Introduction
Chronic inflammation is associated with the
pathogenesis of several human diseases, including cancer and
neurodegenerative diseases (1,2).
Nuclear factor-κB (NF-κB) is the most well-known regulator of
inflammatory signaling (3), and
activated NF-κB is commonly observed in inflammatory lesions during
pathogenesis (4). Evidence to
date demonstrates that the functions of NF-κB are modulated by
diverse post-transcriptional modifications (5). Among these modifications,
acetylation has been shown to enhance the nuclear localization of
NF-κB and lead to the transcription of NF-κB target genes (6,7).
NF-κB is mainly acetylated by p300/CBP and p300/CBP-associated
factor (PCAF) (8). p300/CBP
acetylates multiple lysine residues of NF-κB, including Lys-122,
-123, -218, -221 and -310, activating its transcriptional activity
(7). On the other hand, PCAF
specifically acetylates only Lys-122 of NF-κB (9). The difference between acetylation by
PCAF and p300 on the regulation of NF-κB function, however, is
largely unknown. Conversely, the blocking of NF-κB acetylation by
histone acetyl transferase (HAT) inhibitors diminishes the nuclear
retention and transcriptional activity of NF-κB (10–12). Thus, the pharmacological
inhibition of HAT activity may be a useful method for the treatment
of chronic inflammation.
Neuroinflammation is a hallmark of neurodegenerative
diseases that is linked to glial cell activation (13–15). The abnormal activation of
microglia promotes neuronal injury through the release of
pro-inflammatory and cytotoxic factors, including tumor necrosis
factor-α (TNF-α), interleukin (IL)-1β, inducible nitric oxide
synthase (iNOS), cyclooxygenase-2 (COX-2), nitric oxide and
reactive oxygen species that contribute to localized and more
widespread central nervous system injuries (16–18). NF-κB is also involved in
neuroinflammation and neuronal cell death induced by β-amyloid (Aβ)
(19–21). Consistent with this, the
immunoreactivity of the RelA (p65) subunit of NF-κB in the brains
of patients with Alzheimer’s disease is stronger in the neurons,
astrocytes and microglial cells surrounding amyloid plaques
(22). In a recent study, we
reported the inhibitory effects of gallic acid, a HAT inhibitor, on
NF-κB activity and the activation of microglial inflammation
(10). In addition, we have
previously shown that gallic acid inhibits multiple HAT enzymes,
including p300 and PCAF (23). Of
note, it has recently been demonstrated that PCAF knockout mice
develop resistance to amyloid toxicity and have altered memory
capacities, suggesting the importance of the PCAF enzyme in the
development of neurodegenerative diseases (24,25). However, the detailed mechanisms
through which PCAF promotes Aβ-induced microglial inflammation and
neurotoxicity remain unclear.
In this study, we used computer-based molecular
docking simulations to screen for PCAF-specific inhibitors. We
demonstrate that the inhibition of PCAF abrogates
lipopolysaccharide (LPS)-induced NF-κB activation and the
acetylation of NF-κB at Lys-122. Finally, we used co-culture
analysis, demonstrating that a PCAF inhibitor efficiently prevented
neuronal cell death caused by Aβ-induced neurotoxicity.
Collectively, our results demonstrate that the selective inhibition
of NF-κB acetylation by a PCAF inhibitor shows promise for the
treatment of neurodegenerative diseases.
Materials and methods
Molecular docking analysis and
chemicals
Structure-based artificial screening with
computer-based molecular docking simulations was conducted by
InnoPharmaScreen Inc. (Asan, Korea). The X-ray crystal structure of
the HAT domain of human PCAF bound to coenzyme A (CoA; Protein Data
Bank ID: 1CM0) was used for the initial structure of the target
protein. CoA was removed and atomic hydrogens were added to produce
a complete molecular model of the target protein for use in the
molecular docking simulations. Docking simulations were performed
between the target model and the ChemBridge chemical library that
consists of 450,000 unique compounds. Binding efficiency was based
on the binding force between the target molecule and the test
compound as calculated by the Gold 4.0.1 software package
(Cambridge Crystallographic Data Centre, Cambridge, UK). The top 10
docking positions for each compound were calculated, and the
library search efficiency was 100%.
