Introduction
An estimated 200 million patients worldwide undergo
anesthesia and surgery each year (1,2).
Previous studies have reported that subanesthetic exposure to
individual anesthetic drugs, including ketamine (3), propofol (4) and sevoflurane (5), triggers a significant neuroapoptotic
response in the infant rodent brain. Likewise, isoflurane alone
causes cell damage in various neuronal and non-neuronal tissues and
cells (6,7). The anesthetic action of isoflurane
is thought to be mediated by multiple mechanisms, including actions
on GABAA, glycine and glutamate receptors and potassium
channels (8–10). Isoflurane is also known to exert a
depolarizing action on neuronal mitochondria (11). Isoflurane induces caspase
activation and apoptosis through the mitochondrial-dependent
apoptotic pathway (12).
Resveratrol (RESV; trans-3,5,4′-trihydroxystilbene),
a polyhydroxyphenolic antioxidant, was first isolated in 1940 as an
ingredient of the roots of white hellebore (13) and since then it has been isolated
from a variety of plant species, including grapes (14). The trans-isomeric form of RESV is
the steady form and mediates a broad-spectrum of beneficial health
effects, including anti-infective, antioxidant and cardioprotective
functions (15). In multiple
models of neurological injury, RESV has demonstrated efficacy in
reducing neuropathological and behavioral sequelae, such as
stroke (16,17), spinal cord injury (18,19) and Huntington’s disease (20). Thus, it is important to obtain a
better understanding of the mechanisms behind the neuroprotective
effects of RESV in brain cells.
To our knowledge, there are no related reports to
date on the role of RESV in isoflurane-induced neuroapoptosis. In
this study, we confirm and extend previous findings and demonstrate
the neuroprotective effects of RESV. Importantly, the mechanism(s)
of action of RESV were determined by western blot analysis to
determine the expression of apoptosis-related molecules, such as
caspase-3, -8 and -9.
Materials and methods
Cell culture and treatment
All experiments with animals were performed
according to the guidelines of our University Ethics Committee.
Neuronal cells derived from the cerebral neocortex were harvested
from 16-day-old embryonic mice by caesarean section from pregnant
BALB/c mice. The cells (5×105) were plated on 24-well
plates pre-coated with poly-L-lysine (Sigma Chemical, St. Louis,
MO, USA) and the cultures were maintained at 37°C in a 5% (v/v)
CO2 incubator and supplemented with neurobasal medium
supplemented with B27 (X1) and glutamine (25 mM). Neuronal cell
cultures were ready to use on day 7. O2 (21%),
CO2 (5%) and isoflurane (2%) were delivered from an
anesthesia machine to a sealed plastic box. A Datex infrared gas
analyzer (Puritan-Bennett, Tewksbury, MA, USA) was used to
continuously monitor the delivered carbon dioxide, oxygen and
isoflurane concentrations.
Drugs
RESV (Sigma Chemical) was dissolved in 7:3 saline
(0.9 % NaCl):solutol (BASF Corp., Wyandotte, MI, USA). Butin was
purchased from Wako Pure Chemical Industries, Ltd. (Tokyo, Japan)
and dissolved in dimethylsulfoxide (DMSO); the final concentration
of DMSO did not exceed 0.02%.
Experimental design
The cells were divided into 7 groups as follows:
group 1, control; group 2, 2% isoflurane for 6 h; group 3, RESV 50
μM for 24 h + 2% isoflurane for 6 h; group 4, RESV 100
μM for 24 h + 2% isoflurane for 6 h; group 5, RESV 200
μM for 24 h + 2% isoflurane for 6 h; group 6, butin 10
μg/ml for 24 h + 2% isoflurane for 6 h; group 7, RESV 200
μM for 24 h + siRNA AKT for 24 h + 2% isoflurane for 6
h.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
To investigate the protective effects of RESV on
isoflurane-induced neuronal death, MTT assay was used. MTT (10
μl, at 5 mg/ml) was added to each well at a final
concentration of 500 μg/ml, and the mixture was further
incubated for 1 h at 37°C and the liquid in the wells was removed
thereafter. DMSO (100 μl) was then added to each well and
the absorbance was read using a UV Max microplate reader (Molecular
Devices, Palo Alto, CA, USA) at 560 nm.
TUNEL assay
For apoptosis detection, the cells were washed in
PBS, fixed, permeabilized and subjected to TUNEL labeling using an
in situ Cell Death Detection kit (KeyGen, Nanjing, China)
according to the manufacturer’s instructions. Following
counterstaining with DAPI (1 μg/ml), photographic images
were acquired using an Olympus CX71 fluorescence microscope
(Olympus, Tokyo, Japan). TUNEL-positive nuclei were stained green
and all other nuclei were stained blue as previously described
(21,22).
