Introduction
Atrial fibrillation (AF), a supraventricular
tachyarrhythmia characteristic of the rapid and chaotic electrical
activity of the atria with consequent deterioration of atrial
mechanical function, is the most prevalent type of sustained
cardiac arrhythmia in humans, afflicting roughly one-third of all
hospitalized patients with miscellaneous heart rhythm disturbances
(1). The prevalence of AF is
approximately 1% in the general population and increases
drastically with advancing age, increasing from <1% in
individuals aged <60 years up to ~10% in those >80 years of
age (1). According to the report
from the Framingham Heart Study, the lifetime risk of developing AF
is approximately one in four among subjects older than 40 years
(2). In the United States, the
number of individuals affected by AF is currently estimated at 2.3
million and is projected to exceed 10 million by 2050 (3). This common cardiac dysrhythmia may
lead to poor quality of life, decreased exercise tolerance,
cognitive impairment, tachycardiomyopathy, thromboembolism,
congestive heart failure and even death (1). The mortality rate of patients with
AF is approximately twice that of age- and gender matched patients
with normal heart rhythm, which persists even after the adjustment
for pre-existing cardiovascular conditions (4). Patients with AF are at a 2- to
7-fold increased risk of ischemic stroke compared with those
without AF and 15 to 20% of all strokes are attributed to AF
(5). Notably, the risk of
thromboembolic stroke ascribed to AF also increases steeply with
increasing age, ranging from 1.5% for persons aged 50 to 59 years
to 23.5% for persons aged 80 to 89 years (5). Hence, AF has imposed a heavy
economic burden on patients and health care systems and the
socioeconomic burden is anticipated to increase drastically in the
future with life expectancies increasing (6). Despite the high prevalence and
important clinical significance, the molecular mechanisms
underlying AF remain poorly understood.
Traditionally, AF has been referred to as a
complication of diverse cardiac and systemic disorders, including
coronary heart disease, rheumatic heart disease, congenital heart
disease, pulmonary heart disease, cardiomyopathy, cardiothoracic
surgery, myocarditis, congestive heart failure, essential
hypertension, diabetes mellitus and hyperthyroidism (1). However, in 12 to 30% of all AF
patients and 20 to 45% of younger AF patients, AF occurs separately
in the absence of underlying diseases or precipitating factors, a
condition termed as lone or idiopathic AF, of which at least 15% of
cases have a positive family history, thus defined as familial AF
(1,7). A growing body of epidemiological
research has demonstrated the familial aggregation of AF and the
increased vulnerability to AF in the close relatives of patients
with AF, strongly suggesting that genetic risk factors play a
pivotal role in the pathogenesis of AF in a subset of patients
(8–14). In previous studies, using
genome-wide scanning with polymorphic microsatellite markers and
linkage analysis, AF susceptibility loci were mapped on human
chromosomes 10q22, 6q14-16, 11p15.5, 5p15, 10p11-q21 and 5p13,
where AF-causative mutations in two genes, potassium voltage-gated
channel, KQT-like subfamily, member 1 (KCNQ1) on chromosome
11p15.5 and nucleoporin 155 kDa (NUP155) on chromosome 5p13,
were identified and functionally characterized (15–21). Direct analyses of candidate genes
unveiled a high number of AF-related genes, including potassium
voltage-gated channel, Isk-related family, member 2 (KCNE2),
potassium voltage-gated channel, subfamily H (eag-related), member
2 (KCNH2), potassium inwardly-rectifying channel, subfamily J,
member 2 (KCNJ2), potassium voltage-gated channel, shaker-related
subfamily, member 5 (KCNA5), sodium channel, voltage-gated, type V,
alpha subunit (SCN5A), sodium channel, voltage-gated, type IV, beta
subunit (SCN4B), atrial natriuretic peptide (ANP), gap junction
protein, alpha 5, 40 kDa (GJA5), GATA binding protein (GATA)4,
GATA5, GATA6 and NK2 homeobox 5 (NKX2-5) (22–42). Nevertheless, AF is a genetically
heterogeneous disease and the genetic basis underlying AF in an
overwhelming majority of patients remains to be elucidated.
In a previous study, Gudbjartsson et al
(43) performed a genome-wide
association scan, followed by replication studies in three
populations of European descent and a Chinese population and found
a strong association between two sequence variants (rs2200733 and
rs10033464) on chromosome 4q25 and AF. They demonstrated that ~35%
of individuals of European descent had at least one of the variants
and that the risk of AF increased by 1.72 and 1.39 per copy. The
association with the stronger variant is replicated in the Chinese
population, where it was carried by 75% of individuals, and the
risk of AF was increased by 1.42 per copy. Subsequently, Kääb et
al (44) also confirmed the
strong association of the non-coding polymorphisms, rs2200733 and
rs10033464, with AF in four independent cohorts of European
descent. In two independently collected cardiac surgery cohorts,
the two variants within the chromosome 4q25 region were
independently associated with post-operative AF following coronary
artery bypass graft surgery after adjusting for clinical covariates
(45). Furthermore, the two
sequence variants have been reported to increase the risk of the
early and late recurrence of AF following catheter ablation
(46) and have been shown to be
genetic modifiers of rare ion channel mutations linked to familial
AF (47). The gene closest to
these sequence variants is pituitary homeobox 2 (PITX2), a
member of the PITX family of transcription factors that play an
essential role in embryonic morphogenesis, with PITX2c being the
predominant isoform expressed in the embryonic and adult heart
(48). Emerging evidence
highlights the pivotal role of PITX2c in the embryonic development
of the left atrium, the cardiac conduction system and pulmonary
venous myocardium, a major source of ectopic activity involved in
the initiation and maintenance of AF (49,50); the abnormal expression of PITX2c
has also been implicated in the increased susceptibility to AF
(51–53). These findings warrant the
screening of PITX2c as a preferred candidate gene for lone
AF.
Materials and methods
Study subjects
A cohort of 100 unrelated patients with lone AF was
enlisted from the Han Chinese population. A total of 200 ethnically
matched, unrelated healthy individuals were enrolled as the
controls. Peripheral venous blood samples were prepared and
clinical data, including medical records, electrocardiogram and
echocardiography reports were collected. The study subjects were
clinically classified using a consistently applied set of
definitions (7,34). Briefly, AF was diagnosed by a
standard 12-lead electrocardiogram demonstrating no P waves and
irregular R-R intervals irrespective of clinical symptoms. Lone AF
was defined as AF occurring in individuals <60 years of age
without other cardiac or systemic diseases by physical examination,
electrocardiogram, transthoracic echocardiogram and extensive
laboratory tests. Subjects were classified as ‘healthy’ if they
were asymptomatic and had a normal electrocardiogram with no
history of AF. Paroxysmal AF was defined as AF lasting >30 sec
that terminated spontaneously. Persistent AF was defined as AF
lasting >7 days and requiring either pharmacological therapy or
electrical cardioversion for termination. AF that was refractory to
cardioversion or that was allowed to continue was classified as
permanent. The present study was performed in compliance with the
ethical principles of the revised Declaration of Helsinki (48th WMA
General Assembly, Somerset West, Republic of South Africa, 1996).
