Introduction
Over the last few years, studies have demonstrated
that apoptosis is an early event involved in cardiomyopathy
associated with diabetes mellitus (1). A key pathological consequence of
sustained hyperglycemia is the induction of cardiomyocyte apoptosis
in diabetic patients and animal models of diabetes (2,3).
Several clinical studies have demonstrated that elevated blood
glucose levels are a risk factor for the development of diabetic
cardiomyopathy (4–7). Sustained hyperglycemia can produce a
large number of oxygen free radicals, which can induce the
apoptosis of cardiomyocytes and lead to oxidative myocardial
injury. Cardiomyocyte apoptosis causes a loss of contractile units
which reduces organ function and provokes cardiac remodeling, which
is associated with the hypertrophy of viable cardiomyocytes
(8,9). As such, should myocardial apoptosis
be inhibited, one would expect to prevent or slow the development
of heart failure. Yet, the means by which hyperglycemia induces
apoptosis in cardiomyocytes have not yet been fully elucidated.
Apoptosis is a major mechanism of cell death,
characterized by a series of tightly regulated processes involved
in the activation of a cascade of molecular events leading to cell
death. Cells undergoing apoptosis have been shown to have elevated
levels of cytochrome c in the cytosol and a corresponding
decrease in mitochondrial potential (10). Bcl-2 exerts a pro-survival effect
in response to a wide range of apoptotic stimuli through the
inhibition of mitochondrial cytochrome c release (11,12). Bak is a pro-apoptotic member of
the Bcl-2 family. This protein is located on the outer membrane of
the mitochondria and is an essential component for the transduction
of apoptotic signals through the mitochondrial pathway (13–15). Bax is a key component for
cellular-induced apoptosis through mitochondrial stress. Upon
apoptotic stimulation, Bax forms oligomers and translocates from
the cytosol to the mitochondrial membrane. Through interactions
with pore proteins on the mitochondrial membrane, Bax increases the
permeability of the membrane, which leads to the release of
cytochrome c from the mitochondria, the activation of
caspase-9 and the initiation of the caspase activation pathway for
apoptosis (13–15).
Cardiomyocyte apoptosis is a multifactorial process
involving the activation of several signal transduction pathways,
including the p38 mitogen-activated protein kinase (MAPK) pathway.
It has been reported that p38 activation induces p53-dependent
apoptosis (16). The inhibition
of p38 phosphorylation prevents the activation of p53 (Ser15
phosphorylation and expression), suppresses Bax expression and
inhibits the cleavage of caspase-3 and cardiomyocyte apoptosis.
The flavonoid naringin (NAR;
4′,5,7-trihydroxyflavanone-7-rhamnoglucoside) is a polyphenolic
compound that naturally occurs in citrus fruit. NAR has previously
been shown to have anti-inflammatory (17,18), antioxidant (19,20), anticancer (21), antibacterial (22), antimutagenic (23), antiulcer, antitussive (24,25), neuroprotective (26–28), as well as anti-hypertensive
properties (29), and can reduce
blood cholesterol levels, preventing thrombosis (30). In recent years, it has been
reported that NAR inhibits the apoptosis of hydrogen
peroxide-induced p388 cells (31)
and Rotenone-induced human neuroblastoma SH-SY5Y cells (32). However, whether NAR can inhibit
high glucose (HG)-induced cardiomyocyte apoptosis has not yet been
reported. Therefore, this study aimed to investigate the effects of
NAR on HG-induced cardiomyocyte apoptosis at the cellular and
molecular level. We hypothesized that NAR can inhibit cardiomyocyte
apoptosis by attenuating mitocondrial dysfuntion through he
mitochondrial signaling pathway and inhibiting p38 activation.
Materials and methods
Reagents
All reagents were purchased from Sigma Inc. (St.
Louis, MO, USA) unless otherwise stated. JC-1 was obtained from
Beyotime Biotechnology, Inc. (Nantong, China).
Cell culture and treatments
H9c2 cells were obtained from the Sun Yat-sen
University Experimental Animal Centre (Guangzhou, China). The H9c2
cell line, a subclone of the original clonal cell line, was derived
from embryonic rat heart tissue. The cells were cultured in
Dulbecco’s modified Eagle’s medium (DMEM) medium supplemented with
normal glucose (5.5 mM) and 15% FBS at 37°C under an atmosphere of
5% CO2 and 95% air.
H9c2 cells were incubated in DMEM with HG (16.7 mM)
for 6 h to induce apoptosis. NAR (5 μM) was added into the above
medium (DMEM) to examine its cytoprotective effects. The selective
p38 inhibitor, SB203580 (10 μM), was administered to the H9c2 cells
for 60 min prior to exposure to HG (16.7 mM) and/or treatment with
NAR (5 μM).