Comet assay
A comet assay was performed to assess oxidative DNA
damage (23,24). The cell pellet (1.5×105
cells) was mixed with 100 μl of 0.5% low melting agarose
(LMA) at 39°C and spread on a fully frosted microscopic slide that
was pre-coated with 200 μl of 1% normal melting agarose
(NMA). Following solidification of the agarose, the slide was
covered with an additional 75 μl of 0.5% LMA and then
immersed in lysis solution (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, 1%
Trion X-100 and 10% DMSO; pH 10.0) for 1 h at 4°C. The slides were
then placed in a gel electrophoresis apparatus containing 300 mM
NaOH and 10 mM EDTA (pH 13.0) for 40 min to allow DNA unwinding and
the expression of the alkali-labile damage. Following
electrophoresis, the slides were washed 3 times for 5 min at 4°C in
a neutralizing buffer (0.4 M Tris, pH 7.5) and then stained with 75
μl of ethidium bromide (20 μg/ml). The slides were
observed under an Olympus CX71 fluorescence microscope.
Cell apoptosis assay
Apoptosis was determined using an Apoptosis
Detection kit (KeyGen). Briefly, the cells were collected, washed
twice in ice-cold PBS and then resuspended in binding buffer at a
density of 1×106 cells/ml. The treated cells were
incubated with fluorescein-labeled Annexin V and propidium iodide
(PI) for 20 min. Following the labeling reaction, the expression of
Annexin at the cell surface was analyzed by a FACSCalibur (Model
FACSC 420; BD Biosciences, Baltimore, MD, USA). Data were analyzed
using CellQuest software from BD Biosciences.
Determination of mitochondrial membrane
potential (MMP)
MMP was analyzed using the fluorescent dye,
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolycarbocyanine
iodide (JC-1), following the manufacturer’s instructions (KeyGen).
Briefly, the cells were plated in a 6-well culture plate. Following
treatment for 24 h, the cells were washed twice with PBS, harvested
and incubated with 20 nM JC-1 for 30 min in the dark. MMP was then
analyzed using a FACSCalibur, as described above.
Flow cytometric analysis of mitochondrial
permeability transition pore (mPTP) opening
The opening of mPTPs was determined by flow
cytometry (FCM), using the MitoProbe™ Transition Pore Assay kit
(Invitrogen, Carlsbad, CA, USA). Under normal conditions, the
non-fluorescent acetoxymethyl (AM) ester of calcein dye (calcein
AM) and cobalt enters the cell. The AM groups are cleaved from
calcein via non-specific esterase and calcein can then show
fluorescence signals in the cytosol and mitochondria. Cobalt
quenches the cytosolic calcein signal. However, cobalt cannot enter
healthy mitochondria freely and therefore cannot quench the
mitochondrial calcein signal. When the opening of mPTPs occurs,
cobalt enters through the pores and subsequently quenches the
mitochondrial calcein signal. FCM was used to detect the number of
cells that exhibited quenched calcein signals inside the
mitochondria. The location of the curves indicates the numbers of
such cells, which suggests the opening of mPTPs (25).
Quantification of cellular reactive
oxygen species (ROS)
The levels of cellular ROS were quantified according
to a previously described method (26). Briefly, the cells
(5×105) were cultured in 12-well tissue culture plates
overnight and then co-treated with drugs and
2′,7′-dichlorofluorescein diacetate (DCF-DA), a ROS-sensitive dye.
Following treatment with the drugs, the cells were harvested and
suspended in PBS. Relative fluorescence intensities of the cells
were quantified using a FACSCalibur, as described above.
Catalase (CAT) assay
CAT activity was assayed using the method developed
by Aebi (27) which is based on
the disappearance of hydrogen peroxide (H2O2)
at 240 nm. One unit was defined as 1 μmol of
H2O2 consumed per min, and the specific
activity was reported as U/mg/protein.
Superoxide dismutase (SOD) assay
The mitochondria and cytosol were fractionated using
the Mitochondria/Cytosol Fractionation kit (BioVision, Sheffield,
UK). The amount of proteins was determined using the Bio-Rad
Protein Assay (Bio-Rad, Hercules, CA, USA). SOD activity was
determined using the SOD Activity Assay kit (BioVision). The
relative SOD activity was normalized according to the protein
content and shown as a percentage of SOD activity present in the
control cells.
Measurement of adenosine triphosphate
(ATP) levels
According to the instructions provided by the
company, ATP assays were conducted using either the ATPlite Assay
(PerkinElmer, Waltham, MA, USA) or the ATP Determination kit
(Invitrogen).
Intracellular Ca2+
measurement
The Ca2+ fluorescence intensity in the
cells was also measured using a flow cytometer (Beckman Coulter,
Brea, CA, USA). The cells in 6-well plates were digested with 0.05%
trypsin-EDTA, incubated with 10 μM Fluo-4/AM ester for 30
min at 37°C, centrifuged and washed by PBS 3 times. The cells were
diluted to 5×105 cell suspension with fixation solution
for FCM. For each experiment experiment, 5,000 random cells were
selected by FCM to analyze the fluorescence intensity.
Measurement of caspase-3, -8 and -9
activities
Caspase activity was measured using a Colorimetric
Assay kit according to the manufacturer’s instructions. After
harvesting, the cells were washed in ice-cold PBS and lysed;
proteins were extracted and stored at −80°C until use. Cell lysate
(20 μl) was added to a buffer containing a p-nitroaniline
(pNA)-conjugated substrate (80 μl) for caspase-3
(Ac-DEVD-pNA; KGA203), -8 (Ac-IETD-pNA; KGA302), or -9 (LEHD-pNA;
KGA402; all from KeyGen). Incubation was performed at 37°C with
shaking (500 rpm for 1 min) and then at room temperature for 2 h.
