Introduction
The molecular chaperone heat shock protein 90
(Hsp90) was initially identified as one of the highly conserved
heat shock proteins (1,2). The Hsp90 proteins are ubiquitous
molecular chaperones that have important functions in protein
folding and in renaturation (3,4).
They also play a key role in the stability and function of a number
of client proteins. To date, >200 client proteins are known to
be regulated by Hsp90, including mutated and oncogenic regulatory
and signaling proteins (ErbB2, Akt, Raf-1, mutated p53), cell cycle
regulators (Cdk4, CdK6) and steroid receptors (5). A number of these proteins are
mutated and/or overexpressed in cancers, indicating that the client
proteins are directly associated with oncogenesis upon Hsp90
machinery. For this reason, Hsp90 has emerged as a promising target
in the development of cancer therapeutics.
ATP-competitive Hsp90 inhibitors, including the
benzoquinone ansamycin, 17-allylamino-17-demethoxygeldanamycin
(17-AAG) and its derivatives,
17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG) and
IPI-504, have been developed as antitumor agents (6). We, as well as others have previously
reported that the benzoquinone ansamycins, geldanamycin and 17-AAG,
sensitize tumor cells, but not normal cells, to radiation (7–10).
The mechanisms responsible for the enhancement of the radiation
response in tumor cells have been suggested to induce apoptosis by
inhibiting cell survival signals. We have also previously
demonstrated that 17-AAG inhibits the repair of radiation-induced
DNA double-strand breaks (DSBs) in tumor cell lines, but not in
normal cells (11).
The ansamycin, 17-AAG, was the first Hsp90 inhibitor
to enter clinical trials. In a phase I trial of 17-AAG in patients
with advanced cancer, early clinical results revealed delayed
hepatotoxicity with a twice-weekly continuous dosing schedule
(12). In addition, its limited
solubility and stability have led to the development of derivative
compounds, such as 17-DMAG and IPI-504, which have been
investigated for the treatment of different malignancies. However,
side-effects remain a concern with these newer ansamycins (13). These results prompted the
development of a non-ansamycin Hsp90 inhibitor. Chiosis et
al designed Hsp90 inhibitors using purine as a scaffold, of
which
8-(6-indozenzo[d][1,3]dioxol-5-ylthio)-9-(3-(isopropylamino)propyl)-9H-purine-6-amine
(PU-H71) is the most potent (14). PU-H71 has shown anticancer
activity in pre-clinical trials on triple-negative breast cancer
(15), lymphoma (16) and hepatocellular carcinoma
(17), suggesting that it is a
promising drug for the treatment of cancer.
In this study, we demonstrate that PU-H71 sensitizes
the human lung cancer cell lines, SQ-5 and A549, to radiation. As
shown by our results, PU-H71 inhibited the repair of
radiation-induced DSBs in both cancer cell lines, and significantly
disturbed the homologous recombination (HR) pathway, one of the DSB
repair pathways (18). Therefore,
our data demonstrate that the inhibition of Hsp90 by PU-H71
represents an effective method to enhance the sensitivity of cancer
cells to radiotherapy.
Materials and methods
Cell lines, culture and drugs
The human lung cancer cell lines, SQ-5 and A549, and
the normal fibroblast cell line, HFL-III, were used in this study.
The cells were cultured in α-MEM supplemented with 10% fetal calf
serum, 20 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid
(HEPES), 8 mM NaHCO3, 50 U/ml penicillin and 50 μg/ml
streptomycin. The cells were cultured in a humidified incubator at
37°C with a mixture of 98% air and 2% CO2. PU-H71 was
purchased from Sigma-Aldrich (St. Louis, MO, USA), stored in
aliquots at −20°C at 2 mM solution in DMSO, and diluted in α-MEM
immediately prior to use.
Irradiation
The cells were irradiated at room temperature with
10 MV X-rays from a linear accelerator (EXL-15SP, Mitsubishi
Electric, Tokyo, Japan) at a dose rate of 4 Gy/min. Doses were
measured using an Innax Dosemaster 2590 (NE Technologies, Atlanta,
GA, USA).
Cell survival
Cell survival was measured by a colony formation
assay, as previously described (8). Actively growing cells in 24
cm2 flasks were incubated with 1 μM PU-H71 for 16 h at
37°C, irradiated with X-rays and incubated in the presence of the
drug for a further 8 h. Following treatment with PU-H71 for 24 h,
the cells were washed with Dulbecco’s phosphate-buffered saline
(PBS) and dispersed with 0.05% trypsin containing 0.02% EDTA,
counted, diluted and seeded in 60-mm or 100-mm dishes at various
cell densities. Following 12–14 days of incubation in a
CO2 incubator, the colonies were stained with crystal
violet dissolved in 20% methanol. Colonies of >50 cells were
counted as survivors.
Immunofluorescence
Immunofluorescence was performed as previously
described (19). Briefly, the
cells were grown on glass slides placed in 100-mm dishes and
exposed to PU-H71 at 1 μM or DMSO for 16 h, irradiated with X-rays
of 5 Gy, and incubated in the presence of the drug for a further 8
h at 37°C. The medium was then removed and replaced with fresh
drug-free medium. At various time points until 24 h after X-ray
irradiation, the medium was removed, and the cells were fixed with
cold methanol for 20 min followed by acetone for 5–10 sec,
air-dried and blocked with 10% bovine serum albumin (BSA) in PBS.