Western blot analysis
Following treatment, the H9c2 cells were harvested
and lysed with ice-cold cell lysis solution and the homogenate was
centrifuged at 10,000 × g for 15 min at 4°C. Total protein in the
supernatant was quantified using a BCA protein assay kit. Total
protein (20 μg) from each sample was separated by 12% SDS-PAGE and
transferred onto a PVDF membrane, the PVDF membrane was then placed
in washing buffer containing skimmed milk powder at room
temperature and blocked for 2 h, and then washed three times.
Bcl-2, Bax, Bak, voltage-dependent anion channel (VDAC), cytochrome
c, p38, phosphorylated p38 (p-p38), p53, phosphorylated p53
(p-p53) and β-actin monoclonal antibodies were then added, followed
by incubation at 4°C overnight. Horseradish peroxidase-conjugated
secondary antibody was then added followed by incubation for 1 h.
The membrane was then exposed to X-ray film, and processed using an
AlphaImager HP fluorescence/visible light gel imaging analyzer.
Image analysis software were used to analyze the gray value.
Detection of apoptosis
As stated in the KGI Annexin V-FITC apoptosis
detection kit instruction manual, the cells were digested with
trypsin, and centrifuged at 1,000 rpm for 5 min. After collection,
the cells were washed twice with PBS, and centrifuged at 1,000 rpm
for 5 min. A total of 5×105 cells were collected and
suspensed in 500 μl binding buffer. Subsequently, 5 μl Annexin
V-FITC and 5 μl propidium iodide (PI) were added followed by mixing
at room temperature, in the dark for 15 min. Within 1 h, the
apoptotic cells were detected by flow cytometry. The excitation
wavelength was 488 nm and the emission wavelength was 530 nm. Green
fluorescence of Annexin V-FITC was detected using the FITC channel
(FL1); PI red fluorescence was detected using the PI channel (FL2
or FL3). For fluorescent compensation adjustment, normal cells not
treated with HG to induce apoptosis were used as the controls for
fluorescence compensation settings adjustment.
Caspase-3, -8 and -9 activity
assessment
To measure caspase-3, -8 and -9 enzymatic activity,
the H9c2 cells were cultured for 24 h followed by treatment with HG
(16.7 mM) for 6 h in the presence or absence of NAR (5 μM). APO
LOGIX Carboxyfluorescein Caspase Detection kits (Cell Technology,
Inc., Mountain View, CA, USA) were used to detect active caspase-3,
-8 and -9 according to the manufacturer’s instructions.
Detection of mitochondrial membrane
potential (MMP)
The MMP detection kit (JC-1; Beyotime Biotechnology
Research Institute) was used to detect MMPs. Cells were washed once
with PBS and then 1 ml cell culture medium containing serum and
phenol red was added. JC-1 dye working solution (1 ml) was then
added followed by mixing. The cells were subsequently incubated in
a cell incubator at 37°C for 20 min, and then washed with JC-1
staining buffer (1X) twice. Cell culture medium (2 ml) was then
added and the cells were observed under fluorescence or confocal
microscope. The aggregate JC-1 (red fluorescence) was detected at
an emission wavelength of 590 nm, and the monomeric JC-1 (green
fluorescence) was monitored at 529 nm. The ratio of aggregated and
monomeric JC-1 was used to quantify changes in MMP, and a decreased
JC-1 ratio represented the depolarization of the mitochondria,
indicating a decrease in MMP.
Statistical analysis
The results of each experimental condition were
determined from the mean of triplicate trials. Data are expressed
as the means ± SEM (n=6 unless otherwise stated). A two-tailed
Student’s t-test was used to assess the significance of differences
between two groups. Analysis of variance was used when comparing
more than two groups; differences between two groups within the set
were analyzed by a Fisher’s protected least-significant differences
test. P-values <0.05 were considered to indicate statistically
significant differences.
Results
NAR decreases HG-induced H9c2 cell
apoptosis
To investigate the effect of NAR on HG-induced
cardiomyocyte apoptosis, Annexin V and PI staining was performed in
the H9c2 cells treated with HG (16.7 mM) in the absence or presence
of NAR. Exposure to HG induced apoptosis in H9c2 cells and the
apoptotic rate was 46.5±8.5 in the HG group (Fig. 1). NAR decreased the apoptotic rate
to 25.6±5.9 in the HG + NAR group. No apoptosis was observed in the
cells treated with NAR alone.
Caspase-3, -8 and -9 activity was measured to
evaluate the effect of NAR on HG-induced cardiomyocyte apoptosis.