The released pNA in each well was measured using a plate-reading
luminometer (Thermo Scientific, Beijing, China).
Immunoblotting
Protein extracts were analyzed by western blot
analysis using the antibodies listed in Table I. Cell extracts obtained in
Laemmli buffer were resolved on SDS-PAGE, followed by
electrotransfer onto nitrocellulose membranes. Following a blocking
step in 5% milk in Tween-TBS, the membranes were incubated with
primary and secondary antibodies. The membranes were then developed
and visualized by enhanced chemiluminescence (ECL) (Pierce
Antibodies; Thermo Fisher Scientific, Inc., Rockford, IL, USA).
 | Table IAntibodies used in western blot
analysis. |
Table I
Antibodies used in western blot
analysis.
| Protein | Producer | Catalog no. | Dilution |
|---|
| AKT | Santa Cruza | sc-377457 | 1:200 |
| p-AKT | Santa Cruz | sc-135650 | 1:200 |
| Bax | Santa Cruz | sc-7480 | 1:200 |
| Bcl-xL | Santa Cruz | sc-8392 | 1:200 |
| Bcl-2 | Santa Cruz | sc-783 | 1:200 |
| p-Bcl-2 | Santa Cruz | sc-16323 | 1:200 |
| Caspase-9 | Santa Cruz | sc-56076 | 1:200 |
| Caspase-3 | Santa Cruz | sc-136219 | 1:200 |
| β-actin | Santa Cruz | sc-47778 | 1:1,000 |
Statistical analysis
The statistical significance of the differences
between the control and drug-treated groups was evaluated using the
Student’s t-test and differences were considered statistically
significant at P-values <0.05. Analysis of the data was
performed using GraphPad Prism 4 software (GraphPad Software, Inc.,
San Diego, CA, USA).
Results
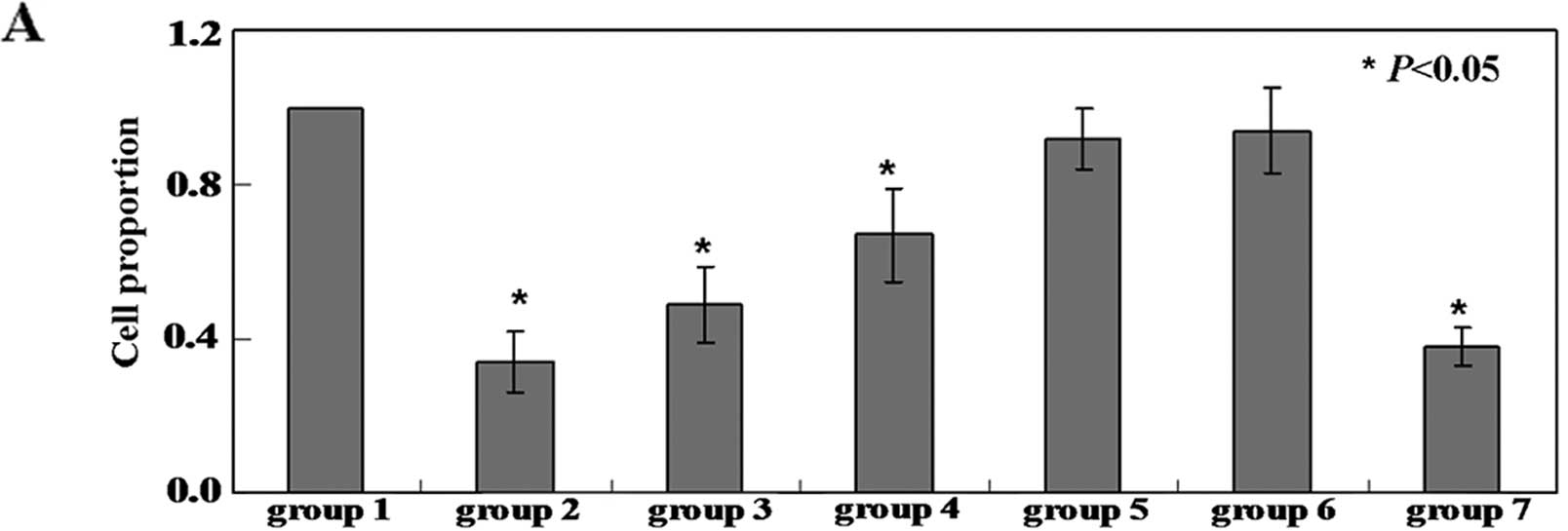
Isoflurane induces apoptosis in neuronal
cells
To determine whether isoflurane induces apoptosis in
neuronal cells, the cells were treated with 2% isoflurane for 6 h.
As shown in Fig. 1A, the
proliferation rate of the neuronal cells was inhibited by
isoflurane (P<0.05). The proliferation of the neuronal cells was
inhibited by isoflurane. The isoflurane-treated cells with damaged
DNA displayed high migration of DNA fragments from the nucleus,
forming a tail in comet form (Fig.
1B). TUNEL assay confirmed that the number of apoptotic cells
in the isoflurane-treated groups was significantly higher than that
in the untreated group (Fig. 1C).