The study protocol was reviewed and approved by the local
institutional ethics committee and written informed consent was
obtained from all participants prior to enrollment in this
study.
Genotyping for PITX2c
Genomic DNA was extracted from the peripheral venous
blood lymphocytes of all the participants using the Wizard Genomic
DNA Purification kit (Promega, Madison, WI, USA). The coding exons
and intron-exon boundaries of PITX2c were sequenced in the
100 unrelated patients with lone AF. The 200 control individuals
were subsequently genotyped for PITX2c due to the identified
mutation in patients with AF. The referential genomic DNA sequence
of PITX2c was derived from GenBank [accession no. NC_000004;
available at the National Center for Biotechnical Information
(NCBI; http://www.ncbi.nlm.nih.gov/)]. With
the aid of the on-line Primer 3 program (http://frodo.wi.mit.edu), the primers used to amplify
the entire coding region and splice junctions of PITX2c by
polymerase chain reaction (PCR) were designed as presented in
Table I. PCR was carried out
using HotStar Taq DNA Polymerase (Qiagen, Hilden, Germany) on a
Veriti Thermal Cycler (Applied Biosystems, Foster City, CA, USA),
with standard conditions and concentrations of reagents. Amplified
products were analyzed on 1% agarose gels stained with ethidium
bromide and purified using the QIAquick Gel Extraction kit
(Qiagen). Both strands of each amplicon were sequenced with a
BigDye® Terminator v3.1 Cycle Sequencing kit under an
ABI PRISM 3130xl DNA Analyzer (both from Applied Biosystems). The
sequencing primers were the same as those previously designed for
specific region amplification. The DNA sequences were viewed using
DNA Sequencing Analysis software v5.1 (Applied Biosystems). The
variant was validated by re-sequencing an independent PCR-generated
amplicon from the subject and met the quality control thresholds
with a call rate >99%. Additionally, for an identified sequence
variant, the Exome Variant Server (EVS; http://evs.gs.washington.edu/EVS) and the NCBI single
nucleotide polymorphism (SNP; http://www.ncbi.nlm.nih.gov/SNP) online databases were
queried to confirm its novelty.
 | Table IIntronic primers used to amplify the
coding exons and exon-intron boundaries of PITX2c. |
Table I
Intronic primers used to amplify the
coding exons and exon-intron boundaries of PITX2c.
| Exon | Forward primer (5′
to 3′) | Reverse primer (5′
to 3′) | Amplicon size
(bp) |
|---|
| 1 | CAG CTT GGC TTG AGA
ACT CG | TGA CTT CCT TGG GGC
GAG AG | 442 |
| 2 | CAG CTC TTC CAC GGC
TTC TG | GCT GCC TTC CAC ATT
CTC TC | 387 |
| 3 | AAT CTG CAC TGT GGC
ATC TG | AGT CTT TCA AGG GCG
GAG TT | 677 |
Multiple alignments of PITX2c protein
sequences
Multiple PITX2c protein sequences across various
species were aligned using an online program MUSCLE, v3.6
(http://www.ncbi.nlm.nih.gov/).
Prediction of the causative potential of
a PITX2c sequence variation
The disease-causing potential of a PITX2c sequence
variation was predicted by the online program MutationTaster
(http://www.mutationtaster.org), which
automatically yielded a probability for the variation to be either
a pathogenic mutation or a benign polymorphism. Of note, the
P-value used in this study is the probability of the correct
prediction rather than the probability of error as used in t-test
statistics (i.e., a value close to 1 indicates a high accuracy of
the prediction). In addition, another online program PolyPhen-2
(http://genetics.bwh.harvard.edu/pph2)
was also used to evaluate the pathogenic likeliness of a
mutation.
Expression plasmids and site-directed
mutagenesis
The recombinant expression plasmid, PITX2c-pcDNA4,
which was constructed by Strungaru et al (54), was a gift from Professor Georges
Christé, Physiopathologie des Troubles du Rythme Cardiaque, Faculté
de Pharmacie de Lyon, Université Lyon 1, Lyon, France. The atrial
natriuretic factor (ANF)-luciferase reporter plasmid, which
contains the 2600-bp 5′-flanking region of the ANF gene,
namely ANF(−2600)-Luc, was kindly provided by Dr Ichiro Shiojima,
from the Department of Cardiovascular Science and Medicine, Chiba
University Graduate School of Medicine, Chuo-ku, Chiba, Japan. The
identified mutation was introduced into the wild-type PITX2c using
the QuickChange II XL Site-Directed Mutagenesis kit (Stratagene, La
Jolla, CA, USA) with a complementary pair of primers. The mutant
was sequenced to confirm the desired mutation and to exclude any
other sequence variations.
Luciferase reporter gene assay
Chinese hamster ovary (CHO) cells were grown in
Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf
serum, 100 U/ml penicillin and 100 μg/ml streptomycin. The
CHO cells were cultured 24 h prior to transfection. The
ANF(−2600)-Luc reporter construct and an internal control reporter
plasmid pGL4.75 (hRluc/CMV; Promega) were used in transient
transfection assays to explore the transactivational activity of
the PITX2c mutant. CHO cells were transfected with 2 μg of
wild-type PITX2c-pcDNA4 or mutant PITX2c-pcDNA4 or empty vector
pcDNA4, 2.0 μg of ANF(−2600)-Luc reporter construct and 0.04
μg of pGL4.75 control reporter vector using
Lipofectamine® 2000 Transfection Reagent (Invitrogen,
Carlsbad, CA, USA). For the co-transfection experiments, 1
μg of wild-type PITX2c-pcDNA4, 1 μg of mutant
PITX2c-pcDNA4, 2.0 μg of ANF(−2600)-Luc and 0.04 μg
of pGL4.75 were used. Transfected cells were incubated for 24 h,
then lysed and assayed for reporter activities. Firefly luciferase
and Renilla luciferase activities were measured using the Dual-Glo
Luciferase Assay System (Promega). The activity of the ANF
promoter was presented as the fold activation of Firefly luciferase
relative to Renilla luciferase. At least three independent
experiments were performed for wild-type or mutant PITX2c.