Caspase-3 activity was significantly higher 6 h following exposure
to HG in the HG group compared with the control group (Fig. 1). Following the addition of 5 μM
NAR, caspase-3 activity markedly decreased in the HG + NAR group
compared with the HG group. Caspase-9 activity showed a significant
increase 6 h following exposure to 16.7 mM glucose, which was
almost completely attenuated in the presence of 5 μM NAR. However,
no evident changes were observed in the activity of caspase-8 6 h
following stimulation with HG and/or NAR.
NAR prevents HG-induced MMP loss in H9c2
cells
The MMP of H9c2 cells was measured by JC-1, an
indicator mitochondrial function, in the H9c2 cells treated with
glucose (16.7 mM) in the absence or presence of NAR (5 μM) for 6 h.
Red fluorescence represents the mitochondrial aggregate JC-1 and
green fluorescence indicates the monomeric JC-1. The exposure of
H9c2 cells to glucose (16.7 mM) for 6 h induced a marked decrease
in MMP, and treatment with NAR prevented the HG-induced loss in MMP
(Fig. 2).
Mitochondrial pathway is involved in the
inhibitory effect of NAR on HG-induced H9c2 cell apoptosis
It has been reported that the HG-induced apoptosis
of cardiomyocytes is a multifactorial process. In this study, we
examined Bcl-2, cytosolic cytochrome c, mitochondrial Bax
and Bak levels in H9c2 cells. Following exposure to 16.7 mM glucose
for 6 h, cytoplasmic cytochrome c, mitochondrial Bax and Bak
levels significantly increased compared with the controls (Fig. 3). By contrast, cytoplasmic
cytochrome c, mitochondrial Bax and Bak levels decreased
following treatment with NAR in the cardiomyocytes exposed to HG.
HG (16.7 mM) reduced Bcl-2 expression in the H9c2 cells, which was
significantly elevated following treatment with NAR. These results
suggest that the mitochondrial pathway is involved in the
inhibitory effects of NAR on HG-induced H9c2 cell apoptosis.
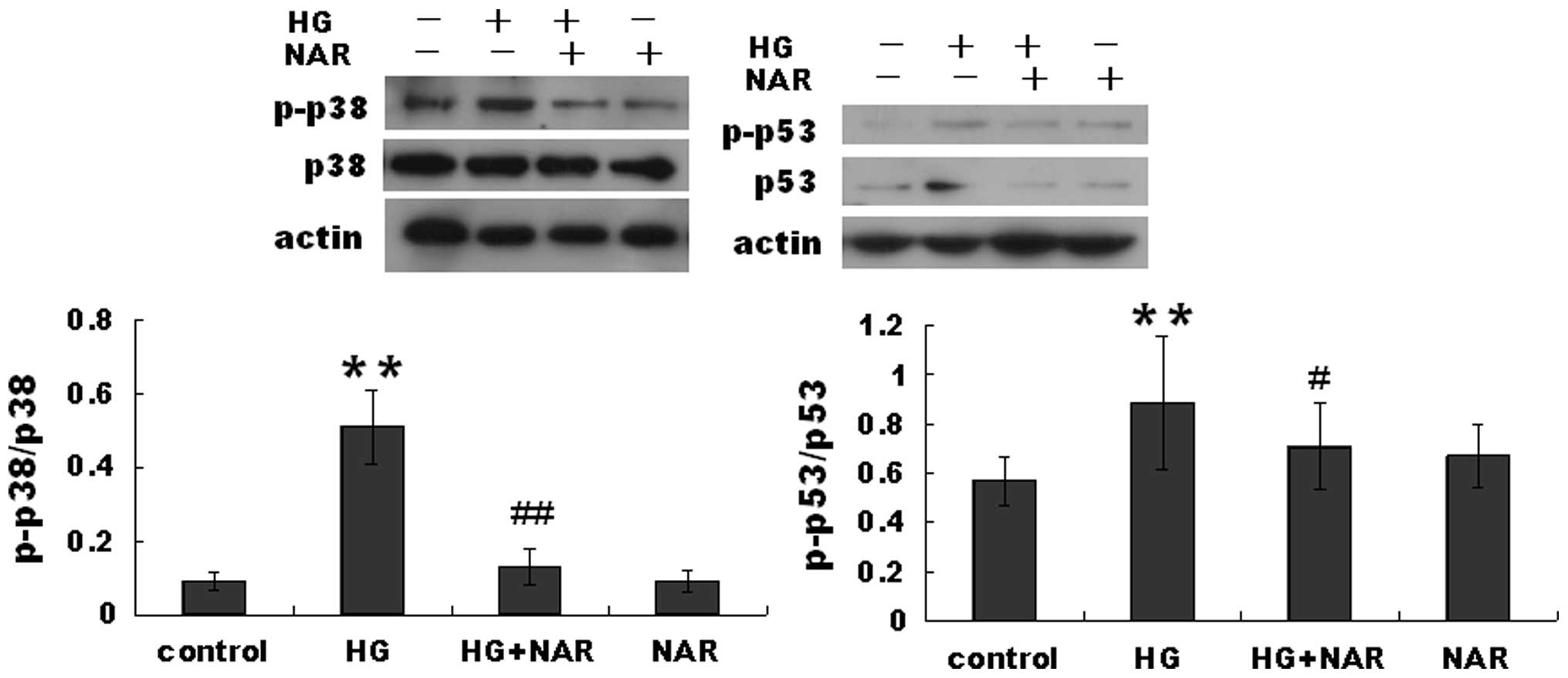
p38 signaling pathway is involved in the
inhibitory effects of NAR on HG-induced H9c2 cell apoptosis
Previous studies have demonstrated that p38 and p53
activation leads to cardiomyocyte apoptosis. Thus, in this study,
we measured the protein levels of p-p53, p53, p38 and p-p38 in H9c2
cells. The p-p53 level was markedly increased in the HG group
compared with the control group, but was decreased in the HG + NAR
group compared with the HG group 6 h following exposure to HG
(Fig. 4). The same tendency in
expression was observed for p-p38.