Cell culture, reagents and
antibodies
Murine BV-2 cells and Neuro-2A cells were obtained
from the American Type Culture Collection (ATCC; Manassas, VA, USA;
CRL no. 2270). Fetal bovine serum (FBS), trypsin-EDTA, penicillin
and streptomycin were purchased from Gibco-BRL (Gaithersburg, MD,
USA). 3-(4,5-Dimethylthiazol-2-ly)-2,5-diphenyl tetrazolium bromide
(MTT) was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Aβ1–42 was purchased from Bachem (Bubendorf,
Switzerland). Other chemicals used were purchased from
Sigma-Aldrich. BV-2 cells were cultured in Dulbecco’s modified
Eagle’s medium (Gibco-BRL) containing 5% heat-inactivated
endotoxin-free FBS, 2 mM glutamine, 100 μg/ml streptomycin and 100
U/ml penicillin in a humidified 5% CO2 atmosphere at
37°C. Neuro-2A cells were cultured in modified Eagle’s medium
(Gibco-BRL) containing 10% heat-inactivated endotoxin-free FBS, 2
mM glutamine, 100 μg/ml streptomycin, and 100 U/ml penicillin in a
humidified 5% CO2 atmosphere at 37°C. Aβ peptides were
dissolved in phosphate-buffered saline (PBS) and pre-incubated at
37°C for 5 days to allow for fibril formation. The peptides were
stored at -20°C until use. Compound C-11 was dissolved in dimethyl
sulfoxide (DMSO) and later diluted with distilled water (the final
concentration of DMSO was <0.5%). For the co-culture
experiments, BV-2 cells were treated with various concentrations of
C-11 (0.1–1.0 μM final concentration) for 12 h prior to stimulation
with 5 μM aggregated Aβ1–42 (the Aβ + C-11 group), 5 μM
non-aggregated Aβ1–42, or medium only (control) for 24
h. Conditioned medium (CM) from the BV-2 cells and primary
microglial cells was collected, centrifuged and transferred to the
Neuro-2A cells for a further 24 h. CM from the medium-only treated
cells was used as the control. Following incubation, cell viability
was measured by MTT assay and western blot analysis was
performed.
HAT, histone deacetylase (HDAC), histone
methyltransferase (HMT) and sirtuin 1 (SIRT1) activity assays
HeLa cell nuclear extract was prepared as previously
described (26). HAT and HDAC
activity assays were performed with nuclear extracts using
commercially available kits according to the manufacturer’s
instructions (BioVision, Inc., Milpitas, CA, USA). For in
vitro HAT activity assays, recombinant HAT proteins were
incubated with HAT assay buffer [50 mM HEPES pH 8.0, 10% glycerol,
1 mM DTT, 1 mM phenylmethanesulfonyl fluoride (PMSF), 10 mM sodium
butyrate and 1 μl [3H]-acetyl-CoA], 5 μg histone H4 tail
peptides, and/or indicated concentration of inhibitors at 30°C for
1 h. Samples were separated on a 15% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel and
analyzed by autoradiography. HMT activity assays were performed
using the assay buffer and core histones from the HMT Assay Reagent
kit (Upstate Biotechnology, Inc., Lake Placid, NY, USA) according
to the manufacturer’s instructions. Core histones were incubated
for 1 h at 30°C in methyltransferase buffer (final concentration 50
mM Tris pH 8.0, 1 mM PMSF, and 0.5 mM DTT), 1 μl (0.55 μCi)
S-adenosyl-L-[methyl-3H]-methionine (Amersham Pharmacia
Biotech, Inc., Piscataway, NJ, USA) and 10 μg/ml HeLa nuclear
extract. The total volume of the reaction mixture was 30 μl. The
reaction was spotted on P-81 paper for scintillation counting. The
P-81 paper was washed 3 times with 10% trichloroacetic acid
(Upstate Biotechnology, Inc.) for 15 min, washed with 95% ethanol
for 5 min at room temperature and allowed to dry. Dry P-81 papers
were counted with a multi-purpose LS 6500 scintillation counter
(Beckman Coulter, Indianapolis, IN, USA). SIRT1 activity was
assayed using a SIRT1/Sir2 Deacetylase Fluorometric assay kit
(CycLex Co., Ltd., Nagano, Japan) according to the manufacturer’s
instructions.
Lentiviral shRNAs
For silencing PCAF expression, 2 pairs of
oligonucleotides that encoded PCAF-specific shRNA were purchased
from MISSION shRNA (Sigma-Aldrich). Lentiviral particles were
prepared using pLKO.1-PURO PCAF shRNA via a 3 plasmid
co-transfection according to the instructions of the manufacturer
(Invitrogen, Carlsbad, CA, USA). The BV-2 cells were transfected
with the lentivirus. After 3 days of incubation, the lentivirus
from the culture medium was collected and concentrated in a
Centricon-plus-20 centrifugal filter device (Millipore, Billerica,
MA, USA). Lentivirus PURO shRNA was generated as the control.
Cell viability assay (MTT assay)
Cell viability was measured to determine the
cytotoxicity of the Aβ peptides on neuronal cells. Neuro-2A cells
were seeded at 1×104 to 1×105 cells in a
96-well plate. Following 12 h of incubation, the medium was
replaced with CM from the BV-2 cell cultures treated with or
without Aβ and different concentrations of C-11. After 24 h, the CM
was replaced with non-CM, 15 μl MTT solution (2 mg/ml final
concentration) was added for 90 min at 37°C, and the absorbance was
recorded at 570 nm. A reference was recorded at 630 nm using a
microplate reader (Model 550; Bio-Rad, Hercules, CA, USA).