Statistical analysis
The continuous data are expressed as the means ±
standard deviation. Continuous variables were tested for normality
of distribution and the Student’s unpaired t-test was used to
compare numeric variables between two groups. The categorical
variables were compared between two groups using Pearson’s
Chi-squared test or Fisher’s exact test, where appropriate. A
two-sided P-value <0.05 was considered to indicate a
statistically significant difference.
Results
Clinical characteristics of the study
population
A cohort of 100 unrelated patients with lone AF was
recruited, clinically evaluated and compared with 200 ethnically
matched, unrelated healthy individuals. None of the participants
had traditional risk factors for AF. There were no significant
differences between the patient and control groups as regards
baseline characteristics, including gender, body mass index, blood
pressure, fasting blood glucose levels, serum lipid levels, left
atrial dimension, left ventricular ejection fraction, heart rate at
rest, as well as life style (data not shown). The baseline clinical
characteristics of the 100 patients with lone AF are summarized in
Table II.
| Table IIBaseline clinical characteristics of
the 100 unrelated patients with lone AF. |
Table II
Baseline clinical characteristics of
the 100 unrelated patients with lone AF.
| Parameter | No. or mean | % or range |
|---|
| Male | 53 | 53 |
| Age at initial
diagnosis of AF (years) | 45.8 | 20–60 |
| Age at the time of
present study (years) | 52.1 | 21–67 |
| Type of AF at
presentation |
| Paroxysmal AF | 62 | 62 |
| Persistent AF | 26 | 26 |
| Permanent AF | 12 | 12 |
| Positive family
history of AF | 5 | 5 |
| History of
cardioversion | 82 | 82 |
| Implanted cardiac
pacemaker | 2 | 2 |
| Resting heart rate
(beats/min) | 78.6 | 56–164 |
| Systolic blood
pressure (mmHg) | 132.7 | 90–138 |
| Diastolic blood
pressure (mmHg) | 81.2 | 70–86 |
| Body mass index
(kg/m2) | 22.4 | 20–24 |
| Left atrial
dimension (mm) | 36 | 30–40 |
| Left ventricular
ejection fraction | 0.6 | 0.5–0.7 |
| Fasting blood
glucose (mmol/l) | 4.6 | 3.5–5.8 |
| Total cholesterol
(mmol/l) | 4.5 | 3.4–6.2 |
| Triglycerides