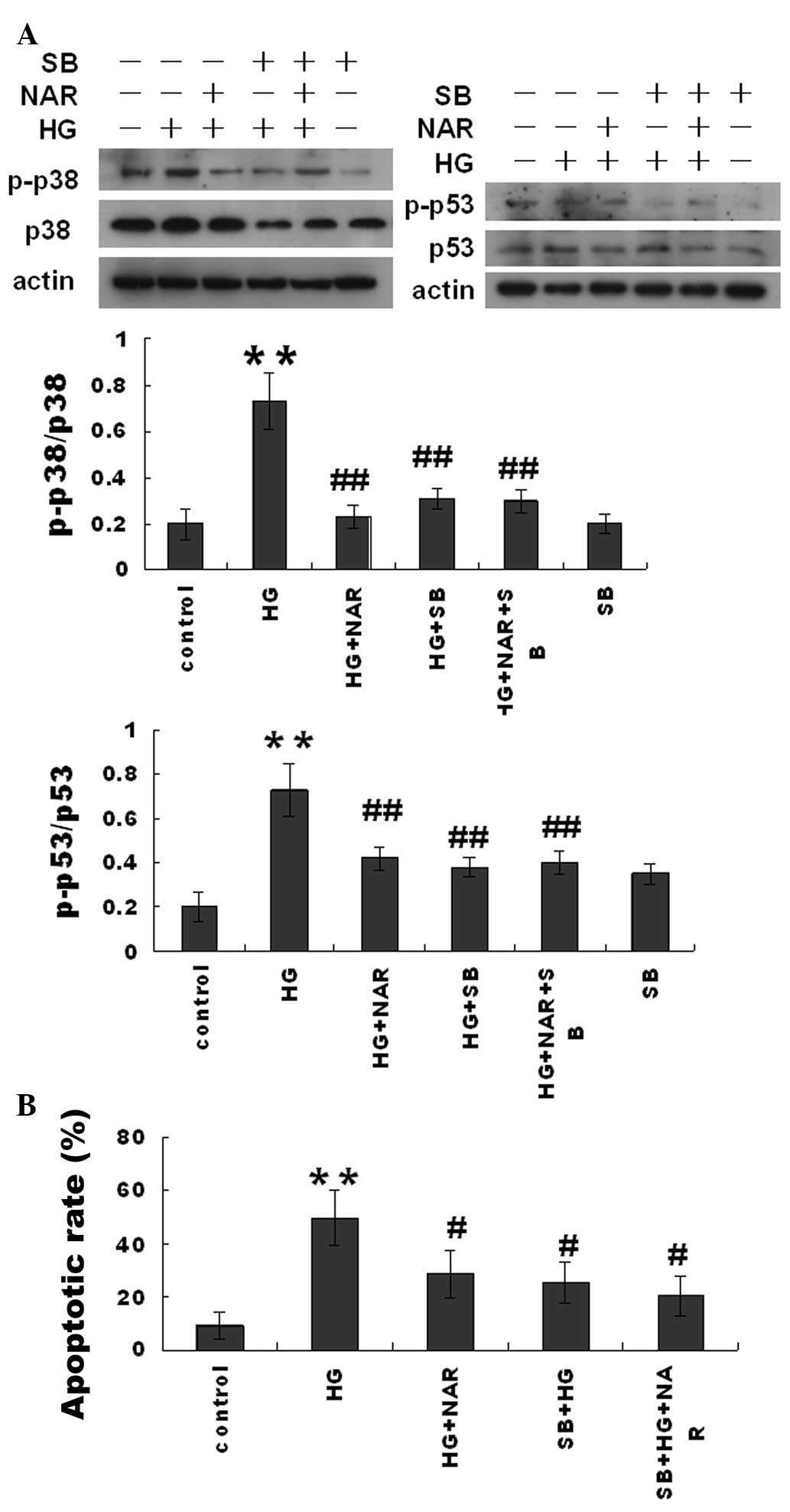
The p38 inhibitor, SB203580, and NAR
inhibit HG-induced H9c2 cell apoptosis and attenuate mitochondrial
dysfunction
In order to determine the role of p38 in HG-induced
H9c2 cell apoptosis, the H9c2 cells were pre-treated with the p38
inhibitor, SB203580 (10 μM), for 90 min and then exposed to HG
(16.7 mM) and/or NAR (5 μM) for 6 h. Cell apoptosis was measured
using Annexin V and PI double staining. SB203580 significantly
inhibited the HG-induced H9c2 cell apoptosis in the HG + SB203580
(SB) group, but did not affect the inhibitory effect of NAR in the
HG + SB + NAR group (Fig. 5). The
results from western blot analysis demonstrated that SB203580
significantly inhibited p38 phosphorylation and in turn, p53
phosphorylation, but did not alter the inhibitory effects of NAR on
p38 and p53 phosphorylation in the HG-challenged H9c2 cells.
