rev. 75:519–560. 1995.PubMed/NCBI
|
|
20
|
Gimbrone MA Jr, Topper JN, Nagel T,
Anderson KR and Garcia-Cardena G: Endothelial dysfunction,
hemodynamic forces, and atherogenesis. Ann NY Acad Sci.
902:230–240. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Boo YC and Jo H: Flow-dependent regulation
of endothelial nitric oxide synthase: role of protein kinases. Am J
Physiol Cell Physiol. 285:C499–C508. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chien S: Mechanotransduction and
endothelial cell homeostasis: the wisdom of the cell. Am J Physiol
Heart Circ Physiol. 292:H1209–H1224. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shyy YJ, Hsieh HJ, Usami S and Chien S:
Fluid shear stress induces a biphasic response of human monocyte
chemotactic protein 1 gene expression in vascular endothelium. Proc
Natl Acad Sci USA. 91:4678–4682. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kraiss LW, Geary RL, Mattsson EJ, Vergel
S, Au YP and Clowes AW: Acute reductions in blood flow and shear
stress induce platelet-derived growth factor-A expression in baboon
prosthetic grafts. Circ Res. 79:45–53. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wilcox JN, Smith KM, Williams LT, Schwartz
SM and Gordon D: Platelet-derived growth factor mRNA detection in
human atherosclerotic plaques by in situ hybridization. J Clin
Invest. 82:1134–1143. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
SenBanerjee S, Lin Z, Atkins GB, Greif DM,
Rao RM, Kumar A, Feinberg MW, Chen Z, Simon DI, Luscinskas FW,
Michel TM, Gimbrone MA Jr, Garcia-Cardena G and Jain MK: KLF2 is a
novel transcriptional regulator of endothelial proinflammatory
activation. J Exp Med. 199:1305–1315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Parmar KM, Larman HB, Dai G, Zhang Y, Wang
ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, Gimbrone MA Jr and
Garcia-Cardena G: Integration of flow-dependent endothelial
phenotypes by Kruppel-like factor 2. J Clin Invest. 116:49–58.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee JS, Yu Q, Shin JT, Sebzda E, Bertozzi
C, Chen M, Mericko P, Stadtfeld M, Zhou D, Cheng L, Graf T, MacRae
CA, Lepore JJ, Lo CW and Kahn ML: Klf2 is an essential regulator of
vascular hemodynamic forces in vivo. Dev Cell. 11:845–857. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Atkins GB and Jain MK: Role of
Kruppel-like transcription factors in endothelial biology. Circ
Res. 100:1686–1695. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dekker RJ, van Soest S, Fontijn RD,
Salamanca S, de Groot PG, VanBavel E, Pannekoek H and Horrevoets
AJ: Prolonged fluid shear stress induces a distinct set of
endothelial cell genes, most specifically lung Kruppel-like factor
(KLF2). Blood. 100:1689–1698. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tsao PS, Buitrago R, Chan JR and Cooke JP:
Fluid flow inhibits endothelial adhesiveness. Nitric oxide and
transcriptional regulation of VCAM-1. Circulation. 94:1682–1689.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fledderus JO, van Thienen JV, Boon RA,
Dekker RJ, Rohlena J, Volger OL, Bijnens AP, Daemen MJ, Kuiper J,
van Berkel TJ, Pannekoek H and Horrevoets AJ: Prolonged shear
stress and KLF2 suppress constitutive proinflammatory transcription
through inhibition of ATF2. Blood. 109:4249–4257. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Passerini AG, Polacek DC, Shi C, Francesco
NM, Manduchi E, Grant GR, Pritchard WF, Powell S, Chang GY,
Stoeckert CJ Jr and Davies PF: Coexisting proinflammatory and
antioxidative endothelial transcription profiles in a disturbed
flow region of the adult porcine aorta. Proc Natl Acad Sci USA.
101:2482–2487. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Berk BC, Abe JI, Min W, Surapisitchat J
and Yan C: Endothelial atheroprotective and anti-inflammatory
mechanisms. Ann NY Acad Sci. 947:93–111. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mullins GE, Sunden-Cullberg J, Johansson
AS, Rouhiainen A, Erlandsson-Harris H, Yang H, Tracey KJ, Rauvala
H, Palmblad J, Andersson J and Treutiger CJ: Activation of human
umbilical vein endothelial cells leads to relocation and release of
high-mobility group box chromosomal protein 1. Scand J Immunol.
60:566–573. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Frommhold D, Kamphues A, Hepper I,
Pruenster M, Lukic IK, Socher I, Zablotskaya V, Buschmann K,
Lange-Sperandio B, Schymeinsky J, Ryschich E, Poeschl J, Kupatt C,
Nawroth PP, Moser M, Walzog B, Bierhaus A and Sperandio M: RAGE and
ICAM-1 cooperate in mediating leukocyte recruitment during acute
inflammation in vivo. Blood. 116:841–849. 2010. View Article : Google Scholar : PubMed/NCBI
|