(mmol/l) | 1.5 | 0.9–1.7 |
| Medications |
| Amiodarone | 78 | 78 |
| Warfarin | 65 | 65 |
| Digoxin | 12 | 12 |
| β-blocker | 10 | 10 |
| Calcium channel
blocker | 6 | 6 |
PITX2c mutation
A heterozygous missense mutation in PITX2c was
identified in one of the 100 unrelated patients with lone AF. The
total population prevalence of PITX2c mutation based on the patient
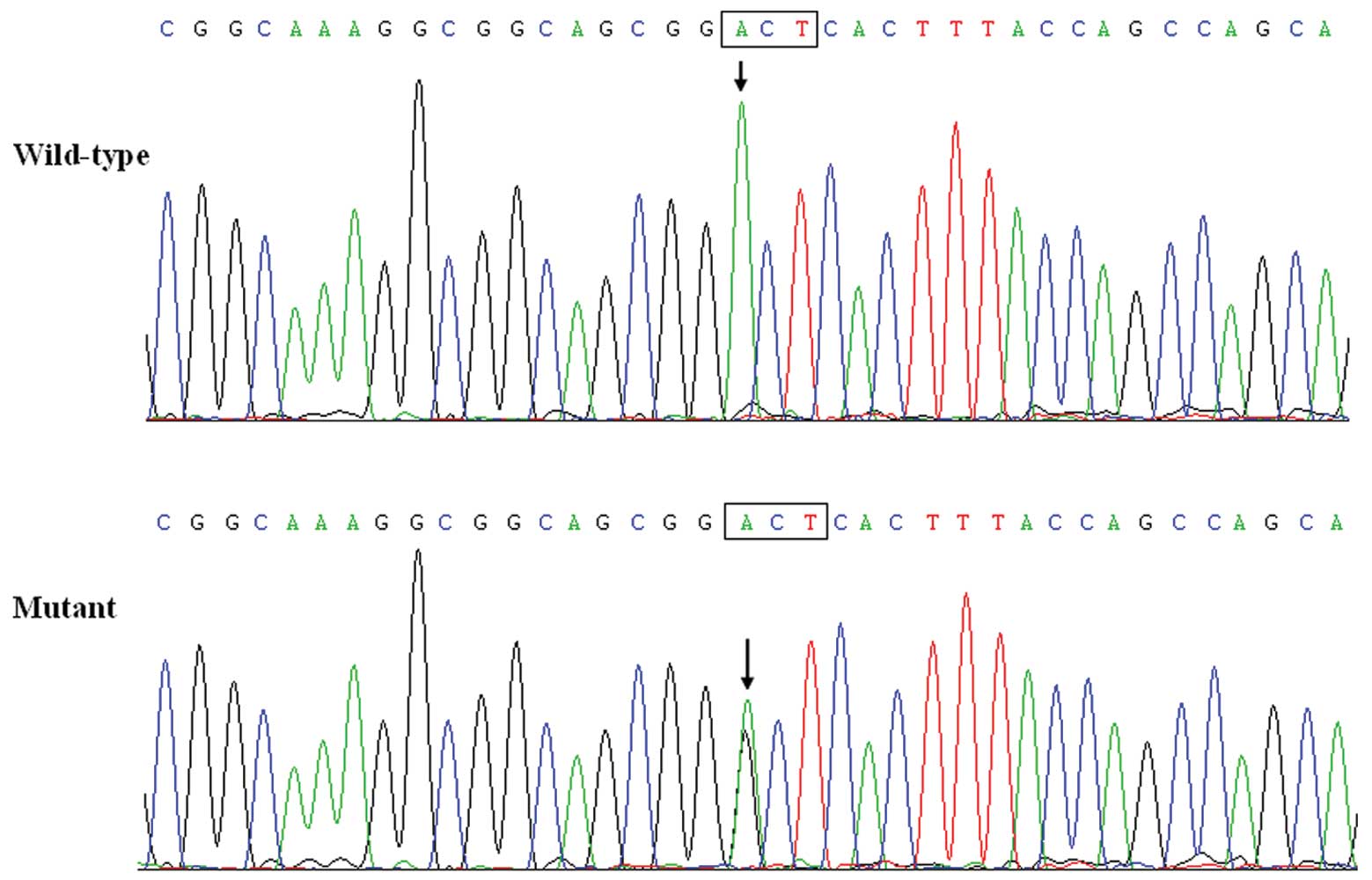
cohort was 1%. Specifically, a substitution of guanine (G) for
adenine (A) in the first nucleotide of codon 97 (c.289A>G),
predicting the transition of threonine (T) into alanine (A) at
amino acid position 97 (p.T97A), was identified in a male patient
aged 46 years, who was initially diagnosed with AF at the age of
32. The mutation carrier had no apparent malformations in the eyes,
teeth, umbilicus, or heart and had no positive family history of
AF. No relatives from the mutation carrier were available for the
genotyping of PITX2c. The sequence chromatograms showing the
detected heterozygous PITX2c mutation of c.289A>G in
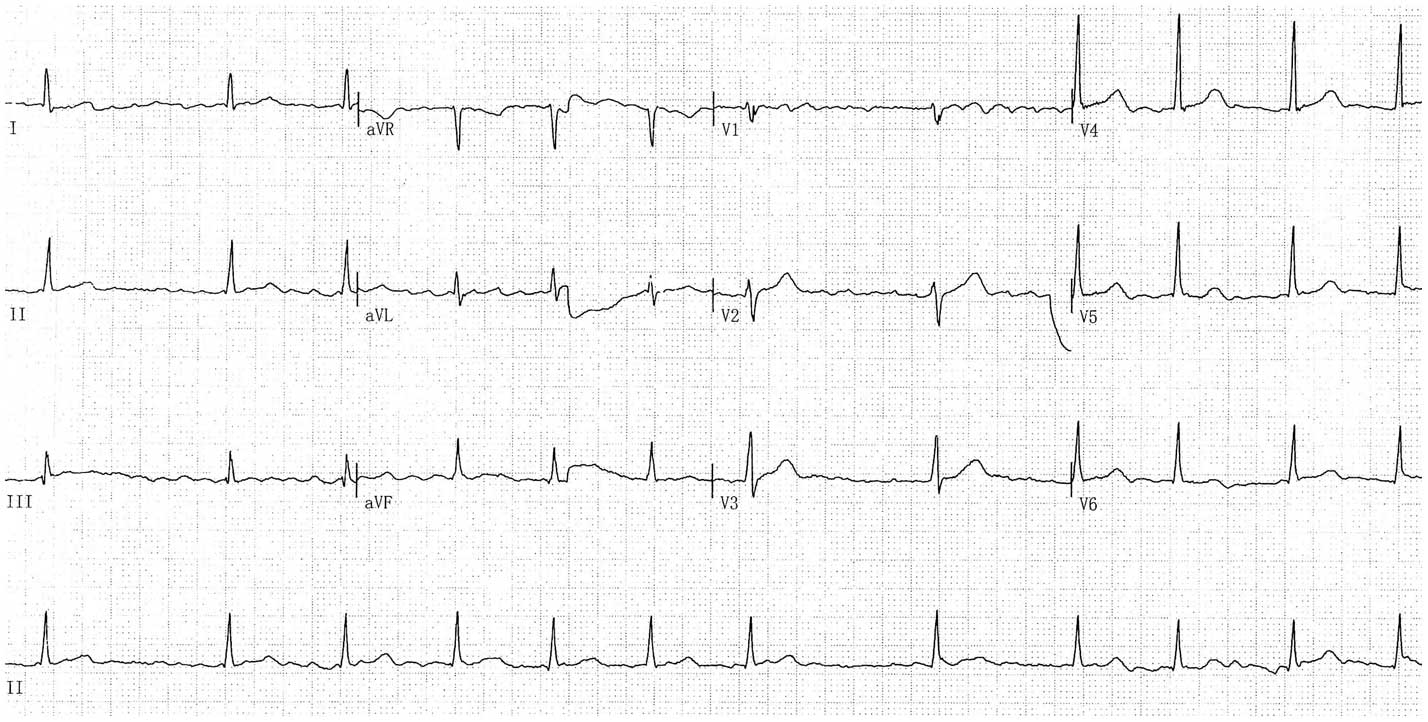
contrast to its control sequence is shown in Fig. 1. A representative
electrocardiogram of the mutation carrier showing AF is displayed
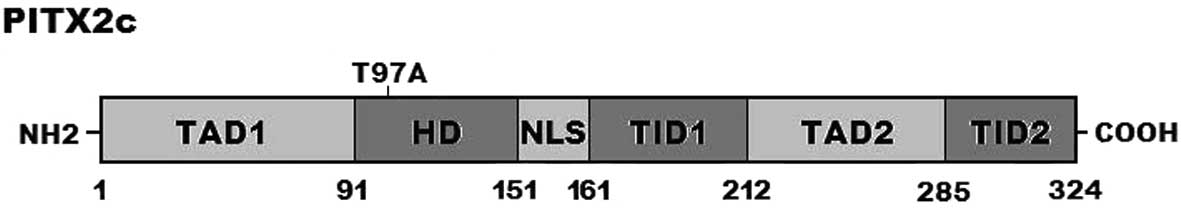
in Fig. 2. A schematic diagram of
PITX2c showing the structural domains and the location of the
identified mutation is exhibited in Fig. 3 [modified from previous studies
(55,56)]. This missense mutation was neither
observed in the control population nor reported in the EVS and NCBI
SNP databases.
Alignment of multiple PITX2c protein
sequences across species
A cross-species alignment of PITX2c protein
sequences revealed that the altered amino acid was completely
conserved evolutionarily (Fig.
4).
Disease-causing potential of the PITX2c
variation
The PITX2c sequence variation of c.289A>G
was automatically predicted by MutationTaster to be a
disease-causing mutation with the P-value of 1.0. No SNP in the
altered region was found in the MutationTaster database. This
PITX2c sequence variation was also predicted by PolyPhen-2 to be
possibly damaging, with a score of 1.000 (sensitivity 0.00;
specificity 1.00).