Moreover, SB203580 inhibited the HG-induced H9c2 MMP loss in the HG
+ SB group, but did not alter the inhibitory effects of NAR in the
HG + NAR + SB group (Fig. 6).
Discussion
Diabetic cardiomyopathy is related directly to
hyperglycemia (6). Cell death
(apoptosis) plays a critical role in cardiac pathogenesis.
Hyperglycemia induces myocardial apoptosis and leads to diabetic
cardiomyopathy (6). Cardiomyocyte
apoptosis and mitochondrial dysfunction are the pathophysiological
basis of diabetic cardiomyopathy, and can significantly increase
the incidence of heart failure (6–8).
Therefore, it is of great significance to elucidate the molecular
mechanisms involved in cardiomyocyte apoptosis, in order to develop
novel and effective clinical methods to prevent this event.
Although a variety of signaling pathways have been confirmed to
participate in the development of cardiomyocyte apoptosis, the
inherent mechanisms involved in cardiomyocyte apoptosis and in the
reversal of mitochondrial dysfunction remain unclear.
NAR (4′,5,7-trihydroxyflavanone-7-rhamnoglucoside)
is derived from grapefruit and related citrus species and has been
evaluated for a wide spectrum of activities, including antioxidant
(19,20,33), anticancer (21,34), anti-inflammatory (35) and cardioprotective activities
(30,36,37). It has been reported to lower
glucose levels and elevate plasma insulin levels in rats with
streptozotocin (STZ)-induced diabetis (33,38). However, the role of NAR in
diabetic complications has not yet been elucidated. In this study,
we examined the hypothesis that NAR inhibits HG-induced apoptotic
cell death in cardiomyocytes by attenuating mitochondrial
dysfunction and modulating the activation of the p38 signaling
pathway.
The mitochondrial pathway of apoptosis is dependent
upon the Bcl-2 family for the release of pro-apoptotic factors
(cytochrome c) from the mitochondrial intermembrane space
(IMS), via the process of mitochondrial outer-membrane
permeabilization (MOMP) (13,15). The Bcl-2 family of proteins are
important regulators of the mitochondrial pathway of apoptosis
(14,39). The multi-BH-domain-containing
pro-apoptotic members, Bax and Bak, are the essential gateways to
MOMP. They directly engage MOMP by creating proteolipid pores
responsible for the release of cytochrome c (13,15). This release of cytochrome c
in turn activates caspase-9, a cysteine protease. Caspase-9 in turn
activates caspase-3 and -7, which are responsible for destroying
the cell from within (40). In
this study, the exposure of cardiomyocytes to HG induced apoptosis
and mitochondrial dysfunction. Treatment with NAR significantly
decreased the expression of apoptosis-related proteins
(mitochondrial Bax and Bak, cytoplasmic cytochrome c), while
it increased the expression of the anti-apoptotic protein, Bcl-2.
In addition, the HG-induced loss of MMP was attenuated in the H9c2
cells following treatment with NAR. Caspase-3 and -9 activity was
also decreased in the H9c2 cells following treatment with NAR.
Previous studies have demonstrated that
hyperglycemia-induced cardiomyocyte apoptosis is associated with
the activation of p53 (41).