To detect apoptotic cells quantitatively, Annexin V-FITC and PI
double staining was performed. In the cells treated with
isoflurane, the apoptotic ratio was 8–10-fold higher than that in
the untreated cells (P<0.05) (Fig.
1D). As caspase-3 activation is one of the final steps of
cellular apoptosis (28), we
assessed the effects of isoflurane on the activation of caspase-3,
-8 and -9 by colorimetric assay and western blot analysis. The
activity of caspase-3, -8 and -9 was significantly increased in the
isoflurane-treated cells compared with the untreated cells
(Figs. 1E and 3).
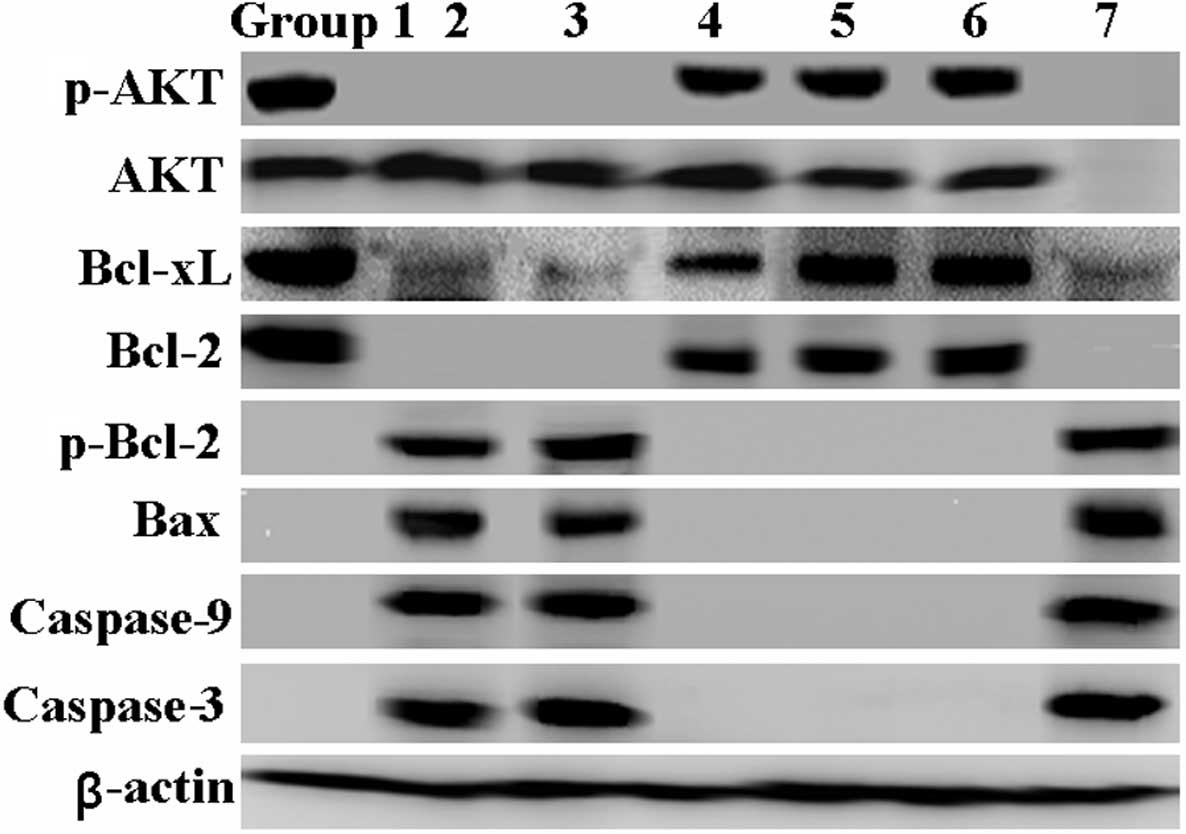 | Figure 3Effects of resveratrol and isoflurane
on mitochondrial apoptosis-related proteins. Cellular protein was
isolated from the neuronal cells following treatment for 24 h.
Western blot analysis was then performed to detect AKT, Bcl-xl,
Bcl-2, Bax, phosphorylated (p)-AKT, phosphorylated (p)-Bcl-2,
actived caspase-9 and caspase-3 proteins using respective specific
antibodies. β-actin was used as an internal control. Group 1,
control; group 2, 2% isoflurane for 6 h; group 3, RESV 50 μM
for 24 h + 2% isoflurane for 6 h; group 4, RESV 100 μM for
24 h + 2% isoflurane for 6 h; group 5, RESV 200 μM for 24 h
+ 2% isoflurane for 6 h; group 6, butin 10 μg/ml for 24 h +
2% isoflurane for 6 h; group 7, RESV 200 μM for 24 h + siRNA
AKT for 24 h + 2% isoflurane for 6 h. |
Isoflurane induces apoptosis in neuronal
cells by destroying the mitochondria
As shown in Fig.