Western blot analysis
The treated cells were washed with cold PBS, scraped
off the culture dishes and harvested. The cells were incubated for
20 min in lysis buffer containing 0.5% Triton X-100, 20 mM HEPES
(pH 7.4), 150 mM NaCl, 2 mM DTT and 1 mM PMSF. The lysates were
centrifuged at 20,000 × g for 10 min at 4°C. The protein
concentrations in the clarified lysates were determined with the
Bradford assay using bovine serum albumin as a reference. Total
cell lysate proteins were separated on 8 or 12% SDS-PAGE gels and
transferred onto nitrocellulose membranes. The membranes were
blocked by incubating for 12 h in 5% (w/v) non-fat Difco™ skim milk
blocking buffer. The blocked membranes were incubated overnight at
4°C with primary antibodies that recognize iNOS (1:1,000 dilution),
COX-2 (1:1,000), IL-1β (1:500), NF-κB (p65; 1:500) or β-actin
(1:5,000). The antibody against acetyl-NF-κB (K122) was raised
against the synthetic peptide, CIHSFQNLGIQCVACKKRDLE, by
GenScript (Piscataway, NJ, USA). After washing 3 times with PBS and
0.1% Tween-20, the membranes were incubated with secondary
horseradish peroxidase-conjugated antibody (1:1,000) for 1 h. The
bands were detected using the enhanced chemiluminescence system
(Amersham Pharmacia Biotech, Inc.) according to the manufacturer’s
instructions.
RNA extraction and real-time PCR
analysis
Total RNA from the Neuro-2A, BV-2 and primary
microglial cells was extracted using TRIzol reagent (Invitrogen)
according to the manufacturer’s instructions. The mRNA levels of
iNOS, COX-2 and IL-1β were determined by real-time PCR (ABI PRISM
500 Sequence Detection System, Applied Biosystems, San Jose, CA,
USA). cDNA amplification was performed in duplicate in 20-μl
reaction mixtures containing 2X SYBR-Green Master Mix (Roche,
Indianapolis, IN, USA) and 10 pM forward and reverse primers. The
initial denaturation step was 5 min at 95°C followed by 40
amplification cycles of 30 sec at 95°C, 30 sec at 58°C and 30 sec
at 72°C with a final 10 min extension at 72°C. The results were
analyzed using ABI sequence detector software version 2.3. Relative
mRNA expression of the target genes was calculated after
normalizing to GAPDH expression and was expressed as the fold
induction. The primers used for amplification were as follows:
human IL-1β gene forward, 5′-GTTGACGGACCCCAAAAGAT-3′ and reverse,
5′-AAGG TCCCACGGGAAAGACAC-3′; human COX-2 gene forward,
5′-GAGTGGGAGGCACTTGCATT-3′ and reverse, 5′-TGGA
GGCGAAGTGGGTTTTA-3′; human iNOS gene forward,
5′-TCTTGGAGCGAGTTGTGGAT-3′ and reverse, 5′-GGGT
GGTAATGTCCAGGAAGT-3′; and human GAPDH gene forward,
5′-GTGTTCCTACCCCCAATGTGT-3′ and reverse,
5′-AGGAGACAACCTGGTCCTCAGT-3′. All reactions were performed in
triplicate. Relative expression levels and standard deviations
(SDs) were calculated using the comparative value method.
Results
Structure-based artificial screening of
PCAF-specific inhibitors
To develop PCAF-specific inhibitors, we performed
structure-based artificial screening using computer-based molecular
docking simulations (Fig. 1A). We
initially selected 50 compounds with a binding force of at least 70
after the molecular docking simulation (Table I). Among these, compound number 11
(C-11) displayed the most significant inhibitory effect against the
PCAF enzyme (Fig. 1B).
Furthermore, C-11 selectively inhibited PCAF, but not p300 and GCN5
(Fig. 1C). Thus, we selected C-11
as the putative PCAF inhibitor for our experiments.
 | Table IGold fitness score for each
compound. |
Table I
Gold fitness score for each
compound.