Hyperglycemia induces the overexpression of p53 and its
phosphorylation on Ser15 (activated). This activation of p53 is
linked to the downregulation of Bcl-2 and the upregulation of Bax,
thus activating the intrinsic apoptotic pathway (42). Consistent with these findings, we
also found that exposure to HG induced the phosphorylation of p53
on Ser15 and increased p53 expression in H9c2 cells. Treatment with
NAR abrogated the expression and phosphorylation of p53 induced by
the exposure to HG. Therefore, our data indicate that the
mechanisms underlying the inhibition of HG-induced cardiomyocyte
apoptosis by NAR are associated with the suppression of p53
overexpression and phosphorylation.
Recently, it was reported that p38 activation
induced p53-dependent apoptosis. In a previous study, treatment
with doxorubicin (DOX) induced p38 (Thr172) phosphorylation in
cardiomyocytes (43). The p38
inhibitor, SB203580, inhibited DOX-induced p53 activation (Ser15
phosphorylation and expression), decreased Bax expression,
inhibited the cleavage of caspase-3, preventing cardiomyocyte
apoptosis. These results indicate that p38 activation contributes
to DOX-induced cardiomyocyte apoptosis by inducing the
phosphorylation of p53 on Ser15 (43,44). In the present study, we also
observed that exposure to HG significantly increased the
phosphorylation of p38 and p53 6 h following stimulation with HG in
the H9c2 cells. SB203580, an inhibitor of p38 phosphorylation,
suppressed the HG-induced p38 and p53 phosphorylation, preventing
cardiomyocyte apoptosis. Similar results were observed in the
NAR-treated H9c2 cells incubated in culture medium containing high
levels of glucose. However, NAR did not completely reverse
HG-induced cardiomyocyte apoptosis. Exposure to HG has been
reported to induce cardiomyocyte apoptosis through the activation
of a variety of signaling pathways associated with oxidative stress
(45), calcium (45), transcription factors (46) and MAPKs (47). Thus, the results from the present
study suggest that NAR inhibits HG-induced cardiomyocyte apoptosis
by inhibiting the activation of p38.
In conclusion, NAR inhibits HG-induced cardiomyocyte
apoptosis by attenuating mitochondrial dysfunction and preventing
the activation of p38. Therefore, NAR exerts cardioprotective
effects, preventing cardiomyocyte apoptosis. This inhibitory effect
of NAR on cardiomyocyte apoptosis may provide insight into the
investigation of the prevention and development of diabetic
cardiomyopathy.
Acknowledgements
The authors are grateful to Dr Qitao Yan and Dr Rui
Zhao (Southern Medical University, Guangzhou, China) for providing
technical assistance. The present study was supported by grants
from the Science and Technology Planning Project of Guangdong in
China (no. 2012A080202020) and the Guangdong Natural Science
Foundation (no. S2011010002620).
References
|
1
|
Cai L and Kang YJ: Cell death and diabetic
cardiomyopathy. Cardiovasc Toxicol. 3:219–228. 2003. View Article : Google Scholar
|
|
2
|
Adeghate E: Molecular and cellular basis
of the aetiology and management of diabetic cardiomyopathy: a short
review. Mol Cell Biochem. 261:187–191. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lakshmanan AP, Harima M, Suzuki K, et al:
The hyperglycemia stimulated myocardial endoplasmic reticulum (ER)
stress contributes to diabetic cardiomyopathy in the transgenic
non-obese type 2 diabetic rats: a differential role of unfolded
protein response (UPR) signaling proteins. Int J Biochem Cell Biol.
45:438–447. 2013. View Article : Google Scholar
|
|
4
|
Jankyova S, Kmecova J, Cernecka H, et al:
Glucose and blood pressure lowering effects of
Pycnogenol® are inefficient to prevent prolongation of
QT interval in experimental diabetic cardiomyopathy. Pathol Res
Pract. 208:452–457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cai L: Diabetic cardiomyopathy and its
prevention by metallothionein: experimental evidence, possible
mechanisms and clinical implications. Curr Med Chem. 14:2193–2203.
2007. View Article : Google Scholar
|
|
6
|
Miki T, Yuda S, Kouzu H and Miura T:
Diabetic cardiomyopathy: pathophysiology and clinical features.
Heart Fail Rev. Mar 28–2012.(Epub ahead of print).
|
|
7
|
Regan TJ, Ahmed S, Haider B, Moschos C and
Weisse A: Diabetic cardiomyopathy: experimental and clinical
observations. N J Med. 91:776–778. 1994.PubMed/NCBI
|
|
8
|
Frustaci A, Kajstura J, Chimenti C, et al:
Myocardial cell death in human diabetes. Circ Res. 87:1123–1132.