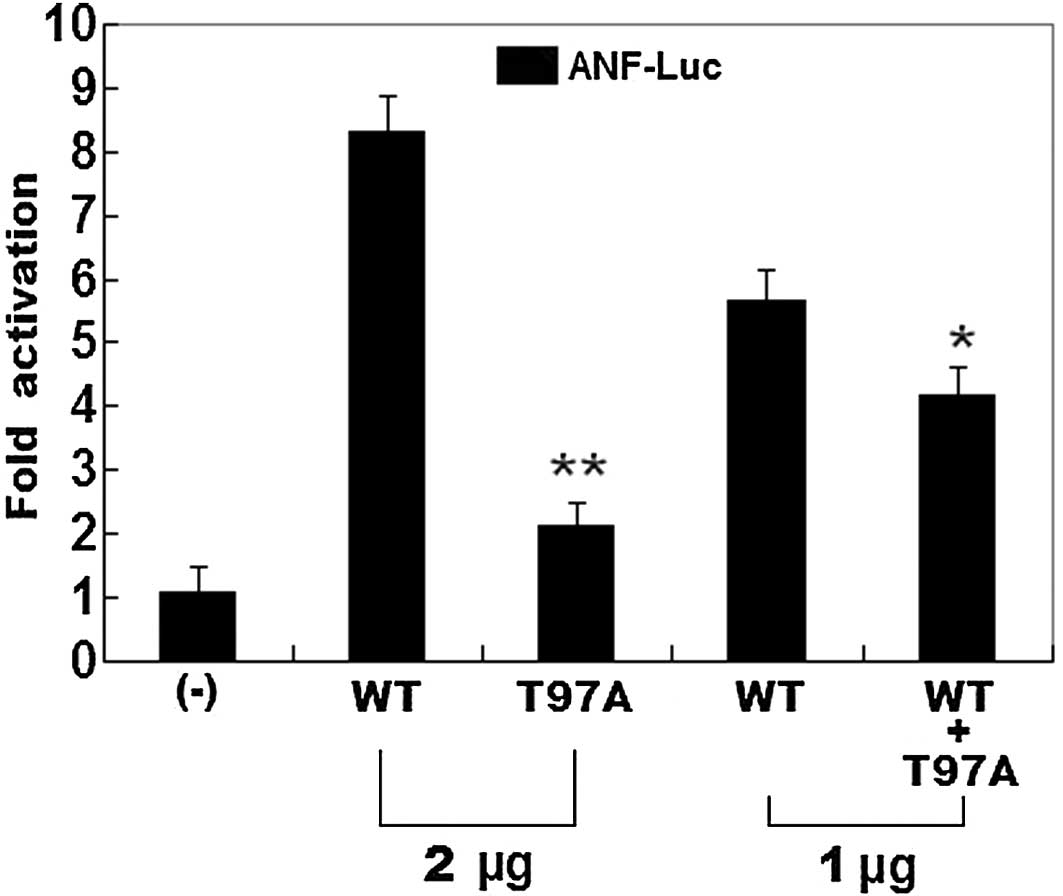
Transcriptional activity of the PITX2c
mutant
The same amounts of wild-type PITX2c (2 μg)
and T97A-mutant PITX2c (2 μg) activated the ANF
promoter (~8- and 2-fold increase, respectively), when compared
with the empty plasmid. When the same amount of wild-type PITX2c (1
μg) was transfected in combination with T97A-mutant PITX2c
(1 μg), the induced activation of the ANF promoter
was increased by ~4-fold, in comparison with the empty plasmid
(Fig. 5).
Discussion
In the present study, a novel heterozygous PITX2c
mutation, p.T97A, was identified in a patient with lone AF, which
was absent in the 400 reference chromosomes and was predicted to be
pathogenic by MutationTaster and PolyPhen-2. A cross-species
alignment of PITX2c protein sequences indicated that the altered
amino acid was completely conserved evolutionarily. Functional
analysis demonstrated that the mutant PITX2c protein was associated
with a significantly reduced transcriptional activity. Therefore,
it is likely that functionally compromised PITX2c predisposes to
AF.
PITX2 is a member of the bicoid-like homeobox
transcription factor family. Four different PITX2 transcripts,
derived from alternative promoter usage and differential mRNA
splicing, have been described. PITX2a, PITX2b and PITX2c differ
only in their amino-termini and have been identified in chicks,
humans, mice, zebrafish and xenopus, while the fourth
isoform, PITX2d, which lacks the amino-terminal domain and most of
the homeodomain, has only been observed in humans. The unique
amino-termini of PITX2a, PITX2b and PITX2c modify their ability to
activate transcription in a cell type- and promoter-dependent
manner. The homeodomain is responsible for recognizing specific DNA
sequences (5′-TAATCC-3′), which is essential for DNA binding,
nuclear translocation and interaction with other transcription
factors (57). The human
PITX2c gene maps to chromosome 4q25, which consists of three
exons coding for a protein of 324 amino acids (58). PITX2c is expressed asymmetrically
in the developing and adult heart, playing a crucial role in normal
cardiovascular genesis and maturation (53). The PITX2c mutation of p.T97A
identified in the present study is located in the homeodomain;
thus, it can be hypothesized that it may affect the transcriptional
activity of PITX2c by interfering with its DNA-binding ability.
Previous studies have validated that PITX2c is an
upstream regulator of multiple target genes expressed in the heart
during embryogenesis, including the gene that encodes ANF (59). Therefore, the functional effect of
a PITX2c mutation can be explored by assay of the transcriptional
activity of the ANF promoter in cells expressing PITX2c
mutant in contrast to its wild-type counterpart. In the present
study, the functional characteristics of the novel PITX2c mutation
identified in a patient with AF were investigated by
transcriptional activity analysis and the results revealed that the
mutation was associated with a significantly diminished
transcriptional activity on a downstream gene, suggesting that
dysfunctional PITX2c caused by mutation is potentially an
alternative pathological mechanism of AF.
The association of functionally impaired PITX2c with
the enhanced susceptibility to AF may be partially attributable to
the abnormal development of the cardiovascular system, particularly
the pulmonary venous myocardium (60). The pulmonary venous vessel is
sheathed by a layer of myocardium known as the pulmonary myocardial
sleeve, which has been implicated in the initiation and maintenance
of AF by several presumably electrophysiological mechanisms,
including increased automaticity, anisotropic arrangement of the
myocardial fibers and delayed conduction that facilitate re-entry
(61). It has been verified that
PITX2c is abundantly expressed in the atria and pulmonary
myocardium and functions as a repressor of the pacemaker lineage
gene program by downregulating the sinoatrial nodal gene program,
such as short stature homeobox 2 (ShOX2), hyperpolarization
activated cyclic nucleotide-gated potassium channel 4 (HCN4) and
Cav3.1, and upregulating a gene program characteristic of a working
myocardium phenotype, i.e. NKX2-5, Cx40, Cx43, ANP and Kir2.1
(48,51,53,60). In PITX2c knockout mice, a
microarray analysis of gene expression profiles revealed that the
sinoatrial nodal genes, ShOX2 and HCN4, were upregulated in the
atrium, as well as a number of ion channel genes, including KCNQ1
(51). Therefore, PITX2c
loss-of-function mutation may contribute to the switch of the
atrial and pulmonary myocardium to a sinoatrial node-like
phenotype, creating an arrhythmogenic substrate prone to AF.
It has been corroberated that there are some
downstream genes transactivated by PITX2c (48) and loss-of-function mutations in
several target molecules have been causally involved in AF,
including NKX2-5, Cx40, Cx43 and ANF (28,29–32,42,62). Therefore, mutated PITX2c may
predispose to AF by downregulating the expression of such target
genes.