2A, the ratio of red/green in the neuronal cells (2.3% green,
97.7% red) was reversed following treatment with isoflurane (73.8%
green, 26.2% red). The fluorescence emission shift from red to
green indicated the loss of membrane potential. The results
indicated that the isoflurane-induced apoptosis was associated with
the loss of MMP in the neuronal cells. Flow cytometric analysis of
calcein AM and cobalt illustrated that treatment with isoflurane
induced an increase the opening of mPTPs in the neuronal cells
(Fig. 2B, peak 2) compared with
the untreated cells (Fig. 2B,
peak 1), as evidenced by the right shift of the curve. We used the
fluorescent dye, DCF-DA, to measure the ROS content in the neuronal
cells following treatment with isoflurane. As shown in Fig. 2C, isoflurane directly induced an
increase in fluorescence intensity in the neuronal cells (48.6%) as
compared with the untreated cells (16.2%; P<0.05). Furthermore,
we evaluated the antioxidant enzyme activity of CAT and SOD and
found that the levels of CAT and SOD were significantly decreased
in the isoflurane-treated neuronal cells (P<0.05) (Fig. 2D and E). We then examined the
cellular ATP levels of the isoflurane-treated neuronal cells and
the untreated cells using an ATP-based luminescent assay. The
untreated cells had 2-fold higher total cellular ATP levels
compared with the isoflurane-treated cells (P<0.05) (Fig. 2F). Furthermore, we observed a
decrease in the intracellular calcium ion concentration in the
isoflurane-treated neuronal cells (Fig. 2G).
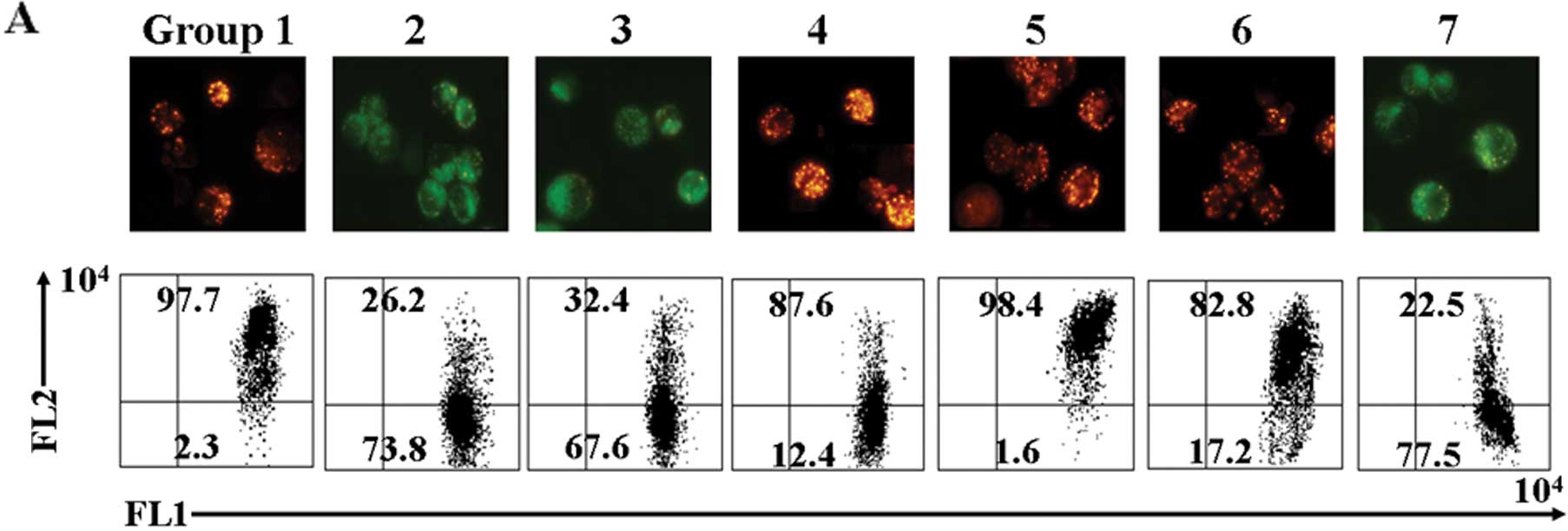 | Figure 2Mitochondrial changes in neuronal
cells following treatment with resveratrol (RESV) and isoflurane.
(A) Following treatment, cells were plated in 6-well culture plates
for 24 h. Mitochondrial membrane potential was then analyzed by
immunofluorescence and flow cytometry. (B) Flow cytometric analysis
revealed changes in calcein levels in the mitochondria of neuronal
cells stained with calcein acetoxymethyl (AM) ester or calcein AM
plus cobalt, which indicates the opening of mitochondrial
permeability transition pores. (C) Cells were dispensed in a 10-cm
culture dish at a density of 1×106 cells/well and
treated as described in ‘Materials and methods’. The DCF-positive
cells (reactive oxygen species production) were then detected using
a FL1 signal detector (525 nm) using a FACSCalibur. (D) Catalase
activity was assayed based on the disappearance of
H2O2 at 240 nm. (E) Mitochondrial and
cytosolic fractions were isolated as described in ‘Materials and
methods’. The superoxide dismutase enzymatic activity in each
fraction was measured and normalized as a ratio relative to the
control activity. (F) ATP levels were measured in the indicated
cells at 24 h using the ATP Determination kit. (G) Following
treatment, cells were labeled with the fluorescent probe Fluo-4-AM.
