Introduction
Alzheimer’s disease (AD), the most common form of
dementia in elder persons, is a neurodegenerative disease which is
characterized by the progressive loss of memory, deterioration of
language, as well as defects in visual and motor coordination
(1). Documented data have shown
that the pathological hallmarks of AD include the accumulation of
intracellular neurofibrillary tangles rich in Tau protein and
extracellular plaques containing β-amyloid protein (Aβ) (2). Despite the fact that the precise
cause of AD remains unknown, toxic Aβ accumulation-induced neuronal
loss and Aβ, the constituent of extracellular plaques have been
observed in patients with AD. This indicates that Aβ may play an
important role in the development of AD (3).
Oxidative stress, an imbalance toward the
pro-oxidant side of pro-oxidant/antioxidant homeostasis, occurs in
some brain neurodegenerative disorders. Although it has been
reported that nicotine possibly promotes lung cancer development
and reduces the efficacy of chemotherapeutic agents (4), the rapid synaptic transmission in
key regions controlling behavior mediated by nicotine via nicotine
acetylcholine receptors (nAChRs) has also reported (5). Notably, nicotine has been confirmed
to improve memory function and reduce amyloid plaque burden in a
transgenic mouse model of AD (6),
which indicates that nicotine may be a survival agonist against
apoptosis induced by various types of stress (7). However, the exact mechanisms of
action of nicotine and its role in the improvement of AD remain
unclear; elucidating these mechanisms pay prove useful in the
treatment of AD.
Bcl-2, Bcl-xL and Mcl-1, which belong to the Bcl-2
family, are localized in the outer mitochondrial membrane and
protect cells against a variety of apoptotic stimuli (8). Despite the fact that nicotine
increases Bcl-2, Bcl-xL and Mcl-1 expression and facilitates
multiple drug resistance in lung cancer (9), the exact effects of the
nicotine-induced upregulation of anti-apoptotic proteins in the
treatment of AD have not yet been elucidated. Mitogen-activated
protein kinase (MAPK) signaling pathways, which included Erk,
SAPK/JNK and p38, play important roles in growth, differentiation,
development and cell survival (10–12). Nicotine has been reported to
activate the MAPK pathway in various tissues and cell types
(13,14). Other studies have also shown that
the inhibition of MAPK pathways is involved in the anti-apoptotic
effects and mediates neuroprotection (15). Hence, the exact roles of Erk,
SAPK/JNK and p38 in nicotine-mediated neuroprotection remain
unelucidated.
In the present study, we aimed to investigate the
protective effects of nicotine on Aβ-induced neurotoxicity. The
effects of nicotine on Aβ-induced SH-SY5Y cell apoptosis were first
determined by flow cytometry and microscopic observation. The
effects of nicotine on Bcl-2, Bcl-xL and Mcl-1 expression, as well
as MAPK kinase activation were further explored by western blot
analysis. Using kinase inhibitors, the levels of Erk, SAPK/JNK and
p38 phosphorylation, as well as caspase-3 activation, Bcl-xL
expression and cell viability were further investigated.
Importantly, the neuroprotective effects of nicotine on the
impairment of spatial working memory by Aβ25–35 and
Bcl-2 expression were investigated by a Morris water maze
navigation test and immunohistochemical analysis of hippocampal
sections using a mouse model of Aβ-induced AD. The results revealed
the following: firstly, Aβ markedly augmented SH-SY5Y cell
apoptosis, the release of cytochrome c and caspase-3
activation in a time- and dose-dependent manner. Secondly,
pre-treatment with nicotine attenuated the Aβ-induced cell
apoptosis, inhibited caspase-3 activation and increased cell
viability. Thirdly, treatment with nicotine markedly upregulated
Bcl-2, Bcl-xL and Mcl-1 expression and MAPK kinase phosphorylation.
Notably, when the activities of Erk, SAPK/JNK and p38 were
inhibited, the nicotine-induced Bcl-xL upregulation and
anti-apoptotic effects were reversed accordingly. Importantly,
in vivo nicotine administration greatly ameliorated the
impairment in spatial working memory induced by Aβ25–35
and upregulated Bcl-2 expression in the hippocampus. The data
presented in this study indicate that nicotine exerts
neuroprotective effects against Aβ-induced neurotoxicity by
activating the MAPK pathway and upregulating the expression of
anti-apoptotic proteins.