The cells were then washed twice with PBS and incubated with
primary antibodies for 1 h. The primary antibody used for
immunostaining was anti-phospho-H2AX (Ser139) (Merck Millipore,
Darmstadt, Germany) and Rad51 (GeneTex, Inc. Irvine, CA, USA). The
cells were again washed twice with PBS and incubated with the
secondary antibody (Alexa Fluor 488 goat anti-mouse IgG; Life
Technologies, Carlsbad, CA, USA) in PBS with 1% BSA, and washed
twice again. Subsequently, the cells were incubated with
4′,6-diamidino-2-phenylindole (DAPI) in PBS for 5 min and again
washed twice. Cover glasses were mounted and fluorescence images
were captured using a fluorescence microscope (Olympus, Tokyo,
Japan). The foci of γ-H2AX and Rad51 were counted in at least 50
cells.
Immunoblotting
The cells were exposed to PU-H71 or DMSO for 16 h,
irradiated with 5 Gy X-rays, and incubated in the presence of the
drug for a further 8 h. The cells were washed with ice-cold PBS,
collected and pelleted by centrifugation. The cells were lysed in
lysis buffer (Cell Signaling Technology, Danvers, MA, USA)
containing 1 mM phenylmethylsulfonyl fluoride (PMSF) for 30 min on
ice. The cell lysates were centrifuged at 15,000 rpm for 10 min at
4°C and the supernatant was recovered. The protein concentration
was measured using a BCA protein assay kit (Thermo Scientific,
Rockford, IL, USA). Lysates of 20 μg protein were boiled in sample
buffer and separated by electrophoresis on 7.5% polyacrylamide gel,
and electrotransferred onto polyvinylidene difluoride (PVDF)
membranes using a semi-dry transfer system. The membranes were
blocked in 5% non-fat milk for 1 h at room temperature, and
incubated overnight at 4°C with primary antibody. Then, membranes
were washed 3 times with PBS-T and incubated with a secondary
antibody conjugated to horseradish peroxidase for 1 h. After
washing 3 times, immunoblots were visualized by an enhanced
chemiluminescence (ECL) detection system (GE Healthcare, Fairfield,
CT, USA).
Detection of apoptosis
The induction of apoptosis was measured by detecting
the number of apoptotic bodies (8). The cells were incubated with PU-H71
at 1 μM or DMSO for 16 h, irradiated with 6 Gy X-rays, and then
incubated in the presence of the drug for a further 8 h. The medium
was removed and replaced with fresh drug-free medium. Twenty-four
hours after X-ray irradiation, both attached and floating cells
were collected by trypsinization and centrifugation, resuspended in
a fixed solution containing 3% paraformaldehyde in PBS and stained
with DAPI. The cells were placed on microscope slides and covered
with glass coverslips. The cells were then photographed using a
fluorescence microscope, and the numbers of apoptotic and
non-apoptotic cells were counted.
Results
PU-H71 inhibits the proliferation of
cancer cells
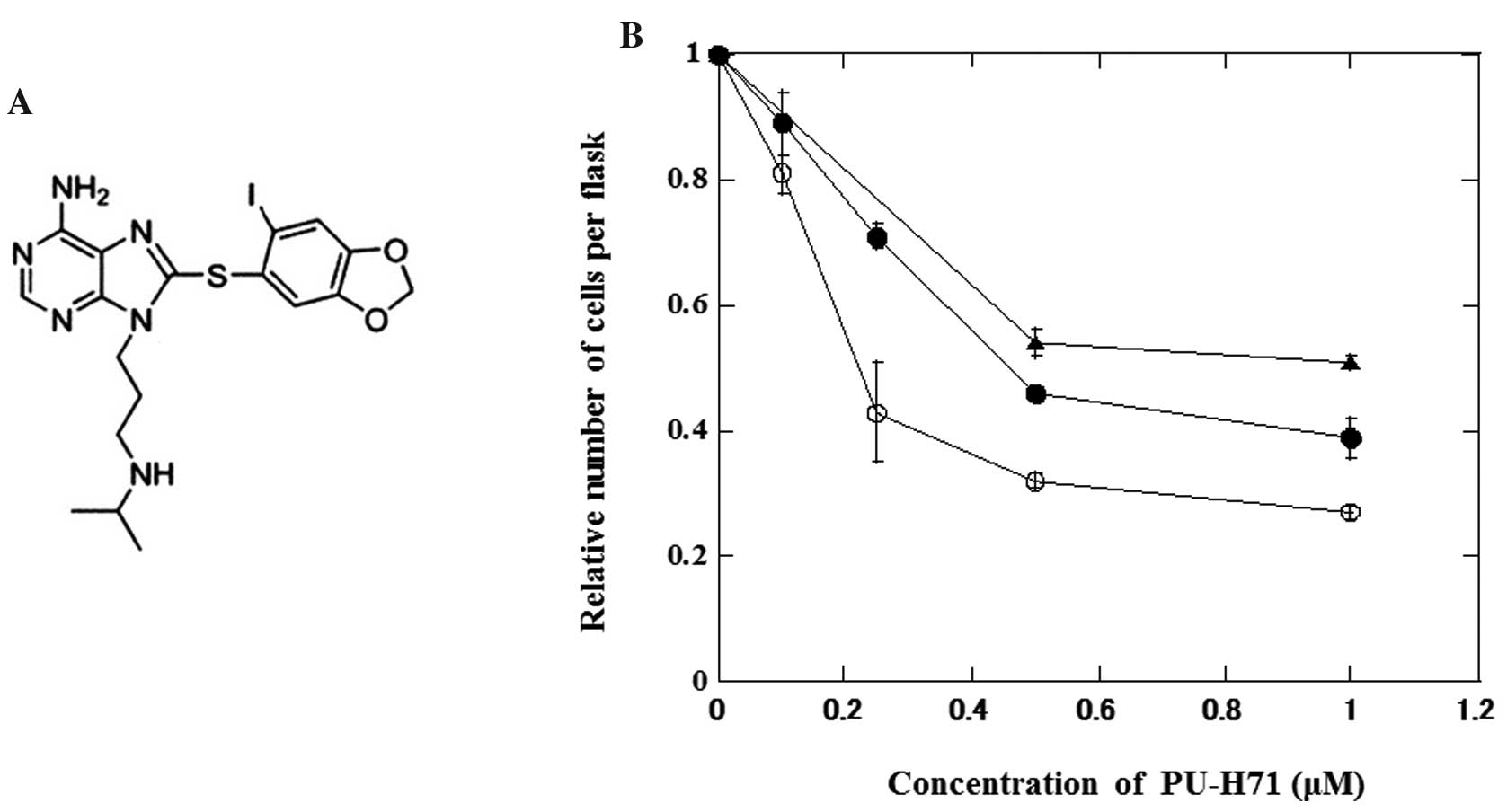
The effects of PU-H71 (Fig. 1A) on the growth of lung cancer and
normal fibroblasts are shown in Fig.