2000. View Article : Google Scholar
|
|
9
|
Cai L, Wang Y, Zhou G, et al: Attenuation
by metallothionein of early cardiac cell death via suppression of
mitochondrial oxidative stress results in a prevention of diabetic
cardiomyopathy. J Am Coll Cardiol. 48:1688–1697. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Condorelli G, Morisco C, Stassi G, et al:
Increased cardiomyocyte apoptosis and changes in proapoptotic and
antiapoptotic genes bax and bcl-2 during left ventricular
adaptations to chronic pressure overload in the rat. Circulation.
99:3071–3078. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lv X, Yu X, Wang Y, et al: Berberine
inhibits doxorubicin-triggered cardiomyocyte apoptosis via
attenuating mitochondrial dysfunction and increasing Bcl-2
expression. PLoS One. 7:e473512012. View Article : Google Scholar
|
|
13
|
Ruffolo SC and Shore GC: BCL-2 selectively
interacts with the BID-induced open conformer of BAK, inhibiting
BAK auto-oligomerization. J Biol Chem. 278:25039–25045. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Youle RJ and Strasser A: The BCL-2 protein
family: opposing activities that mediate cell death. Nat Rev Mol
Cell Biol. 9:47–59. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yi X, Yin XM and Dong Z: Inhibition of
Bid-induced apoptosis by Bcl-2. tBid insertion, Bax translocation,
and Bax/Bak oligomerization suppressed. J Biol Chem.
278:16992–16999. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim SJ, Hwang SG, Shin DY, Kang SS and
Chun JS: p38 kinase regulates nitric oxide-induced apoptosis of
articular chondrocytes by accumulating p53 via NFkappa B-dependent
transcription and stabilization by serine 15 phosphorylation. J
Biol Chem. 277:33501–33508. 2002. View Article : Google Scholar
|
|
17
|
Jain M and Parmar HS: Evaluation of
antioxidative and anti-inflammatory potential of hesperidin and
naringin on the rat air pouch model of inflammation. Inflamm Res.
60:483–491. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kawaguchi K, Maruyama H, Hasunuma R and
Kumazawa Y: Suppression of inflammatory responses after onset of
collagen-induced arthritis in mice by oral administration of the
Citrus flavanone naringin. Immunopharmacol Immunotoxicol.
33:723–729. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cavia-Saiz M, Busto MD, Pilar-Izquierdo
MC, Ortega N, Perez-Mateos M and Muniz P: Antioxidant properties,
radical scavenging activity and biomolecule protection capacity of
flavonoid naringenin and its glycoside naringin: a comparative
study. J Sci Food Agric. 90:1238–1244. 2010. View Article : Google Scholar
|
|
20
|
Jagetia GC and Reddy TK: Alleviation of
iron induced oxidative stress by the grape fruit flavanone naringin
in vitro. Chem Biol Interact. 190:121–128. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Camargo CA, Gomes-Marcondes MC, Wutzki NC
and Aoyama H: Naringin inhibits tumor growth and reduces
interleukin-6 and tumor necrosis factor alpha levels in rats with
Walker 256 carcinosarcoma. Anticancer Res. 32:129–133.
2012.PubMed/NCBI
|
|
22
|
Celiz G, Daz M and Audisio MC:
Antibacterial activity of naringin derivatives against pathogenic
strains. J Appl Microbiol. 111:731–738. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Carino-Cortes R, Alvarez-Gonzalez I,
Martino-Roaro L and Madrigal-Bujaidar E: Effect of naringin on the
DNA damage induced by daunorubicin in mouse hepatocytes and
cardiocytes. Biol Pharm Bull. 33:697–701. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gao S, Li P, Yang H, Fang S and Su W:
Antitussive effect of naringin on experimentally induced cough in
Guinea pigs. Planta Med. 77:16–21. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu M, Zou W, Yang C, Peng W and Su W:
Metabolism and excretion studies of oral administered naringin, a
putative antitussive, in rats and dogs. Biopharm Drug Dispos.
33:123–134. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gopinath K, Prakash D and Sudhandiran G:
Neuroprotective effect of naringin, a dietary flavonoid against
3-nitropropionic acid-induced neuronal apoptosis. Neurochem Int.
59:1066–1073. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gopinath K and Sudhandiran G: Naringin
modulates oxidative stress and inflammation in 3-nitropropionic
acid-induced neurodegeneration through the activation of nuclear
factor-erythroid 2-related factor-2 signalling pathway.
Neuroscience. 227:134–143. 2012. View Article : Google Scholar
|
|
28
|
Kandhare AD, Raygude KS, Ghosh P, Ghule AE
and Bodhankar SL: Neuroprotective effect of naringin by modulation
of endogenous biomarkers in streptozotocin induced painful diabetic
neuropathy. Fitoterapia. 83:650–659. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ikemura M, Sasaki Y, Giddings JC and
Yamamoto J: Preventive effects of hesperidin, glucosyl hesperidin
and naringin on hypertension and cerebral thrombosis in
stroke-prone spontaneously hypertensive rats. Phytother Res.