Materials and methods
Reagents
Nicotine and the Aβ25–35 peptide were
purchased from Sigma-Aldrich (St. Louis, MO, USA).
Aβ25–35 was dissolved in deionized distilled water at a
concentration of 5 mg/ml. The stock solution was diluted to the
desired concentrations immediately before use. Dulbecco’s modified
Eagle’s medium (DMEM) and fetal bovine serum (FBS) were obtained
from HyClone (Logan, USA). The Annexin V/PI Apoptosis detection kit
was obtained from Promega (Madison, WI, USA). Erk1/2 inhibitor
(U0126), p38 inhibitor (SB203580) and JNK inhibitor (SP600125), as
well as antibodies to tubulin, Bcl-2, Bc-xL, Mcl-1, cleaved
caspase-3, cytochrome c, phospho-p38, phospho-Erk1/2,
phospho-Mek1/2, phospho-p90Rsk, phospho-Msk, phospho-c-Jun,
phospho-SAPK/JNK and phospho-MKK3/6 were purchased from Cell
Signaling Technology Inc. (Beverly, MA, USA).
Animals
Pathogen-free C57BL/6 mice (male, 4 weeks old,
weighing 18–22 g) were purchased from the Shanghai Laboratory
Animal Center of the Chinese Academy of Sciences (Shanghai, China)
and kept at the Animal Center of Xiamen University (Xiamen, China).
All animal experiments were approved by the Review Board of the
Medical College of Xiamen University.
Cell lines
Human SH-SY5Y neuroblastoma cells were obtained from
the Shanghai Cell Bank (Shanghai, China). Cells were cultured in
DMEM with 10% FBS at 37°C in 5% CO2 and passaged every
1–2 days to maintain logarithmic growth. Cells were synchronized by
serum starvation for at least 12 h prior to treatment with nicotine
or Aβ25–35 for the indicated periods of time and
concentrations.
Cell apoptosis assay
Cell apoptosis assay was determined by flow
cytometry according to a previously described method (16). Briefly, 8×104 SH-SY5Y
cells seeded in 24-well plates were pre-treated with 0.1 μM
nicotine and further treated with Aβ25–35. The cells
were then removed by trypsinization, rinsed with PBS and
re-suspended in binding buffer containing Annexin V and propidium
iodide (PI) for 20 min at room temperature. The samples were
analyzed on a FACSCalibur flow cytometer and data were analyzed
using CellQuest software.
Western blot analysis
Proteins were obtained in lysis buffer as previously
described (13). Proteins were
loaded onto SDS-PAGE gels for electrophoresis and transferred onto
PVDF membranes. After blocking in 5% fat-free milk in TBST for 90
min, the membranes were incubated with primary antibodies at 4°C
overnight. Subsequently, the membranes were incubated with
corresponding horseradish peroxidase (HRP)-conjugated secondary
antibodies at room temperature for 90 min. After washing 6 times
with TBST (for 10 min each), the bound antibodies were visualized
using enhanced chemiluminescence (ECL). Tubulin was used as a
loading control.
Grouping of mice and establishment of
animal model of AD
To investigate the effects of nicotine on AD
symptoms, a mouse model of AD induced by Aβ was established
according to previously described method (17). Briefly, 4-week-old male C57BL/6
mice were randomly divided into the control group, the AD group and
AD group pre-treated with nicotine (n=7 per group). The mice in the
AD group pre-treated with nicotine were administered nicotine
(0.1125 mg/kg) subcutaneously twice a day. Animals that were
injected with the vehicle served as the controls. After 2 weeks of
nicotine administration, the mice were anesthetized with 4% chloral
hydrate and the scalps were incised and retracted to expose the
skull. The lambda and bregma were aligned in the same horizontal
plane. The AD group and AD group pre-treated with nicotine were
injected with 5 μl (5 mg/ml) Aβ25–35 using a stereotaxic
instrument (RWD Life Science Co., Ltd., Shenzhen, China) and a
microinjector (Kd Scientific, Holliston, MA, USA) into the
hippocampal CA1 region of the right hemisphere (coordinates:
anteroposterior, −2.0 mm from the bregma; lateral, −2.0 mm;
dorsovental, −3.5 mm) with a 10 μl Hamilton syringe driven by a
microinjector at a speed of 1 μl/min. After the injection, the
needle was kept in the injection site for a further 10 min and then
slowly withdrawn in 5 min; simultaneously, mice in the control
group were injected with identical doses of sodium chloride into
the same area. On the 17th day after the microinjection, the mice
were subjected to a Morris water maze navigation test. On the 21st
day after the microinjection, all mice in the 3 groups were firstly
anesthetized, and then the mouse brain tissues were removed and
fixed in 4% paraformaldehyde in phosphate buffer for 24 h, and
embedded in paraffin for H&E staining, Congo red staining and
immunohistochemical staining.