1B. Actively growing cells were treated with PU-H71 at various
concentrations for 24 h. PU-H71 inhibited the growth of cancer
cells more effectively than that of normal cells.
PU-H71 sensitizes cancer cells to
radiation
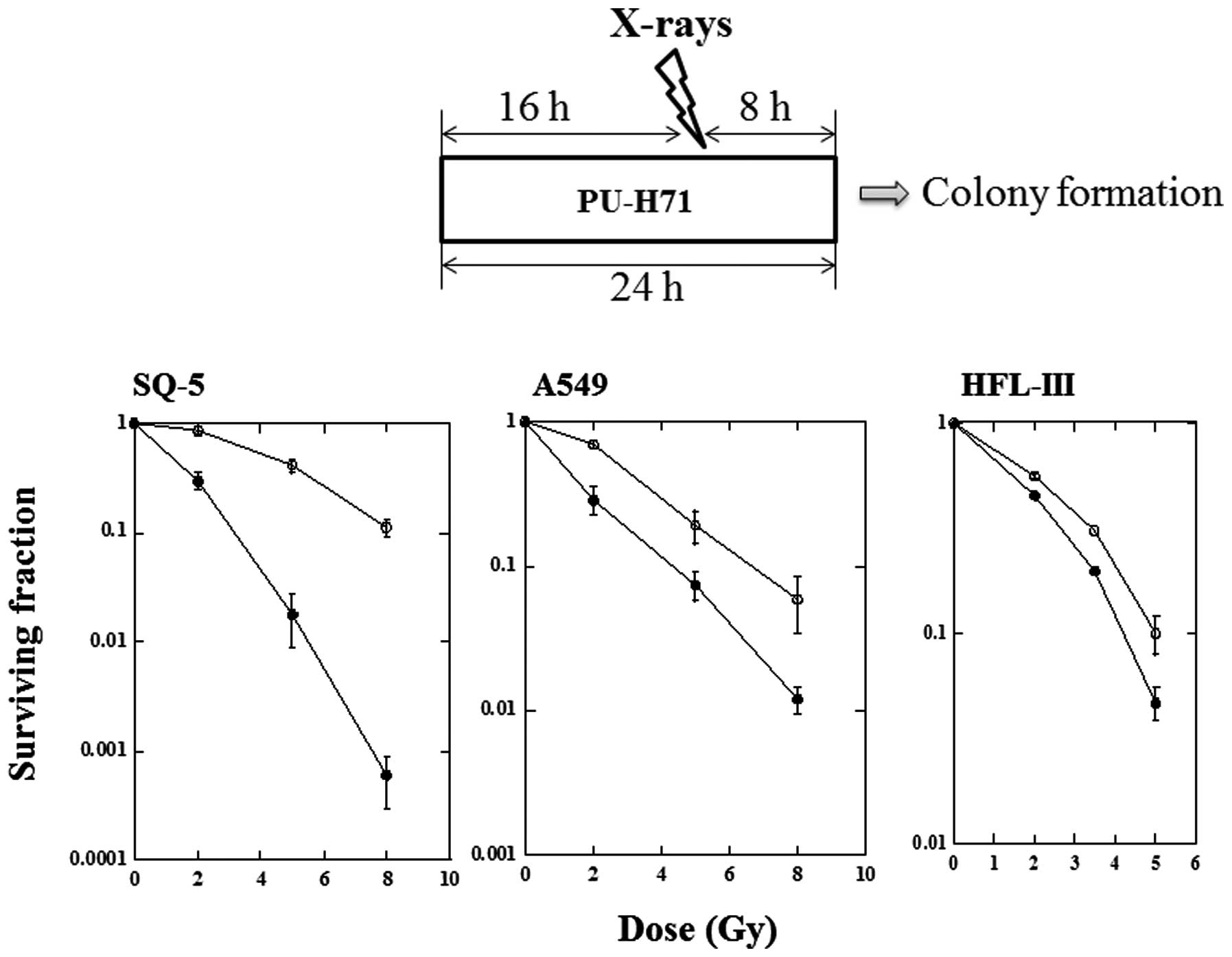
The ability of PU-H71 to enhance the sensitivity of
the SQ-5, A549 and HFL-III cells to radiation in vitro was
assessed by a clonogenic assay (Fig.