In conclusion, to our knowledge, this is the first
study on the association of PITX2c loss-of-function mutation with
lone AF, which provides novel insight into the molecular mechanisms
underlying AF, indicating potential implications for the early
prophylaxis and allele-specific treatment of this prevalent type of
arrhythmia.
Acknowledgements
The authors are really grateful to the participants
for their dedication to the study. The present study was supported
by grants from the National Natural Science Foundation of China
(81070086, 81070153, 81270161 and 60103223) and the Natural Science
Foundation of Shandong Province of China (ZR2010HM134 and
Y2010C36).
References
|
1
|
Fuster V, Rydén LE, Cannom DS, Crijns HJ,
Curtis AB, Ellenbogen KA, Halperin JL, Kay GN, Le Huezey JY, Lowe
JE, Olsson SB, Prystowsky EN, Tamargo JL, Wann LS, Smith SC Jr,
Priori SG, Estes NA III, Ezekowitz MD, Jackman WM, January CT, Lowe
JE, Page RL, Slotwiner DJ, Stevenson WG, Tracy CM, Jacobs AK,
Anderson JL, Albert N, Buller CE, Creager MA, Ettinger SM, Guyton
RA, Halperin JL, Hochman JS, Kushner FG, Ohman EM, Stevenson WG,
Tarkington LG and Yancy CW; American College of Cardiology
Foundation/American Heart Association Task Force. 2011 ACCF/AHA/HRS
focused updates incorporated into the ACC/AHA/ESC 2006 guidelines
for the management of patients with atrial fibrillation: a report
of the American College of Cardiology Foundation/American Heart
Association Task Force on practice guidelines. Circulation.
123:e269–e367. 2011.
|
|
2
|
Lloyd-Jones DM, Wang TJ, Leip EP, Larson
MG, Levy D, Vasan RS, D’Agostino RB, Massaro JM, Beiser A, Wolf PA
and Benjamin EJ: Lifetime risk for development of atrial
fibrillation: the Framingham Heart Study. Circulation.
110:1042–1046. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Miyasaka Y, Barnes ME, Gersh BJ, Cha SS,
Bailey KR, Abhayaratna WP, Seward JB and Tsang TS: Secular trends
in incidence of atrial fibrillation in Olmsted County, Minnesota,
1980 to 2000, and implications on the projections for future
prevalence. Circulation. 114:119–125. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Benjamin EJ, Wolf PA, D’Agostino RB,
Silbershatz H, Kannel WB and Levy D: Impact of atrial fibrillation
on the risk of death: the Framingham Heart Study. Circulation.
98:946–952. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wolf PA, Abbott RD and Kannel WB: Atrial
fibrillation as an independent risk factor for stroke: the
Framingham Study. Stroke. 22:983–988. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wolowacz SE, Samuel M, Brennan VK,
Jasso-Mosqueda JG and Van Gelder IC: The cost of illness of atrial
fibrillation: a systematic review of the recent literature.
Europace. 13:1375–1385. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Darbar D, Herron KJ, Ballew JD, Jahangir
A, Gersh BJ, Shen WK, Hammill SC, Packer DL and Olson TM: Familial
atrial fibrillation is a genetically heterogeneous disorder. J Am
Coll Cardiol. 41:2185–2192. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ellinor PT, Yoerger DM, Ruskin JN and
MacRae CA: Familial aggregation in lone atrial fibrillation. Hum
Genet. 118:179–184. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Arnar DO, Thorvaldsson S, Manolio TA,
Thorgeirsson G, Kristjansson K, Hakonarson H and Stefansson K:
Familial aggregation of atrial fibrillation in Iceland. Eur Heart
J. 27:708–712. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Junttila MJ, Raatikainen MJ, Perkiömäki
JS, Hong K, Brugada R and Huikuri HV: Familial clustering of lone
atrial fibrillation in patients with saddleback-type ST-segment
elevation in right precordial leads. Eur Heart J. 28:463–468. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Christophersen IE, Ravn LS,
Budtz-Joergensen E, Skytthe A, Haunsoe S, Svendsen JH and
Christensen K: Familial aggregation of atrial fibrillation: a study
in Danish twins. Circ Arrhythm Electrophysiol. 2:378–383. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang YQ, Zhang XL, Wang XH, Tan HW, Shi
HF, Fang WY and Liu X: Familial aggregation of lone atrial
fibrillation in the Chinese population. Intern Med. 49:2385–2391.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lubitz SA, Yin X, Fontes JD, Magnani JW,
Rienstra M, Pai M, Villalon ML, Vasan RS, Pencina MJ, Levy D,
Larson MG, Ellinor PT and Benjamin EJ: Association between familial
atrial fibrillation and risk of new-onset atrial fibrillation.
JAMA. 304:2263–2269. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fox CS, Parise H, D’Agostino RB Sr,
Lloyd-Jones DM, Vasan RS, Wang TJ, Levy D, Wolf PA and Benjamin EJ:
Parental atrial fibrillation as a risk factor for atrial
fibrillation in offspring. JAMA. 291:2851–2855. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brugada R, Tapscott T, Czernuszewicz GZ,
Marian AJ, Iglesias A, Mont L, Brugada J, Girona J, Domingo A,
Bachinski LL and Roberts R: Identification of a genetic locus for
familial atrial fibrillation. N Engl J Med. 336:905–911. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ellinor PT, Shin JT, Moore RK, Yoerger DM
and MacRae CA: Locus for atrial fibrillation maps to chromosome
6q14-16. Circulation. 107:2880–2883. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang
Y, Xu WY, Jin HW, Sun H, Su XY, Zhuang QN, Yang YQ, Li YB, Liu Y,
Xu HJ, Li XF, Ma N, Mou CP, Chen Z, Barhanin J and Huang W: KCNQ1
gain-of-function mutation in familial atrial fibrillation. Science.
299:251–254. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oberti C, Wang L, Li L, Dong J, Rao S, Du
W and Wang Q: Genome-wide linkage scan identifies a novel genetic
locus on chromosome 5p13 for neonatal atrial fibrillation
associated with sudden death and variable cardiomyopathy.
Circulation. 110:3753–3759. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang X, Chen S, Yoo S, Chakrabarti S,
Zhang T, Ke T, Oberti C, Yong SL, Fang F, Li L, de la Fuente R,
Wang L, Chen Q and Wang QK: Mutation in nuclear pore component
NUP155 leads to atrial fibrillation and early sudden cardiac death.
Cell. 135:1017–1027. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Volders PG, Zhu Q, Timmermans C, Eurlings
PM, Su X, Arens YH, Li L, Jongbloed RJ, Xia M, Rodriguez LM and
Chen YH: Mapping a novel locus for familial atrial fibrillation on
chromosome 10p11-q21. Heart Rhythm. 4:469–475. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Darbar D, Hardy A, Haines JL and Roden DM:
Prolonged signal-averaged P-wave duration as an intermediate
phenotype for familial atrial fibrillation. J Am Coll Cardiol.