H&E staining and Congo red
staining
Mice were sacrificed by CO2 asphyxiation,
and the brains were fixed for 48 h in 4% paraformaldehyde in PBS.
Free-floating sections (50 μM) were obtained using a vibratome
slicing system. Sections were deparaffinized with various
concentrations of ethanol. For H&E staining, the sections were
stained with hematoxylin for 15 min and washed in running tap water
for 20 min. Counterstaining was performed with eosin. For Congo red
staining, the sections were stained with methanol Congo red for
10–20 min and following by 0.2% alkaline ethanol staining. Finally,
the sections were dehydrated in 95% and absolute alcohols for 2
changes of 2 min each and observed under a microscope.
Immunohistochemitry
Mice were sacrificed by CO2 asphyxiation,
and the brains were fixed for 48 h in 4% paraformaldehyde in PBS.
Free-floating sections (50 μm) were obtained using a vibratome
slicing system. Bcl-2 expression in the hippocampus was determined
by immunohistochemitry according to a previously described method
(18). Briefly, endogenous
peroxidase activity was quenched for 30 min in
H2O2, and the sections were subsequently
incubated in 90% formic acid for 7 min to expose the epitope. The
primary Bcl-2 antibody was applied, and the sections were incubated
overnight at 4°C. The sections were subsequently washed in TBS to
remove the excess primary antibody. The sections were then
incubated in biotinylated secondary antibody for 1 h at 20°C. After
a final wash of 20 min, slides were developed with diaminobenzidine
substrate by using the avidin-biotin HRP system (Vector
Laboratories, Burlingame, CA, USA).
Morris water maze navigation test
To investigate the effects of nicotine
administration on spatial learning and memory following exposure to
Aβ, the Morris water maze navigation test was performed according
as previously described (19).
Briefly, the water maze consisted of a circular water tank (120 cm
in diameter) filled with water (26°C) in which an escape platform
(10 cm in diameter) was hidden 0.5 cm below the surface of the
30-cm-deep water. The water was made opaque by addition of milk
powder, thereby rendering the platform invisible. A video camera
was placed above the centre of the pool to capture images of the
swimming animal. Mice were trained in a 4-trial per day task for 5
consecutive days. Each mouse was allowed a maximum of 60 sec to
find the hidden platform and allowed to remain on it for 15 sec. If
a mouse failed to find the platform within 60 sec, the mouse was
placed on the platform for 30 sec by the investigator. The time
required to find the hidden platform during these 4 acquisition
trials was averaged. The navigation of the mice was tracked by a
video camera. The escape latency of the mice was recorded. The
experiment lasted 5 days, and each day was divided into morning and
afternoon blocks, 4 trials in each block.
Statistical analysis
Each experiment was repeated at least 3 times and
similar data were obtained. All data are presented as the means ±
standard error of the means (SEM). Statistical significance was
examined using one-way or two-way ANOVA with a post hoc
Newman-Keuls test. A value of p<0.05 was considered to indicate
a statistically significant difference.