2). Cell survival curves for SQ-5, A549 and HFL-III cells after
X-irradiation in the presence or absence of PU-H71 were
constructed. Cells were exposed to PU-H71 at 1 μM for 16 h,
irradiated with X-rays and incubated for a further 8 h in the
presence of the drug. Each radiation survival curve after a
combination of X-rays and PU-H71 was corrected for drug
cytotoxicity. Irradiation in combination with PU-H71 had a strong
radiosensitizing effect on the SQ-5 cells. In the A549 cells, a
moderate sensitizing effect of PU-H71 was observed. The HFL-III
cells showed a lesser degree of radiosensitization. The
radiosensitivity enhancement ratios at a survival rate of 10% were
2.5, 1.5 and 1.2 in the SQ-5, A549 and HFL-III cells,
respectively.
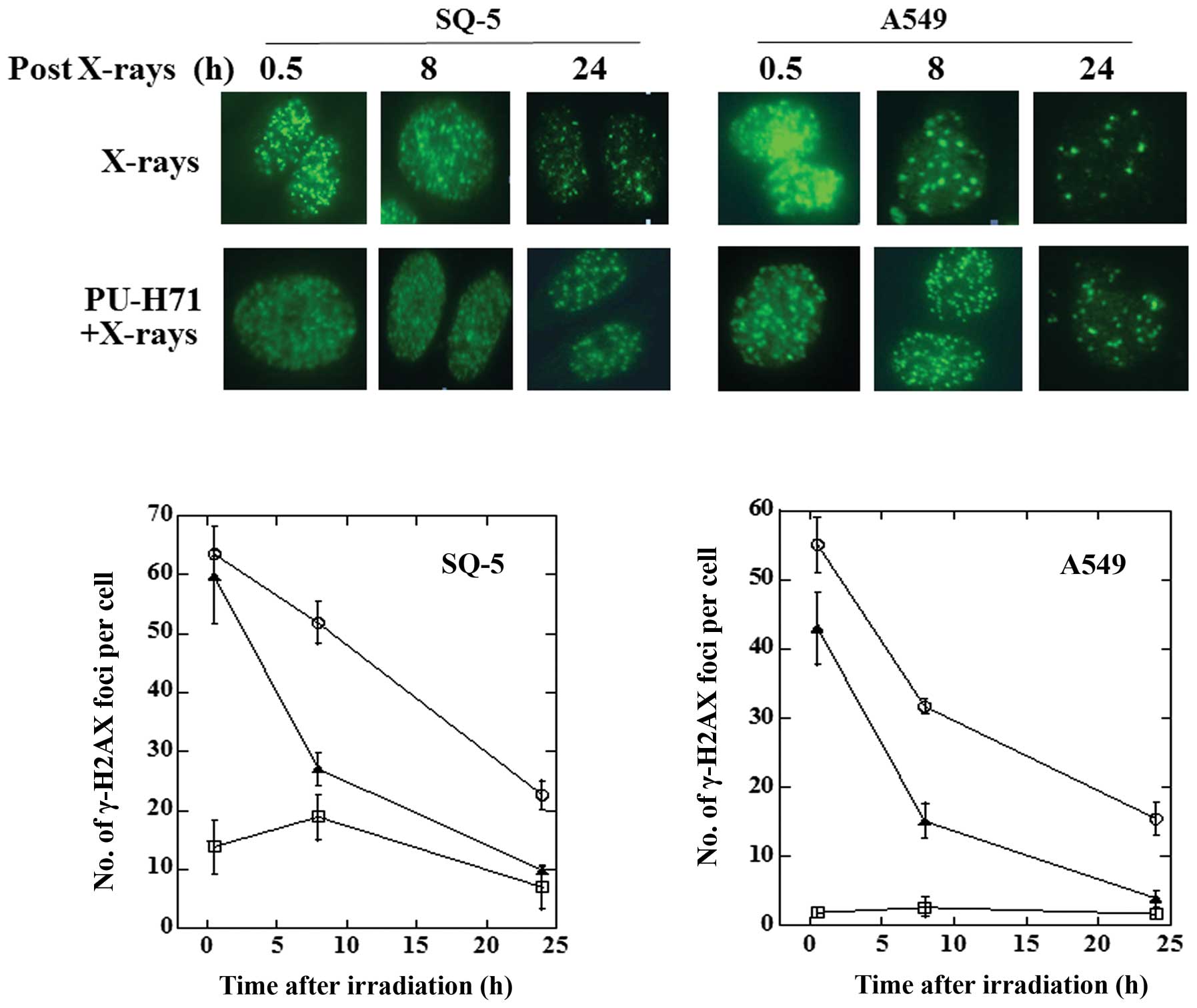
PU-H71 inhibits radiation-induced DNA
DSBs in cancer cells
With respect to radiation-induced cell death, DSBs
are believed to be the most severe damage caused to cells and, if
not repaired, can lead to genomic instability and cell death. To
elucidate the molecular mechanisms behind the radiosensitizing
effects of PU-H71 on cancer cells, we measured the repair of DSBs
by evaluating the foci of phosphorylated histone H2AX (γ-H2AX), an
indicator of DSBs. The results are presented in Fig. 3. In the SQ-5 cells, the number of
foci at 30 min after X-rays of 5 Gy alone was almost identical to
that following the combination of PU-H71 and X-rays. In the A549
cells, there were slightly more foci at 30 min following the
combination of PU-H71 and X-rays compared with X-rays alone. In
both cell lines, the number of foci in the cells exposed to both
PU-H71 and X-rays was significantly higher compared with X-rays
alone at 8 and 24 h, indicating that PU-H71 inhibits the repair of
DSBs in cancer cells. Our observations suggest that PU-H71
radiosensitizes cancer cells by inhibiting the repair of
radiation-induced DSBs.