| No. | Score | No. | Score | No. | Score | No. | Score |
|---|
| C-1 | 85.55 | C-26 | 73.43 | C-51 | 72.12 | C-76 | 70.86 |
| C-2 | 84.11 | C-27 | 73.42 | C-52 | 71.94 | C-77 | 70.81 |
| C-3 | 81.48 | C-28 | 73.33 | C-53 | 71.89 | C-78 | 70.77 |
| C-4 | 81.00 | C-29 | 73.29 | C-54 | 71.87 | C-79 | 70.65 |
| C-5 | 80.75 | C-30 | 73.18 | C-55 | 71.76 | C-80 | 70.58 |
| C-6 | 80.58 | C-31 | 73.14 | C-56 | 71.69 | C-81 | 70.53 |
| C-7 | 80.21 | C-32 | 73.10 | C-57 | 71.59 | C-82 | 70.42 |
| C-8 | 77.79 | C-33 | 73.05 | C-58 | 71.51 | C-83 | 70.40 |
| C-9 | 77.56 | C-34 | 72.97 | C-59 | 71.51 | C-84 | 70.39 |
| C-10 | 77.45 | C-35 | 72.95 | C-60 | 71.38 | C-85 | 70.32 |
| C-11 | 76.83 | C-36 | 72.92 | C-61 | 71.37 | C-86 | 70.28 |
| C-12 | 76.35 | C-37 | 72.90 | C-62 | 71.36 | C-87 | 70.28 |
| C-13 | 76.27 | C-38 | 72.84 | C-63 | 71.36 | C-88 | 70.20 |
| C-14 | 75.87 | C-39 | 72.83 | C-64 | 71.36 | C-89 | 70.20 |
| C-15 | 75.42 | C-40 | 72.80 | C-65 | 71.33 | C-90 | 70.19 |
| C-16 | 74.93 | C-41 | 72.64 | C-66 | 71.30 | C-91 | 70.16 |
| C-17 | 74.73 | C-42 | 72.55 | C-67 | 71.28 | C-92 | 70.15 |
| C-18 | 74.33 | C-43 | 72.46 | C-68 | 71.15 | C-93 | 70.07 |
| C-19 | 74.20 | C-44 | 72.42 | C-69 | 71.09 | C-94 | 70.04 |
| C-20 | 74.10 | C-45 | 72.37 | C-70 | 71.06 | C-95 | 69.98 |
| C-21 | 74.04 | C-46 | 72.36 | C-71 | 71.03 | C-96 | 69.94 |
| C-22 | 73.91 | C-47 | 72.30 | C-72 | 71.00 | C-97 | 69.94 |
| C-23 | 73.71 | C-48 | 72.21 | C-73 | 71.00 | C-98 | 69.93 |
| C-24 | 73.62 | C-49 | 72.19 | C-74 | 70.98 | C-99 | 69.92 |
| C-25 | 73.47 | C-50 | 72.16 | C-75 | 70.86 | C-100 | 69.84 |
To examine the enzyme specificity of C-11, we
examined whether C-11 inhibits other epigenetic enzymes, including
HDAC, HMT and SIRT1. As shown in Fig.
2A, C-11 failed to inhibit the enzyme activities of HDAC and
SIRT1. Furthermore, C-11 had no effect on HMT activity (Fig. 2B). These results demonstrate that
C-11 specifically inhibits PCAF acetyltransferase activity but not
the activity of other epigenetic enzymes. As several HAT inhibitors
(HATi) have recently been shown to possess anti-PCAF activity
(12,23,27), we compared the relative anti-PCAF