51:1083–1089. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang Y, Xia M, Jin Q, Bendahhou S, Shi J
and Chen Y, Liang B, Lin J, Liu Y, Liu B, Zhou Q, Zhang D, Wang R,
Ma N, Su X, Niu K, Pei Y, Xu W, Chen Z, Wan H, Cui J, Barhanin J
and Chen Y: Identification of a KCNE2 gain-of-function mutation in
patients with familial atrial fibrillation. Am J Hum Genet.
75:899–905. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hong K, Bjerregaard P, Gussak I and
Brugada R: Short QT syndrome and atrial fibrillation caused by
mutation in KCNH2. J Cardiovasc Electrophysiol. 16:394–396. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xia M, Jin Q, Bendahhou S, He Y, Larroque
MM and Chen Y, Zhou Q, Yang Y, Liu Y, Liu B, Zhu Q, Zhou Y, Lin J,
Liang B, Li L, Dong X, Pan Z, Wang R, Wan H, Qiu W, Xu W, Eurlings
P, Barhanin J and Chen Y: A Kir2.1 gain-of-function mutation
underlies familial atrial fibrillation. Biochem Biophys Res Commun.
332:1012–1019. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Olson TM, Alekseev AE, Liu XK, Park S,
Zingman LV, Bienengraeber M, Sattiraju S, Ballew JD, Jahangir A and
Terzic A: Kv1.5 channelopathy due to KCNA5 loss-of-function
mutation causes human atrial fibrillation. Hum Mol Genet.
15:2185–2191. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Olson TM, Michels VV, Ballew JD, Reyna SP,
Karst ML, Herron KJ, Horton SC, Rodeheffer RJ and Anderson JL:
Sodium channel mutations and susceptibility to heart failure and
atrial fibrillation. JAMA. 293:447–454. 2005. View Article : Google Scholar
|
|
27
|
Li RG, Wang Q, Xu YJ, Zhang M, Qu XK, Liu
X, Fang WY and Yang YQ: Mutations of SCN4B-encoded sodium
channel β4 subunit in familial atrial fibrillation. Int J Mol Med.
32:144–150. 2013.
|
|
28
|
Hodgson-Zingman DM, Karst ML, Zingman LV,
Heublein DM, Darbar D, Herron KJ, Ballew JD, de Andrade M, Burnett
JC Jr and Olson TM: Atrial natriuretic peptide frameshift mutation
in familial atrial fibrillation. N Engl J Med. 359:158–165. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gollob MH, Jones DL, Krahn AD, Danis L,
Gong XQ, Shao Q, Liu X, Veinot JP, Tang AS, Stewart AF, Tesson F,
Klein GJ, Yee R, Skanes AC, Guiraudon GM, Ebihara L and Bai D:
Somatic mutations in the connexin 40 gene (GJA5) in atrial
fibrillation. N Engl J Med. 354:2677–2688. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang YQ, Zhang XL, Wang XH, Tan HW, Shi
HF, Jiang WF, Fang WY and Liu X: Connexin40 nonsense mutation in
familial atrial fibrillation. Int J Mol Med. 26:605–610.
2010.PubMed/NCBI
|
|
31
|
Sun Y, Yang YQ, Gong XQ, Wang XH, Li RG,
Tan HW, Liu X, Fang WY and Bai D: Novel germline GJA5/connexin40
mutations associated with lone atrial fibrillation impair gap
junctional intercellular communication. Hum Mutat. 34:603–609.
2013.PubMed/NCBI
|
|
32
|
Shi HF, Yang JF, Wang Q, Li RG, Xu YJ, Qu
XK, Fang WY, Liu X and Yang YQ: Prevalence and spectrum of GJA5
mutations associated with lone atrial fibrillation. Mol Med Rep.
7:767–774. 2013.PubMed/NCBI
|
|
33
|
Yang YQ, Wang MY, Zhang XL, Tan HW, Shi
HF, Jiang WF, Wang XH, Fang WY and Liu X: GATA4 loss-of-function
mutations in familial atrial fibrillation. Clin Chim Acta.
412:1825–1830. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jiang JQ, Shen FF, Fang WY, Liu X and Yang
YQ: Novel GATA4 mutations in lone atrial fibrillation. Int J Mol
Med. 28:1025–1032. 2011.PubMed/NCBI
|
|
35
|
Wang J, Sun YM and Yang YQ: utation
spectrum of the GATA4 gene in patients with idiopathic atrial
fibrillation. Mol Biol Rep. 39:8127–8135. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang YQ, Wang J, Wang XH, Wang Q, Tan HW,
Zhang M, Shen FF, Jiang JQ, Fang WY and Liu X: Mutational spectrum
of the GATA5 gene associated with familial atrial fibrillation. Int
J Cardiol. 157:305–307. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang XH, Huang CX, Wang Q, Li RG, Xu YJ,
Liu X, Fang WY and Yang YQ: A novel GATA5 loss-of-function mutation
underlies lone atrial fibrillation. Int J Mol Med. 31:43–50.
2013.PubMed/NCBI
|
|
38
|
Gu JY, Xu JH, Yu H and Yang YQ: Novel
GATA5 loss-of-function mutations underlie familial atrial
fibrillation. Clinics (Sao Paulo). 67:1393–1399. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang YQ, Wang XH, Tan HW, Jiang WF, Fang
WY and Liu X: Prevalence and spectrum of GATA6 mutations associated
with familial atrial fibrillation. Int J Cardiol. 155:494–496.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang YQ, Li L, Wang J, Zhang XL, Li RG, Xu
YJ, Tan HW, Wang XH, Jiang JQ, Fang WY and Liu X: GATA6
loss-of-function mutation in atrial fibrillation. Eur J Med Genet.
55:520–526. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li J, Liu WD, Yang ZL and Yang YQ: Novel
GATA6 loss-of-function mutation responsible for familial atrial
fibrillation. Int J Mol Med. 30:783–790. 2012.PubMed/NCBI
|
|
42
|
Huang RT, Xue S, Xu YJ, Zhou M and Yang
YQ: A novel NKX2.5 loss-of-function mutation responsible for
familial atrial fibrillation. Int J Mol Med. 31:1119–1126.