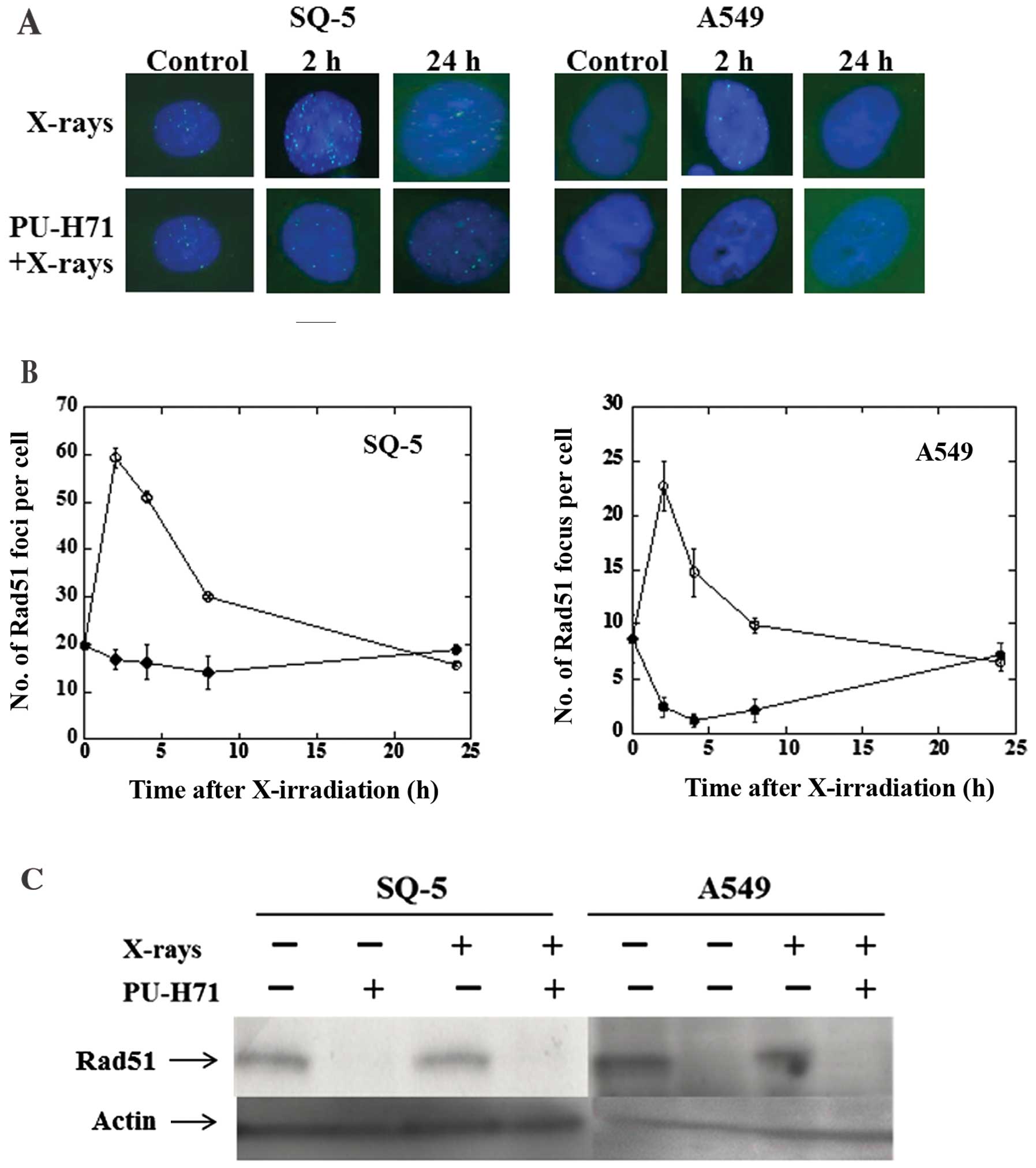
PU-H71 inhibits radiation-induced Rad51
foci formation in tumor cells
In response to ionizing radiation, the Rad51
protein, which is essential for the HR pathway of DSB repair,
relocalizes within the nucleus to form distinct foci that can be
visualized by fluorescence microscopy and that are thought to
represent sites where DSB repair reactions occur (20). To investigate the mechanisms by
which PU-H71 inhibits the repair of radiation-induced DSBs, we
examined its effects at 1 μM on radiation-induced Rad51 foci
formation in SQ-5 and A549 cells (Fig. 4A and B). A distinct difference was
observed between the effect of X-rays alone and the combination of
PU-H71 treatment and radiation in the cancer cell lines. Upon
treatment with X-ray irradiation alone, the formation of Rad51
nuclear foci was observed at 2 h, and then gradually decreased.
However, in the presence of PU-H71, there was little Rad51 foci
formation in response to radiation. We then investigated the
expression levels of the Rad51 protein in both cell lines treated
with PU-H71. Rad51 protein levels were decreased following
treatment with PU-H71 at 1 μM for 24 h (Fig. 4C). These results reveal that
PU-H71 exerts a marked effect on the HR pathway.