activity of C-11 with other HATi. As shown in Fig. 2C, C-11 had the highest anti-PCAF
activity among the reported HATi with a half-maximal inhibitory
concentration of approximately 0.25 μM (Fig. 2C and D).
Inhibition of PCAF suppresses Aβ-induced
NF-κB acetylation at Lys-122
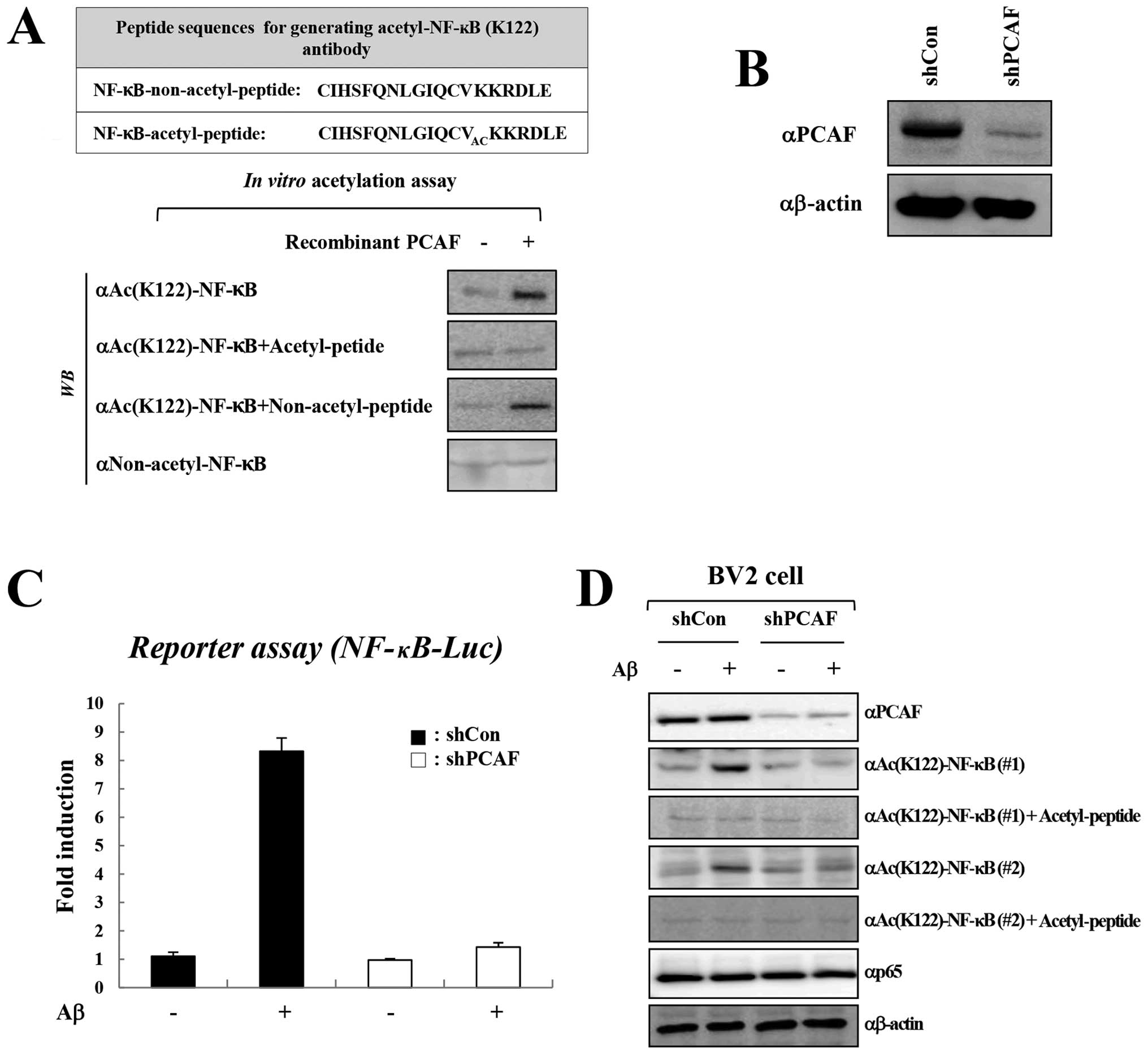
Since PCAF was found to activate NF-κB-mediated
transcription in response to cytokines (6), we first examined whether the
inhibition of PCAF by C-11 suppresses NF-κB activity by inhibiting
the acetylation of NF-κB. Since PCAF is known to acetylate NF-κB at
Lys-122 (6), we first generated
an acetyl-specific NF-κB antibody that recognizes acetylated NF-κB
at Lys-122. Polyclonal antibodies were generated against the NF-κB
acetyl-peptide,
109CIHSFQNLGIQCVACKKRDLE127, and
the antisera were examined by western blot analysis following in
vitro acetylation reactions with recombinant PCAF enzyme and
GST-p65 substrate (Fig. 3A, upper
panel). A peptide competition assay clearly demonstrated the
specificity of the NF-κB Lys(Ac)-122 antibody, as the complete
blocking of NF-κB acetylation was observed by the antibody raised
against the acetyl-peptide, but not by an antibody raised against a
non-acetyl-peptide (Fig. 3A,
lower panel). In addition, non-acetyl-NF-κB antibody failed to
detect the acetylation that resulted from an in vitro HAT
reaction and thus confirmed the specificity of the acetyl-specific
NF-κB antibody for the PCAF-mediated acetylation of NF-κB at
Lys-122 (Fig. 3A, lower
panel).
We then examined whether Aβ treatment induces NF-κB
activation through the acetylation of NF-κB at Lys-122. Aβ
treatment efficiently induced the activation of NF-κB, and this was
inhibited by the knockdown of PCAF (Fig. 3B and C). Importantly, Aβ treatment
also increased the acetylation of NF-κB at Lys-122, which was
verified by 2 antibodies that recognize acetylated NF-κB at Lys-122
(Fig. 3D). The acetylation of
NF-κB at Lys-122 was specifically mediated by PCAF, as shRNA
against PCAF, but not control shRNA, abrogated the Aβ-induced
acetylation of NF-κB. Collectively, these data suggest that PCAF
mediates the Aβ-induced activation of NF-κB through the acetylation
of NF-κB at Lys-122.