Ca2+ levels were detected by flow cytometry. Group 1,
control; group 2, 2% isoflurane for 6 h; group 3, RESV 50 μM
for 24 h + 2% isoflurane for 6 h; group 4, RESV 100 μM for
24 h + 2% isoflurane for 6 h; group 5, RESV 200 μM for 24 h
+ 2% isoflurane for 6 h; group 6, butin 10 μg/ml for 24 h +
2% isoflurane for 6 h; group 7, RESV 200 μM for 24 h + siRNA
AKT for 24 h + 2% isoflurane for 6 h. |
RESV protects neuronal cells against
isoflurane-induced apoptosis
Following treatment with isoflurane, the neuronal
cells were cultured in the presence of increasing concentrations of
RESV (50, 100 and 200 μM) for 24 h. This led to an increase
in cell viability in a concentration-dependent manner (Fig. 1A). Co-treatment of the neuronal
cells with isoflurane and RESV reduced isoflurane-induced cell
death in a concentration-dependent manner, as evidenced by comet
assay and TUNEL assay (Fig. 1B and
C). Subsequently, we investigated the changes in plasma
membrane asymmetry (using Annexin V and PI double staining) to
quantify the population of dead cells. In the cells treated with
RESV and isoflurane, the apoptotic ratio was 1.5-6-fold lower than
that of the cells treated with isoflurane alone (P<0.05)
(Fig. 1D). The activity of
caspase-3, -8 and -9 was significantly decreased in the RESV- and
isoflurane-treated cells compared with the isoflurane-treated cells
(Figs. 1E and 3). Hence, we further investigated the
effects of RESV on isoflurane-induced changes in mitochondrial
function and morphology. Co-treatment with RESV almost completely
prevented the isoflurane-induced loss of MMP (Fig. 2A). We also found that treatment
with RESV led to a decrease in the isoflurane-induced opening of
mPTPs (Fig. 2B, peak 3–5). When
DCF-DA was used as a ROS-sensitive fluorescence indicator, the
accumulation of ROS in the neuronal cells treated with isoflurane
was markedly downregulated by RESV (P<0.05) (Fig. 2C). The presence of RESV abrogated
the decreased levels of CAT and SOD in the isoflurane-treated
neuronal cells (P<0.05) (Fig. 2D
and E). As expected, RESV also increased ATP levels and the
intracellular calcium ion concentration in the isoflurane-treated
neuronal cells (P<0.05) (Fig. 2F
and G). Butin was used as a positive control (Figs. 1 and 2).
Role of the Akt signaling pathway in
mediating the protective effects of RESV against isoflurane-induced
cytotoxicity
In the present study, we examined the roles of
related signaling molecules in mediating isoflurane-induced
neuronal death and the protective effects of RESV in neuronal cells
by western blot analysis. As shown in Fig. 3, p-AKT was downregulated by
isoflurane and the levels of total AKT showed no changes. However,
p-AKT levels were restored following treatment with RESV. By
contrast, the protective effects of RESV were markedly diminished
by specific siRNA targeting AKT (Figs. 1 and 2). In addition, we found that butin
inhibited the activation of the mitochondrial-dependent apoptotic
pathway induced by isoflurane.
Discussion
The results of the present study demonstrate that
RESV effectively protects neuronal cells from isoflurane-induced
cytotoxicity by activating the Akt signaling pathway. Anesthesia
has been associated with widespread apoptotic neurodegeneration in
the neonatal rat brain with persistent functional neurocognitive
impairment, exemplified by impaired memory formation (29,30). Isoflurane, a halogenated volatile
anesthetic, is frequently used in pediatric general anesthesia and
is particularly useful for maintaining a surgical plane of
anesthesia for several hours (10). Isoflurane has been shown to induce
widespread cerebral neuroapoptosis in neonatal rat pups with
subsequent long-term neurocognitive impairment of the animals
(31). A previous study also
showed that the common inhalation anesthetic, isoflurane, may
induce neurotoxicity in vitro (32). In the present study, we
successfully established a model of isoflurane-induced apoptosis
using neuronal cells, as evidenced by the activation of caspase-3
and caspase-9.
Although the underlying molecular mechanisms of
neurotoxicity are not yet completely understood, mitochondrial
dysfunction, altered calcium homeostasis and apoptosis-related
proteins have been implicated. A previous study demonstrated that
isoflurane induces the release of calcium from the endoplasmic
reticulum (ER) in cerebrocortical and hippocampal neurons (33). In this study, we confirmed a
decrease in the intracellular calcium ion concentration in
isoflurane-treated neuronal cells. These findings suggest that
isoflurane induces cellular apoptosis by facilitating the release
of calcium from cells. Wei et al (34) reported that isoflurane induced
cytotoxicity, which was characterized by nuclear condensation and
fragmentation and the activation of caspase-3 and -9, by affecting
the Bcl-2/Bax ratio. We also observed changes in the levels of
Bcl-2 and Bax in neuronal cells following treatment with
isoflurane.