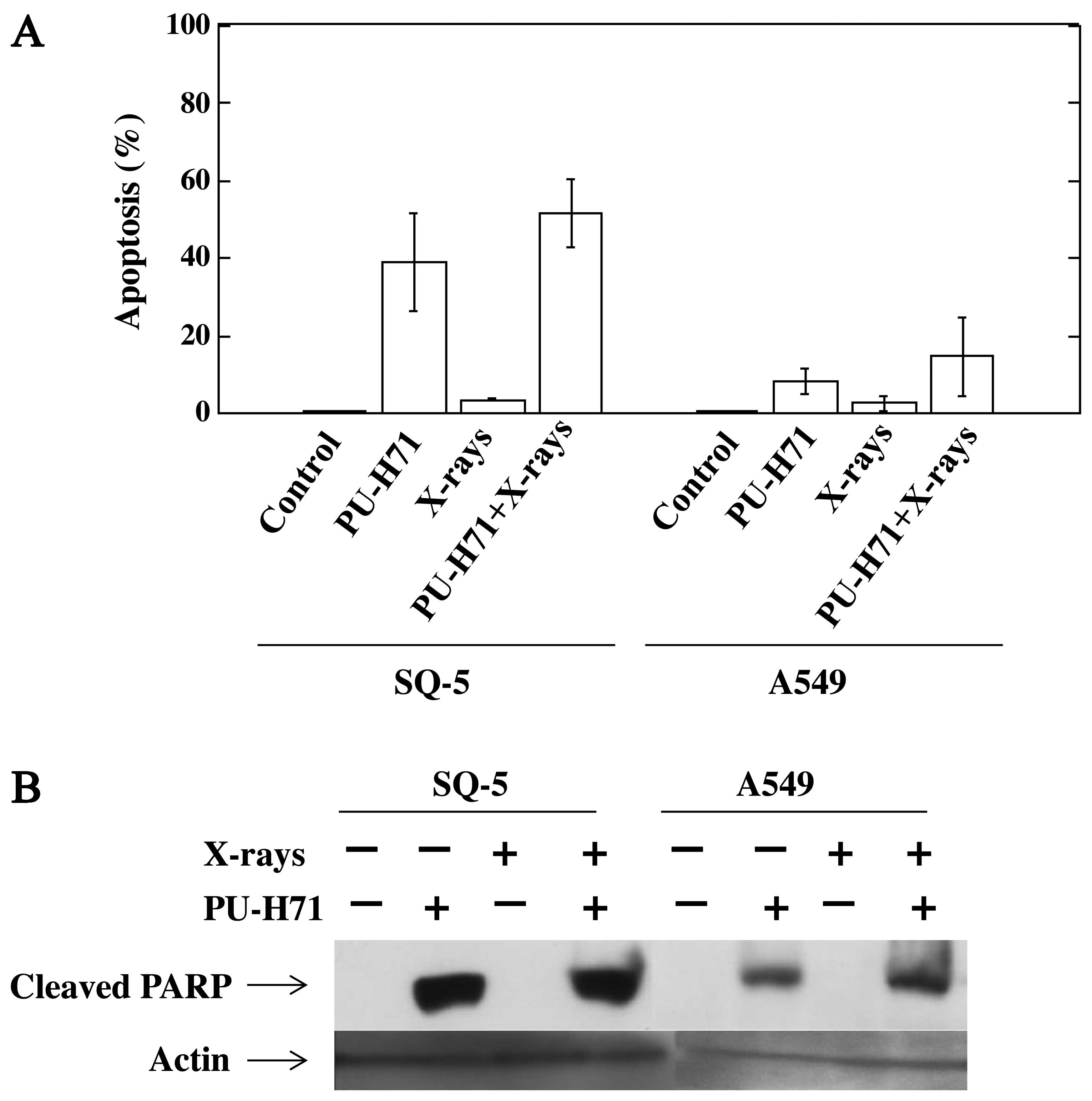
PU-H71 induces apoptosis in cancer
cells
The apoptosis-inducing effect of PU-H71 was examined
using DAPI staining. Treatment with X-rays alone did not induce
apoptosis in the SQ-5 and A549 cells. The combination of PU-H71 and
X-rays induced a marked increase in the number of apoptotic SQ-5
cells and a slight increase in the number of apoptotic A549 cells
(Fig. 5A). We then examined the
cleavage of PARP by western blot analysis using cleaved PARP
antibody. Similar to apoptosis induction, PU-H71 treatment with or
without X-rays induced the cleavage of PARP protein in both cells;
however, this was not observed with X-rays alone (Fig. 5B), indicating that caspase-3 is
activated following treatment with PU-H71 in both cell lines.
Discussion
In clinical radiotherapy, tumor radioresistance is
one of the causes of local failure after radiotherapy. The
development of drugs that enhance the sensitivity of tumor cells to
radiation is thus of great importance. Although a number of studies
have focused on radiosensitizers, no truly clinically effective
treatments have been developed. Several groups have shown that
Hsp90 inhibitors enhance the radiation sensitivity of human cancer
cell lines of different origin (8–11,21–25). These sensitizing effects are the
result of the Hsp90 inhibitor-mediated abrogation of the
G2 checkpoint, apoptosis and the inhibition of DNA
repair. These data strongly suggest that targeting Hsp90 with its
inhibitors represents a promising strategy for enhancing the
sensitivity of cancer cells to radiation.