Pharmacological inhibition of PCAF
abrogates the Aβ-induced activation of NF-κB
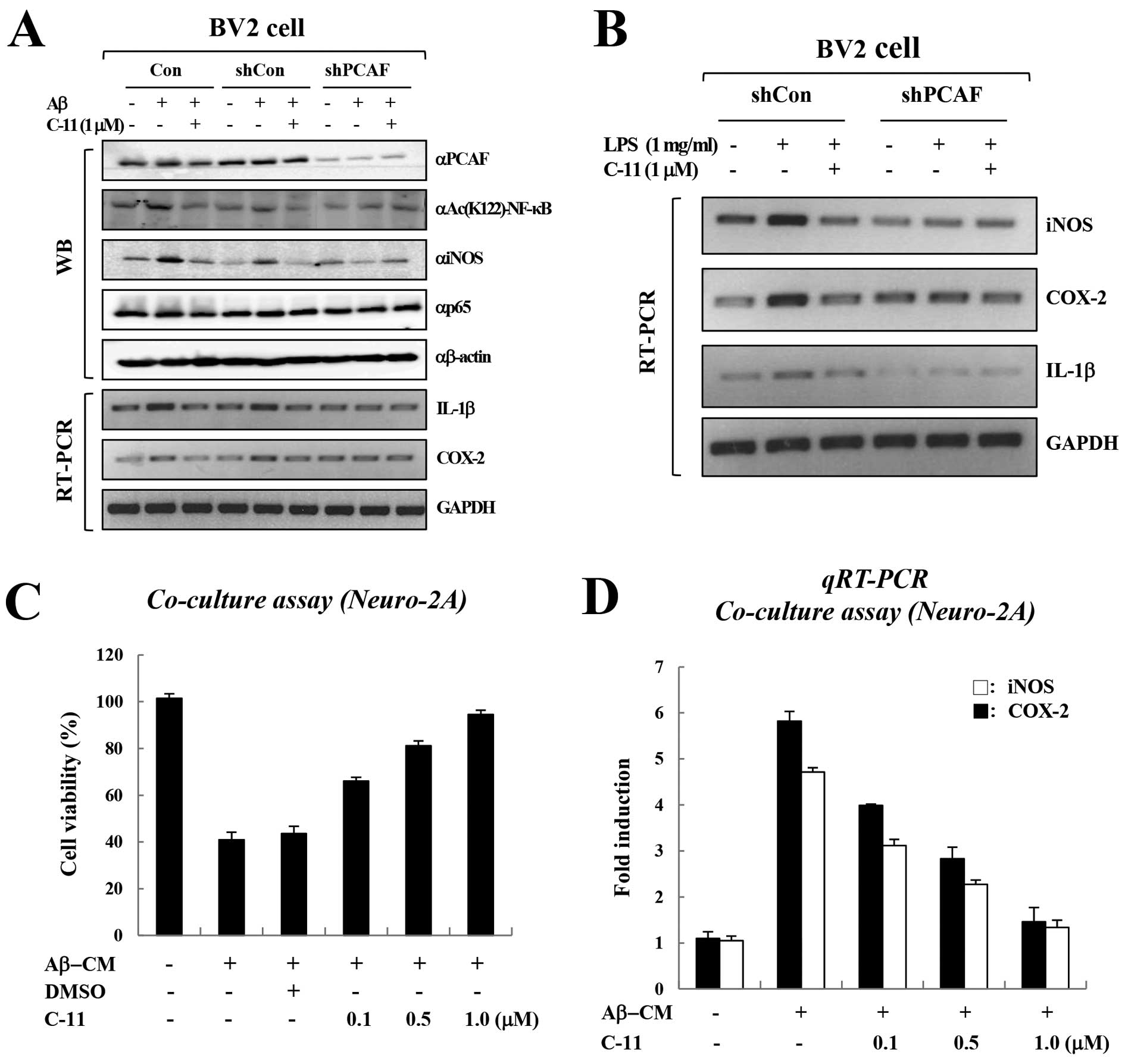
We then investigated the inhibitory effects of PCAF
on NF-κB activity and cytokine production. We first assessed the
effect of knocking down PCAF on cytokine production by BV-2 cells.
Consistent the results from our previous study (10), Aβ treatment significantly
increased the expression of cytokine genes in BV-2 cells (Fig. 4A). Of note, treatment with shRNA
against PCAF markedly diminished the Aβ-induced cytokine production
in BV-2 cells when compared with control shRNA (Fig. 4A). Importantly, PCAF inhibition by
C-11 markedly reduced the Aβ-induced cytokine production in BV-2
cells. Furthermore, we also observed similar results with LPS
treatment (Fig. 4B). These data
collectively demonstrate that the inhibition of PCAF abolishes
NF-κB-mediated cytokine production by blocking the acetylation of
NF-κB at Lys-122.
Inhibition of PCAF prevents Aβ-induced
neuronal cell death
Since PCAF inhibition suppresses the expression of
pro-inflammatory cytokines, we examined the neuroprotective effects
of PCAF inhibition on BV-2 microglial-mediated Aβ neurotoxicity.