RESV has gained considerable attention due to its
potential cancer chemopreventive and anticancer properties
(35). In addition, RESV has the
potential to control atherosclerosis, heart disease, arthritis and
autoimmune disorders (36). RESV
scavenges superoxide anions generated from the rat forebrain
mitochondria in a concentration dependent manner (37). In the present study, we also
confirmed treatment with RESV reversed the production of ROS
scavenged ROS that were produced. While SOD contributes to reducing
the burden of intracellular ROS, previous studies have shown that
SOD plays an important role in neuronal cells against
oxidant-induced mitochondrial oxidative stress and cytotoxicity
(38,39). Consistent with the results from
previous studies, our results confirmed that RESV increased the
levels of CAT and SOD in the isoflurane-treated neuronal cells.
Earlier studies have shown that RESV alters the
activity of PI3K/Akt signaling molecules (40–44), which are regulated by
phosphorylation. We found that the treatment of neuronal cells with
RESV alone or RESV plus isoflurane resulted in Akt activation. The
protective effects of RESV were markedly diminished by specific
siRNA targeting AKT. These results indicate that RESV effectively
protects neuronal cells from isoflurane-induced cytotoxicity by
activating the Akt signaling pathway.
In conclusion, the present study provides strong
evidence that RESV positively controls neurotoxicity triggered by
isoflurane in vitro. Experiments using cell cultures
revealed that RESV acted, at least in part, by activating the Akt
signaling pathway. These findings provide further support for
current clinical trials aimed at assessing the beneficial effects
of RESV administration against isoflurane-induced
neurodegeneration.
Acknowledgements
We thank Dr Li Ming-Yu for carefully proofreading
the manuscript and providing valuable comments.
References
|
1
|
Moonesinghe SR, Mythen MG and Grocott MP:
High-risk surgery: epidemiology and outcomes. Anesth Analg.
112:891–901. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Weiser TG, Regenbogen SE, Thompson KD, et
al: An estimation of the global volume of surgery: a modelling
strategy based on available data. Lancet. 372:139–144.
2008.PubMed/NCBI
|
|
3
|
Wei H, Liang G, Yang H, et al: The common
inhalational anesthetic isoflurane induces apoptosis via activation
of inositol 1,4,5-trisphosphate receptors. Anesthesiology.
108:251–260. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Loop T and Priebe HJ: Costs of
anaesthesia. Eur J Anaesthesiol. 22:1622005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fournier N, Ducet G and Crevat A: Action
of cyclosporine on mitochondrial calcium fluxes. J Bioenerg
Biomembr. 19:297–303. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jevtovic-Todorovic V, Hartman RE, Izumi Y,
et al: Early exposure to common anesthetic agents causes widespread
neurodegeneration in the developing rat brain and persistent
learning deficits. J Neurosci. 23:876–882. 2003.
|
|
7
|
Wang S, Peretich K, Zhao Y, et al:
Anesthesia-induced neurodegeneration in fetal rat brains. Pediatr
Res. 66:435–440. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Winegar BD and Yost CS: Volatile
anesthetics directly activate baseline S K+ channels in
Aplysia neurons. Brain Res. 807:255–262. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Talley EM and Bayliss DA: Modulation of
TASK-1 (Kcnk3) and TASK-3 (Kcnk9) potassium channels: volatile
anesthetics and neurotransmitters share a molecular site of action.
J Biol Chem. 277:17733–17742. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Franks NP: Molecular targets underlying
general anesthesia. Br J Pharmacol. 147(Suppl 1): S72–S81. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Loepke AW, McCann JC, Kurth C and
McAuliffe JJ: The physiologic effects of isoflurane anesthesia in
neonatal mice. Anesth Analg. 102:75–80. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fulda S, Galluzzi L and Kroemer G:
Targeting mitochondria for cancer therapy. Nat Rev Drug Discov.
9:447–464. 2010. View
Article : Google Scholar
|
|
13
|
Csiszar A, Labinskyy N, Pinto JT, et al:
Resveratrol induces mitochondrial biogenesis in endothelial cells.
Am J Physiol Heart Circ Physiol. 297:H13–H20. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pervaiz S: Resveratrol: from grapevines to
mammalian biology. FASEB J. 17:1975–1985. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Frémont L: Biological effects of
resveratrol. Life Sci. 66:663–673. 2000.
|
|
16
|
Wang Q, Xu J, Rottinghaus GE, et al:
Resveratrol protects against global cerebral ischemic injury in
gerbils. Brain Res. 958:439–447. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
West T, Atzeva M and Holtzman DM:
Pomegranate polyphenols and resveratrol protect the neonatal brain
against hypoxic-ischemic injury. Dev Neurosci. 29:363–372. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ates O, Cayli S, Altinoz E, et al: Effects
of resveratrol and methylprednisolone on biochemical,
neurobehavioral and histopathological recovery after experimental
spinal cord injury. Acta Pharmacol Sin. 27:1317–1325. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kaplan S, Bisleri G, Morgan JA, et al:
Resveratrol, a natural red wine polyphenol, reduces
ischemia-reperfusion-induced spinal cord injury. Ann Thorac Surg.
80:2242–2249. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Parker JA, Arango M, Abderrahmane S, et
al: Resveratrol rescues mutant polyglutamine cytotoxicity in
nematode and mammalian neurons. Nat Genet. 37:349–350. 2005.
View Article : Google Scholar
|
|
21
|
Markaryan A, Nelson EG, Tretiakova M and
Hinojosa R: Technical report: immunofluorescence and TUNEL staining
of celloidin embedded human temporal bone tissues. Hear Res.