The first generation of geldanamycin-based Hsp90
inhibitors, including the benzoquinone ansamycins, 17-AAG, 17-DMAG
and IPI-504, have been developed and investigated for the treatment
of various malignancies (6).
Although the geldanamycin derivatives have shown significant
antitumor activity against a spectrum of cancers in pre-clinical
studies, early clinical results with these Hsp90 inhibitors
revealed dose-limiting delayed hepatotoxicity (26), which prompted the discovery of
novel non-ansamycin Hsp90 inhibitors with less toxicity. Thus,
Chiosis et al designed Hsp90 inhibitors using purine as a
scaffold (27,28). PU-H71 is the most potent compound
in its class (29), and has shown
activity in triple-negative breast cancer (15) and diffuse large-cell lymphoma
(16). In addition, Breinig et
al, following in vivo hepatocellular carcinoma xenograft
mouse model experiments, demonstrated that PU-H71 was retained in
tumors at pharmacologically relevant concentrations, while being
rapidly cleared from non-tumorous liver. Thus, PU-H71 was effective
in the treatment of hepatocellular carcinoma, and was well
tolerated, indicating a lack of significant hepatotoxicity
(17). Collectively, these data
indicate that the non-quinone Hsp90 inhibitor, PU-H71, may be a
promising anticancer agent.
In this study, we examined the radiosensitizing
effects of PU-H71 on human lung cancer cells and observed that,
similar to 17-AAG and 17-DMAG, PU-H71 had synergistic effects with
radiation. Certain studies have demonstrated that the
radiosensitizing effects of Hsp90 inhibitors are mediated by the
inhibition of DSB repair (25,30). In our study, we demonstrated that
PU-H71 inhibited the repair of DSBs and sensitized human lung
cancer cell lines to radiation (Fig.
3). PU-H71 reduced the protein levels of Rad51 and inhibited
Rad51 foci formation following X-irradiation. Rad51 is a key
component of the HR, an evolutionarily conserved machinery for DSB
repair. Previously, we reported that 17-AAG, one of the first
generation of geldanamycin-based Hsp90 inhibitors, exerts a marked
inhibitory effect on the HR process (11). Thus, PU-H71 sensitizes human
cancer cells to radiation in a manner similar to that of
17-AAG.
In conclusion, our data demonstrate that PU-H71
inhibits the repair of radiation-induced DSBs by affecting the HR
pathway, and potentially sensitizes human lung cancer cells to
radiation. By contrast, PU-H71 had only slight radiosensitizing
effects on normal human fibroblasts. The combination of PU-H71 and
radiotherapy may be a promising therapeutic strategy for certain
radioresistant solid tumors.
Acknowledgements
We thank Dr Y. Matsumoto and Ms. Huizi Li (National
Institute of Radiological Sciences, Chiba, Japan) for their
providing valuable comments on our manuscript. This study was
supported, in part, by Grants-in-Aid for Scientific Research (no.
23591846) from the Japan Society for the Promotion of Science.
References
|
1
|
Ritossa F: Discovery of the heat shock
response. Cell Stress Chaperones. 1:97–98. 1996. View Article : Google Scholar
|
|
2
|
Westerheide SD and Morimoto RI: Heat shock
response modulators as therapeutic tools for the diseases of
protein conformation. J Biol Chem. 280:33097–33100. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wiech H, Buchner J, Zimmerman R and Jakob
U: Hsp90 chaperones protein folding in vitro. Nature. 358:169–170.
1992. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schneider C, Sepp-Lorenzino I, Nimmersgern
E, Ouerfelli O, Danishefsky S, Rosen N and Harti FU: Pharmacologic
shifting of a balance between protein refolding and degradation
mediated by Hsp90. Proc Natl Acad Sci USA. 93:14536–14541. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhao R, Davey M, Hsu Y, Kaplanek P, Tong
A, Parsons AB, Krogan N, Cagney G, Mal D, Greenblatt J, Boone C,
Emili A and Houry WA: Navigating the chaperone network: an
integrative map of physical and genetic interactions mediated by
the hsp90 chaperone. Cell. 120:715–727. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Neckers L: Hsp90 inhibitors as novel
cancer chemotherapeutic agents. Trends Mol Med. 8(suppl 4):
S55–S61. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Russell JS, Burgan WE, Oswald KA,
Camphausen K and Tofilon PJ: Enhanced cell killing induced by the
combination of radiation and the heat shock protein 90 inhibitor
17-allylamino-17-demethoxygeldanamycin: a multitarget approach to
radiosensitization. Clin Cancer Res. 37:3749–3755. 2003.PubMed/NCBI
|
|
8
|
Machida H, Matsumoto Y, Shirai M and
Kubota N: Geldanamycin, an inhibitor of Hsp90, sensitizes human
tumor cells to radiation. Int J Radiat Biol. 79:973–980. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Machida H, Nakajima S, Shikano N, Nishio
J, Okada S, Asayama M, Shirai M and Kubota N: Heat shock protein 90
inhibitor 17-allylamino-17-demethoxygeldamamycin potentiates the
radiation response of tumor cells grown as monolayer cultures and
spheroids by inducing apoptosis. Cancer Sci. 96:911–917. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Matsumoto Y, Machida H and Kubota N:
Preferential sensitization of tumor cells to radiation by heat
shock protein 90 inhibitor geldanamycin. J Radiat Res. 46:215–221.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Noguchi M, Yu D, Hirayama R, Ninomiya Y,
Sekine E, Kubota N, Ando K and Okayasu R: Inhibition of homologous
recombination repair in irradiated tumor cells pretreated with
Hsp90 inhibitor 17-allylamino-17-demethoxygeldamamycin. Biochem
Biophys Res Commun. 351:658–663. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Solit DB, Ivy SP, Kopil C, Sikorski R,
Morris MJ, SF, Slovin SF, Kelly WK, DeLaCruz A, Curley T, Heller G,
Larson S, Schwartz L, Egorin MJ, Rosen N and Scher HI: Phase I
trial of 17-allylamino-17-demethoxygeldanamycin in patients with
advanced cancer. Clin Cancer Res. 13:1775–1782. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Taldone T, Gozman Maharaj R and Chiosis G:
Targeting Hsp90: small-molecule inhibitors and their clinical
development. Curr Opin Pharmacol. 8:370–374. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chiosis G, Kang Y and Sun W: Discovery and
development of purine-scaffold Hsp90 inhibitors. Expert Opin Drug
Discov. 3:99–114. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Caldas-Lopes E, Cerchietti L, Ahn JH,
Clement CC, Robles AI, Rodina A, Moulick K, Taldone T, Gozman A,
Guo Y, Wu N, de Stanchina E, White J, Gross SS, Ma Y, Varticovski
L, Melnick A and Chiosis G: Hsp90 inhibitor PU-H71, a multimodal
inhibitor of malignancy, induces complete responses in
triple-negative breast cancer models. Proc Natl Acad Sci USA.