Consistent with the results from our previous study showing the
dose-dependent cytotoxicity of gallic acid in the same system
(10), treatment with
Aβ-conditioned medium (CM) from BV-2 microglial cells reduced the
viability of Neuro-2A cells, and C-11 reversed the Aβ-CM-mediated
neuronal cell death in a dose-dependent manner (Fig. 4C). Real-time PCR demonstrated that
C-11 consistently suppressed the Aβ-CM-induced cytokine production
in Neuro-2A cells (Fig. 4D).
Collectively, these data demonstrate that the inhibition of PCAF by
C-11 inhibits NF-κB acetylation, which in turn suppresses
Aβ-induced neuroinflammation and Aβ-mediated neurotoxicity.
Discussion
In this study, we demonstrated a role for PCAF in
microglial inflammation through acetylation-dependent NF-κB
activation, and provide a rationale for the use of PCAF as a
promising therapeutic target for the treatment of Alzheimer’s
disease. Previous studies have shown that PCAF acetylates NF-κB at
Lys-122 in response to cytokine signaling and that this in turn,
leads to increased NF-κB activity (8). However, due to the unavailability of
an acetyl-specific NF-κB antibody, it is still unclear as to
whether PCAF indeed acetylates NF-κB at Lys-122. In the current
study, we utilized an acetyl (Lys-122)-specific NF-κB antibody,
clearly demonstrating that PCAF mediates the Aβ-induced activation
of NF-κB through the acetylation of NF-κB at Lys-122. Furthermore,
we did not observe any change in the acetylation of NF-κB at
Lys-122 after knocking down p300 (data not shown). Thus, our study
confirms the specific acetylation of NF-κB at Lys-122 by PCAF.
Accumulating evidence suggests that microglial
inflammation by cytokines aggravates Aβ-induced neurotoxicity
(28,29). Furthermore, we have previously
demonstrated that the inhibition of NF-κB acetylation with a HAT
inhibitor, such as gallate significantly reduces Aβ-mediated
neuronal cell death through the suppression of inflammatory
signaling (10). The
pharmacological inhibition of NF-κB acetylation may, therefore,
prove useful as a therapeutic intervention in Alzheimer’s disease.
In this context, a previous studies have demonstrated that PCAF
knockout mice are insensitive to Aβ neurotoxicity (24,25), suggesting a pivotal role for PCAF
in the regulation of Aβ-induced neurotoxicity and the memory
capacity of the brain. In support of these findings, to our
knowledge, in this study, we demonstrate for the first time that
the pharmacological inhibition of PCAF prevents Aβ-induced cytokine
production and toxicity in neuronal cells. The most promising
compound selected by our molecular docking analysis was compound
C-11, a PCAF-specific inhibitor. C-11 treatment specifically
inhibited the cytokine-induced activation of NF-κB by preventing
the acetylation of NF-κB at Lys-122. Importantly, C-11 had no
effect on the activity of any of the other epigenetic enzymes
examined. More importantly, the knockdown of PCAF with shRNA had a
similar effect on cytokine production and the viability of neuronal
cells as did C-11 treatment.
In conclusion, our data suggest that the selective
inhibition of NF-κB acetylation by PCAF inhibition is a possible
therapeutic approach for alleviating the inflammatory progression
of Alzheimer’s disease.
Acknowledgements
This study was supported by a grant from the Korea
Health Care Technology R&D Project, Ministry for Health,
Welfare and Family Affairs, Republic of Korea (no. A092039).
References
|
1
|
Ferguson LR and Laing WA: Chronic
inflammation, mutation and human disease. Mutat Res. 690:1–2. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dinarello CA: Inflammation in human
disease: anticytokine therapy. Biol Blood Marrow Transplant.
15(Suppl 1): 134–136. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hayden MS and Ghosh S: Signaling to
NF-kappaB. Genes Dev. 18:2195–2224. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Aradhya S and Nelson DL: NF-kappaB
signaling and human disease. Curr Opin Genet Dev. 11:300–306. 2001.
View Article : Google Scholar
|
|
5
|
Campbell KJ and Perkins ND:
Post-translational modification of RelA (p65) NF-kappaB. Biochem
Soc Trans. 32:1087–1089. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen LF, Mu Y and Greene WC: Acetylation
of RelA at discrete sites regulates distinct nuclear functions of
NF-kappaB. EMBO J. 21:6539–6548. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen LF, Fischle W, Verdin E and Greene
WC: Duration of nuclear NF-kappaB action regulated by reversible
acetylation. Science. 293:1653–1657. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen LF and Greene WC: Shaping the nuclear
action of NF-kappaB. Nat Rev Mol Cell Biol. 5:392–401. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Greene WC and Chen LF: Regulation of
NF-kappaB action by reversible acetylation. Novartis Found Symp.
259:208–217. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim MJ, Seong AR, Yoo JY, et al: Gallic
acid, a histone acetyltransferase inhibitor, suppresses
beta-amyloid neurotoxicity by inhibiting microglial-mediated
neuroinflammation. Mol Nutr Food Res. 55:1798–1808. 2011.