241:1–6. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hurst PR, Mora JM and Fenwick MA:
Caspase-3, TUNEL and ultrastructural studies of small follicles in
adult human ovarian biopsies. Hum Reprod. 21:1974–1980. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rajagopalan R, Ranjan S and Nair CK:
Effect of vinblastine sulfate on gamma-radiation-induced DNA
single-strand breaks in murine tissues. Mutat Res. 536:15–25. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Singh NP: Microgels for estimation of DNA
strand breaks, DNA protein crosslinks and apoptosis. Mutat Res.
455:111–127. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang Y, Dong Y, Xu Z and Xie Z: Propofol
and magnesium attenuate isoflurane-induced caspase-3 activation via
inhibiting mitochondrial permeability transition pore. Med Gas Res.
2:202012. View Article : Google Scholar
|
|
26
|
Chang HC, Lin KH, Tai YT, et al:
Lipoteichoic acid-induced TNF-α and IL-6 gene expressions and
oxidative stress production in macrophages are suppressed by
ketamine through downregulating Toll-like receptor 2-mediated
activation of ERK1/2 and NFκB. Shock. 33:485–492. 2010.
|
|
27
|
Aebi H: Catalase in vitro. Methods
Enzymol. 105:121–126. 1984. View Article : Google Scholar
|
|
28
|
Thornberry NA: Caspases: key mediators of
apoptosis. Chem Biol. 5:R97–R103. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hansen HH, Briem T, Dzietko M, et al:
Mechanisms leading to disseminated apoptosis following NMDA
receptor blockade in the developing rat brain. Neurobiol Dis.
16:440–453. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lu LX, Yon JH, Carter LB and
Jevtovic-Todorovic V: General anesthesia activates BDNF-dependent
neuroapoptosis in the developing rat brain. Apoptosis.
11:1603–1615. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Quinn JJ, Loya F, Ma QD and Fanselow MS:
Dorsal hippocampus NMDA receptors differentially mediate trace and
contextual fear conditioning. Hippocampus. 15:665–674. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xie Z, Dong Y, Maeda U, et al: The common
inhalation anesthetic isoflurane induces apoptosis and increases
amyloid beta protein levels. Anesthesiology. 104:988–994. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kindler CH, Eilers H, Donohoe P, et al:
Volatile anesthetics increase intracellular calcium in
cerebrocortical and hippocampal neurons. Anesthesiology.
90:1137–1145. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wei H, Kang B, Wei W, et al: Isoflurane
and sevoflurane affect cell survival and BCL-2/BAX ratio
differently. Brain Res. 1037:139–147. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Baur JA and Sinclair DA: Therapeutic
potential of resveratrol: the in vivo evidence. Nat Rev Drug
Discov. 5:493–506. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yoshida Y, Shioi T and Izumi T:
Resveratrol ameliorates experimental autoimmune myocarditis. Circ
J. 71:397–404. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zini R, Morin C, Bertelli A, et al:
Effects of resveratrol on the rat brain respiratory chain. Drugs
Exp Clin Res. 25:87–97. 1999.PubMed/NCBI
|
|
38
|
Ookawara T, Imazeki N, Matsubata O, et al:
Tissue distribution of immunoreactive mouse extracellular
superoxide dismutase. Am J Physiol. 275:840–847. 1998.PubMed/NCBI
|
|
39
|
Hinerfeld D, Traini MD, Weinberger RP, et
al: Endogenous mitochondrial oxidative stress: neurodegeneration,
proteomic analysis, specific respiratory chain defects, and
efficacious antioxidant therapy in superoxide dismutase 2 null
mice. J Neurochem. 88:657–667. 2004. View Article : Google Scholar
|
|
40
|
Aziz MH, Nihal M, Fu VX, et al:
Resveratrol-caused apoptosis of human prostate carcinoma LNCaP
cells in mediated via modulation of phosphatidilinositol
3′-kinase/Akt pathway and Bcl-2 family proteins. Mol Cancer Ther.
5:1335–1341. 2006.PubMed/NCBI
|
|
41
|
Pozo-Guisado E, Merino JM, Mulero-Navarro
S, et al: Resveratrol-induced apoptosis in MCF-7 human breast
cancer cells involves a caspase-independent mechanism with
downregulation of Bcl-2 and NF-kappaB. Int J Cancer. 115:74–84.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Alkhalaf M: Resveratrol-induced growth
inhibition in MDA-MB-231 breast cancer cells is associated with
mitogen-activated protein kinase signaling and protein translation.
Eur J Cancer Pre. 16:334–341. 2007. View Article : Google Scholar
|
|
43
|
She QB, Bode AM, Ma WY, et al:
Resveratrol-induced activation of p53 and apoptosis is mediated by
extracellular-signal-regulated protein kinase and p38 kinase.
Cancer Res. 61:1604–1610. 2001.PubMed/NCBI
|
|
44
|
She QB, Huang C, Zhang Y and Dong Z:
Involvement of c-jun NH(2)-terminal kinases in resveratrol-induced
activation of p53 and apoptosis. Mol Carcinog. 33:244–250. 2002.
View Article : Google Scholar : PubMed/NCBI
|