106:8368–8373. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Briones J: Targeted therapy of
BCL6-dependent diffuse large B-cell lymphoma by heat-shock protein
90 inhibition. Expert Rev Hematol. 3:157–159. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Breinig M, Caldas-Lopes E, Goeppert B,
Malz M, Rieker R, Bergmann F, Schirmacher P, Mayer M, Chiosis G and
Kern MA: Targeting heat shock protein 90 with non-quinone
inhibitors: a novel chemotherapeutic approach in human
hepatocellular carcinoma. Hepatology. 50:102–112. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li X and Heyer WD: Homologous
recombination in DNA repair and DNA damage tolerance. Cell Res.
18:99–113. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kuribayashi T, Ohara M, Sora S and Kubota
N: Scriptaid, a novel histone deacetylase inhibitor, enhances the
response of human tumor cells to radiation. Int J Mol Med.
25:25–29. 2010.PubMed/NCBI
|
|
20
|
Bee L, Fabris S, Cherubini R, Mognato M
and Celotti L: The efficiency of homologous recombination and
non-homologous end joining systems in repairing double-strand
breaks during cell cycle progression. PloS One. 8:e690612013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bull EE, Dote H, Brady KJ, Burgan WE,
Carter DJ, Cerra MA, Oswald KA, Hollingshead MG, Camphausen K and
Tofilon PJ: Enhanced tumor cell radiosensitivity and abrogation of
G2 and S phase arrest by the Hsp90 inhibitor
17-(dimethylaminoethylamino)-17-demethoxygeldanamycin. Clin Cancer
Res. 10:8077–8084. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bisht KS, Bradbury CM, Mattson D, Kaushal
A, Sowers A, Markovina S, Ortiz KL, Sieck LK, Isaacs JS, Brechbiel
MW, Mitchell JB, Neckers LM and Gius D: Geldanamycin and
17-allylamino-17-demethoxygeldanamycin potentiate the in vitro and
in vivo radiation response of cervical tumor cells via the heat
shock protein 90-mediated intracellular signaling and cytotoxicity.
Cancer Res. 63:8984–8995. 2003.
|
|
23
|
Koll TT, Feis SS, Wright MH, Teniola MM,
Richardson MM, Robles AI, Bradsher J, Capala J and Varticovski L:
HSP90 inhibitor, DMAG, synergizes with radiation of lung cancer
cells by interfering with base excision and ATM-mediated DNA
repair. Mol Cancer Ther. 7:1985–1992. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stingl L, Stühmer T, Chatterjee M, Jensen
MR, Flentje M and Djuzenova CS: Novel HSP90 inhibitors, NVP-AUY922
and NVP-BEP800, radiosensitise tumour cells through cell-cycle
impairment, increased DNA damage and repair protraction. Br J
Cancer. 102:1578–1591. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kubota N and Matsumoto Y: Hsp90 inhibitors
are promising radiosensitizers for radiotherapy. Thermal Med.
28:53–62. 2012. View Article : Google Scholar
|
|
26
|
Sharp S and Workman P: Inhibitors of the
HSP90 molecular chaperone: current status. Adv Cancer Res.
95:323–348. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chiosis G, Timaul MN, Lucas B, Munster PN,
Zheng FF, Sepp-Lorenzino L and Rosen N: A small molecule designed
to bind to the adenine nucleotide pocket of Hsp90 causes Her2
degradation and the growth arrest and differentiation of breast
cancer cells. Chem Biol. 8:289–299. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chiosis G: Discovery and development of
purine-scaffold Hsp90 inhibitors. Curr Top Med Chem. 6:1183–1191.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chiosis G, Rodina A and Moulick K:
Emerging Hsp90 inhibitors: from discovery to clinic. Anticancer
Agents Med Chem. 6:1–8. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zaidi S, McLaughlin M, Bhide SA, Eccles
SA, Workman P, Nutting CM, Huddart RA and Harrington KJ: The HSP90
inhibitor NVP-AUY922 radiosensitizes by abrogation of homologous
recombination resulting in mitotic entry with unresolved DNA
damage. PLos One. 7:e354362012. View Article : Google Scholar : PubMed/NCBI
|