View Article : Google Scholar
|
|
11
|
Seong AR, Yoo JY, Choi K, et al:
Delphinidin, a specific inhibitor of histone acetyltransferase,
suppresses inflammatory signaling via prevention of NF-κB
acetylation in fibroblast-like synoviocyte MH7A cells. Biochem
Biophys Res Commun. 410:581–586. 2011.PubMed/NCBI
|
|
12
|
Choi KC, Jung MG, Lee YH, et al:
Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor,
inhibits EBV-induced B lymphocyte transformation via suppression of
RelA acetylation. Cancer Res. 69:583–592. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Holmes C: Systemic inflammation and
Alzheimer’s Disease. Neuropathol Appl Neurobiol. 39:51–68.
2012.
|
|
14
|
Cameron B and Landreth GE: Inflammation,
microglia, and Alzheimer’s disease. Neurobiol Dis. 37:503–509.
2010.
|
|
15
|
Jones RW: Inflammation and Alzheimer’s
disease. Lancet. 358:436–437. 2001.
|
|
16
|
Ekdahl CT, Kokaia Z and Lindvall O: Brain
inflammation and adult neurogenesis: the dual role of microglia.
Neuroscience. 158:1021–1029. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim YS and Joh TH: Microglia, major player
in the brain inflammation: their roles in the pathogenesis of
Parkinson’s disease. Exp Mol Med. 38:333–347. 2006.PubMed/NCBI
|
|
18
|
Min KJ, Yang MS, Kim SU, Jou I and Joe EH:
Astrocytes induce hemeoxygenase-1 expression in microglia: a
feasible mechanism for preventing excessive brain inflammation. J
Neurosci. 26:1880–1887. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jayasooriya RG, Kang CH, Seo MJ, Choi YH,
Jeong YK and Kim GY: Exopolysaccharide of Laetiporus
sulphureus var miniatus downregulates LPS-induced
production of NO, PGE(2), and TNF-alpha in BV2 microglia cells via
suppression of the NF-kappaB pathway. Food Chem Toxicol.
49:2758–2764. 2011.PubMed/NCBI
|
|
20
|
Ha SK, Moon E and Kim SY: Chrysin
suppresses LPS-stimulated proinflammatory responses by blocking
NF-kappaB and JNK activations in microglia cells. Neurosci Lett.
485:143–147. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wilms H, Rosenstiel P, Sievers J, Deuschl
G, Zecca L and Lucius R: Activation of microglia by human
neuromelanin is NF-kappaB dependent and involves p38
mitogen-activated protein kinase: implications for Parkinson’s
disease. FASEB J. 17:500–502. 2003.PubMed/NCBI
|
|
22
|
Kaltschmidt B, Uherek M, Volk B, Baeuerle
PA and Kaltschmidt C: Transcription factor NF-kappaB is activated
in primary neurons by amyloid beta peptides and in neurons
surrounding early plaques from patients with Alzheimer disease.
Proc Natl Acad Sci USA. 94:2642–2647. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Choi KC, Lee YH, Jung MG, et al: Gallic
acid suppresses lipopolysaccharide-induced nuclear factor-kappaB
signaling by preventing RelA acetylation in A549 lung cancer cells.
Mol Cancer Res. 7:2011–2021. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Duclot F, Jacquet C, Gongora C and Maurice
T: Alteration of working memory but not in anxiety or stress
response in p300/CBP associated factor (PCAF) histone acetylase
knockout mice bred on a C57BL/6 background. Neurosci Lett.
475:179–183. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Maurice T, Duclot F, Meunier J, et al:
Altered memory capacities and response to stress in
p300/CBP-associated factor (PCAF) histone acetylase knockout mice.
Neuropsychopharmacology. 33:1584–1602. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yoon HG, Chan DW, Huang ZQ, et al:
Purification and functional characterization of the human N-CoR
complex: the roles of HDAC3, TBL1 and TBLR1. EMBO J. 22:1336–1346.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kusio-Kobialka M, Dudka-Ruszkowska W,
Ghizzoni M, et al: Inhibition of PCAF by anacardic acid derivative
leads to apoptosis and breaks resistance to DNA damage in
BCR-ABL-expressing cells. Anticancer Agents Med Chem. Nov
13–2012.(Epub ahead of print).
|
|
28
|
Harry GJ and Kraft AD: Neuroinflammation
and microglia: considerations and approaches for neurotoxicity
assessment. Expert Opin Drug Metab Toxicol. 4:1265–1277. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Block ML, Zecca L and Hong JS:
Microglia-mediated neurotoxicity: uncovering the molecular
mechanisms. Nat Rev Neurosci. 8:57–69. 2007. View Article : Google Scholar : PubMed/NCBI
|