2013.PubMed/NCBI
|
|
43
|
Gudbjartsson DF, Arnar DO, Helgadottir A,
Gretarsdottir S, Holm H, Sigurdsson A, Jonasdottir A, Baker A,
Thorleifsson G, Kristjansson K, Palsson A, Blondal T, Sulem P,
Backman VM, Hardarson GA, Palsdottir E, Helgason A, Sigurjonsdottir
R, Sverrisson JT, Kostulas K, Ng MC, Baum L, So WY, Wong KS, Chan
JC, Furie KL, Greenberg SM, Sale M, Kelly P, MacRae CA, Smith EE,
Rosand J, Hillert J, Ma RC, Ellinor PT, Thorgeirsson G, Gulcher JR,
Kong A, Thorsteinsdottir U and Stefansson K: Variants conferring
risk of atrial fibrillation on chromosome 4q25. Nature.
448:353–357. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kääb S, Darbar D, van Noord C, Dupuis J,
Pfeufer A, Newton-Cheh C, Schnabel R, Makino S, Sinner MF,
Kannankeril PJ, Beckmann BM, Choudry S, Donahue BS, Heeringa J,
Perz S, Lunetta KL, Larson MG, Levy D, MacRae CA, Ruskin JN, Wacker
A, Schömig A, Wichmann HE, Steinbeck G, Meitinger T, Uitterlinden
AG, Witteman JC, Roden DM, Benjamin EJ and Ellinor PT: Large scale
replication and meta-analysis of variants on chromosome 4q25
associated with atrial fibrillation. Eur Heart J. 30:813–819.
2009.PubMed/NCBI
|
|
45
|
Body SC, Collard CD, Shernan SK, Fox AA,
Liu KY, Ritchie MD, Perry TE, Muehlschlegel JD, Aranki S, Donahue
BS, Pretorius M, Estrada JC, Ellinor PT, Newton-Cheh C, Seidman CE,
Seidman JG, Herman DS, Lichtner P, Meitinger T, Pfeufer A, Kääb S,
Brown NJ, Roden DM and Darbar D: Variation in the 4q25 chromosomal
locus predicts atrial fibrillation after coronary artery bypass
graft surgery. Circ Cardiovasc Genet. 2:499–506. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Husser D, Adams V, Piorkowski C, Hindricks
G and Bollmann A: Chromosome 4q25 variants and atrial fibrillation
recurrence after catheter ablation. J Am Coll Cardiol. 55:747–753.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ritchie MD, Rowan S, Kucera G,
Stubblefield T, Blair M, Carter S, Roden DM and Darbar D:
Chromosome 4q25 variants are genetic modifiers of rare ion channel
mutations associated with familial atrial fibrillation. J Am Coll
Cardiol. 60:1173–1181. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Clauss S and Kääb S: Is Pitx2 growing up?
Circ Cardiovasc Genet. 4:105–107. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Haïssaguerre M, Jaïs P, Shah DC, Takahashi
A, Hocini M, Quiniou G, Garrigue S, Le Mouroux A, Le Métayer P and
Clémenty J: Spontaneous initiation of atrial fibrillation by
ectopic beats originating in the pulmonary veins. N Engl J Med.
339:659–666. 1998.PubMed/NCBI
|
|
50
|
Douglas YL, Jongbloed MR, Deruiter MC and
Gittenberger-de Groot AC: Normal and abnormal development of
pulmonary veins: state of the art and correlation with clinical
entities. Int J Cardiol. 147:13–24. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang J, Klysik E, Sood S, Johnson RL,
Wehrens XH and Martin JF: Pitx2 prevents susceptibility to atrial
arrhythmias by inhibiting left-sided pacemaker specification. Proc
Natl Acad Sci USA. 107:9753–9758. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chinchilla A, Daimi H, Lozano-Velasco E,
Dominguez JN, Caballero R, Delpón E, Tamargo J, Cinca J,
Hove-Madsen L, Aranega AE and Franco D: PITX2 insufficiency leads
to atrial electrical and structural remodeling linked to
arrhythmogenesis. Circ Cardiovasc Genet. 4:269–279. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kirchhof P, Kahr PC, Kaese S, Piccini I,
Vokshi I, Scheld HH, Rotering H, Fortmueller L, Laakmann S,
Verheule S, Schotten U, Fabritz L and Brown NA: PITX2c is expressed
in the adult left atrium, and reducing Pitx2c expression promotes
atrial fibrillation inducibility and complex changes in gene
expression. Circ Cardiovasc Genet. 4:123–133. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Strungaru MH, Footz T, Liu Y, Berry FB,
Belleau P, Semina EV, Raymond V and Walter MA: PITX2 is involved in
stress response in cultured human trabecular meshwork cells through
regulation of SLC13A3. Invest Ophthalmol Vis Sci. 52:7625–7633.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Footz T, Idrees F, Acharya M, Kozlowski K
and Walter MA: Analysis of mutations of the PITX2 transcription
factor found in patients with Axenfeld-Rieger syndrome. Invest
Ophthalmol Vis Sci. 50:2599–2606. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Acharya M, Lingenfelter DJ, Huang L, Gage
PJ and Walter MA: Human PRKC apoptosis WT1 regulator is a novel
PITX2-interacting protein that regulates PITX2 transcriptional
activity in ocular cells. J Biol Chem. 284:34829–34838. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Simard A, Di Giorgio L, Amen M, Westwood
A, Amendt BA and Ryan AK: The Pitx2c N-terminal domain is a
critical interaction domain required for asymmetric morphogenesis.
Dev Dyn. 238:2459–2470. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Semina EV, Reiter R, Leysens NJ, Alward
WL, Small KW, Datson NA, Siegel-Bartelt J, Bierke-Nelson D, Bitoun
P, Zabel BU, Carey JC and Murray JC: Cloning and characterization
of a novel bicoid-related homeobox transcription factor gene, RIEG,
involved in Rieger syndrome. Nat Genet. 14:392–399. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ganga M, Espinoza HM, Cox CJ, Morton L,
Hjalt TA, Lee Y and Amendt BA: PITX2 isoform-specific regulation of
atrial natriuretic factor expression: synergism and repression with
Nkx2.5. J Biol Chem. 278:22437–22445. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Mommersteeg MT, Brown NA, Prall OW, de
Gier-de Vries C, Harvey RP, Moorman AF and Christoffels VM: Pitx2c
and Nkx2-5 are required for the formation and identity of the
pulmonary myocardium. Circ Res. 101:902–909. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Mommersteeg MT, Christoffels VM, Anderson
RH and Moorman AF: Atrial fibrillation: a developmental point of
view. Heart Rhythm. 6:1818–1824. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Thibodeau IL, Xu J, Li Q, Liu G, Lam K,
Veinot JP, Birnie DH, Jones DL, Krahn AD, Lemery R, Nicholson BJ
and Gollob MH: Paradigm of genetic mosaicism and lone atrial
fibrillation: physiological characterization of a connexin
43-deletion mutant identified from atrial tissue. Circulation.
122:236–244. 2010. View Article : Google Scholar
|