26:1272–1277. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chanet A, Milenkovic D, Deval C, et al:
Naringin, the major grapefruit flavonoid, specifically affects
atherosclerosis development in diet-induced hypercholesterolemia in
mice. J Nutr Biochem. 23:469–477. 2012. View Article : Google Scholar
|
|
31
|
Kanno S, Shouji A, Asou K and Ishikawa M:
Effects of naringin on hydrogen peroxide-induced cytotoxicity and
apoptosis in P388 cells. J Pharmacol Sci. 92:166–170. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim HJ, Song JY, Park HJ, Park HK, Yun DH
and Chung JH: Naringin protects against rotenone-induced apoptosis
in human neuroblastoma SH-SY5Y cells. Korean J Physiol Pharmacol.
13:281–285. 2009. View Article : Google Scholar
|
|
33
|
Mahmoud AM, Ashour MB, Abdel-Moneim A and
Ahmed OM: Hesperidin and naringin attenuate hyperglycemia-mediated
oxidative stress and proinflammatory cytokine production in high
fat fed/streptozotocin-induced type 2 diabetic rats. J Diabetes
Complications. 26:483–490. 2012. View Article : Google Scholar
|
|
34
|
Ramesh E and Alshatwi AA: Naringin induces
death receptor and mitochondria-mediated apoptosis in human
cervical cancer (SiHa) cells. Food Chem Toxicol. 51:97–105. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nie YC, Wu H, Li PB, et al:
Anti-inflammatory effects of naringin in chronic pulmonary
neutrophilic inflammation in cigarette smoke-exposed rats. J Med
Food. 15:894–900. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xulu S and Oroma Owira PM: Naringin
ameliorates atherogenic dyslipidemia but not hyperglycemia in rats
with type 1 diabetes. J Cardiovasc Pharmacol. 59:133–141. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rajadurai M and Prince PS: Naringin
ameliorates mitochondrial lipid peroxides, antioxidants and lipids
in isoproterenol-induced myocardial infarction in Wistar rats.
Phytother Res. 23:358–362. 2009. View
Article : Google Scholar
|
|
38
|
Punithavathi VR, Anuthama R and Prince PS:
Combined treatment with naringin and vitamin C ameliorates
streptozotocin-induced diabetes in male Wistar rats. J Appl
Toxicol. 28:806–813. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shore GC and Nguyen M: Bcl-2 proteins and
apoptosis: choose your partner. Cell. 135:1004–1006. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cai L, Li W, Wang G, Guo L, Jiang Y and
Kang YJ: Hyperglycemia-induced apoptosis in mouse myocardium:
mitochondrial cytochrome C-mediated caspase-3 activation pathway.
Diabetes. 51:1938–1948. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fiordaliso F, Leri A, Cesselli D, et al:
Hyperglycemia activates p53 and p53-regulated genes leading to
myocyte cell death. Diabetes. 50:2363–2375. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Miyashita T and Reed JC: Tumor suppressor
p53 is a direct transcriptional activator of the human bax gene.
Cell. 80:293–299. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhu W, Soonpaa MH, Chen H, et al: Acute
doxorubicin cardiotoxicity is associated with p53-induced
inhibition of the mammalian target of rapamycin pathway.
Circulation. 119:99–106. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Childs AC, Phaneuf SL, Dirks AJ, Phillips
T and Leeuwenburgh C: Doxorubicin treatment in vivo causes
cytochrome C release and cardiomyocyte apoptosis, as well as
increased mitochondrial efficiency, superoxide dismutase activity,
and Bcl-2:Bax ratio. Cancer Res. 62:4592–4598. 2002.
|
|
45
|
Younce CW, Burmeister MA and Ayala JE:
Exendin-4 attenuates high glucose-induced cardiomyocyte apoptosis
via inhibition of endoplasmic reticulum stress and activation of
SERCA2a. Am J Physiol Cell Physiol. 304:C508–C518. 2013. View Article : Google Scholar
|
|
46
|
Kuo WW, Wang WJ, Tsai CY, Way CL, Hsu HH
and Chen LM: Diallyl trisufide (DATS) suppresses high
glucose-induced cardiomyocyte apoptosis by inhibiting JNK/NFkappaB
signaling via attenuating ROS generation. Int J Cardiol. View Article : Google Scholar : 2012.PubMed/NCBI
|
|
47
|
Yu XY, Geng YJ, Liang JL, et al: High
levels of glucose induce apoptosis in cardiomyocyte via epigenetic
regulation of the insulin-like growth factor receptor. Exp Cell
Res. 316:2903–2909. 2010. View Article : Google Scholar : PubMed/NCBI
|