Results
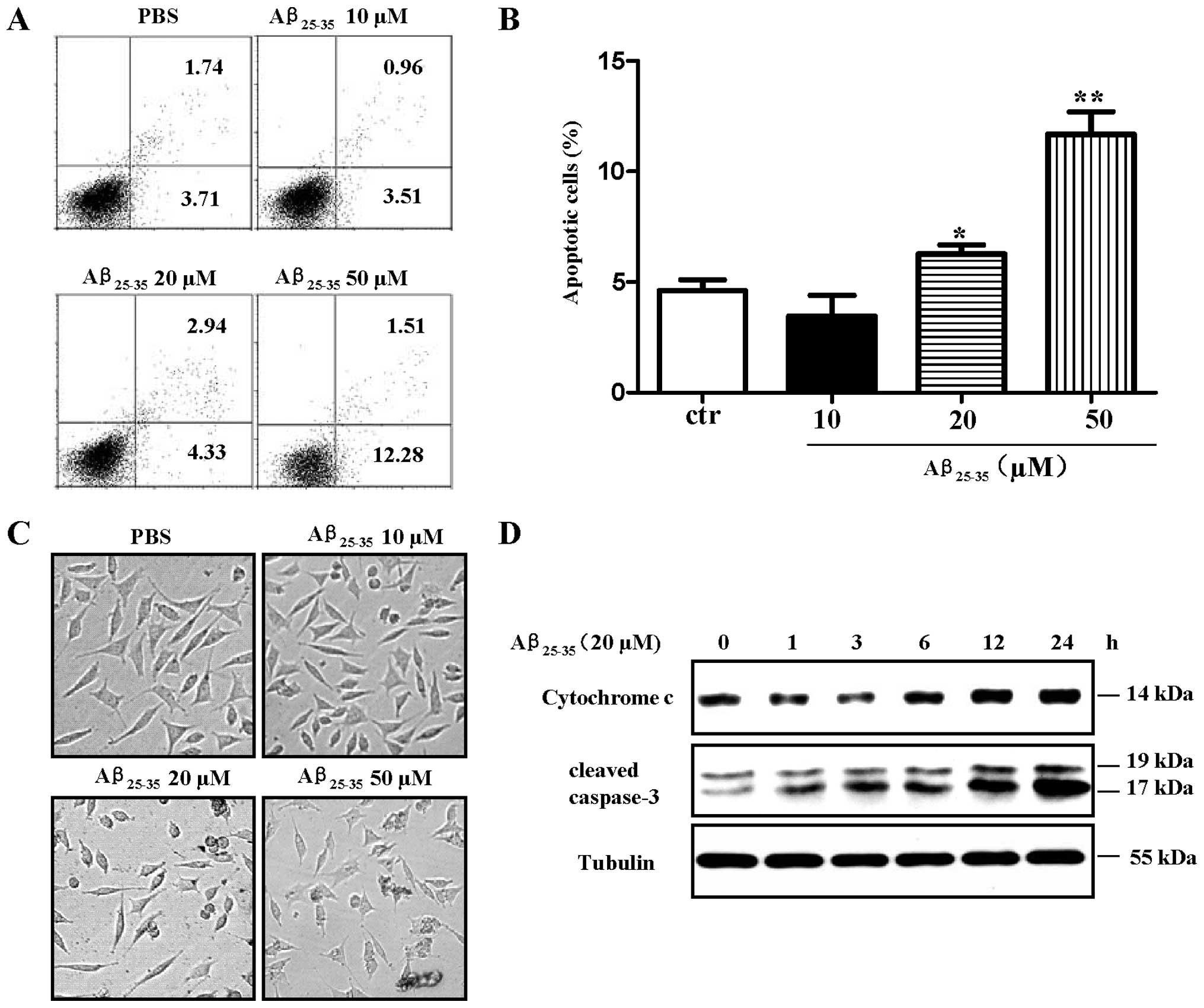
Aβ25–35 induces apoptosis of
SH-SY5Y cells in a dose- and time-dependent manner
Aβ-induced neurotoxicity is thought to be a critical
event in the pathogenesis of AD (2). In this study, to assess the
neurotoxicity of Aβ, SH-SY5Y cells were treated with
Aβ25–35 and cell viability was determined by flow
cytometry, microscopic observation. Western blot analysis was also
performed to determine the release of cytochrome c and the
levels of cleaved caspase-3. The results revealed that treatment
with Aβ25–35 (20 and 50 μM) induced approximately 7.27
and 13.8% SH-SY5Y cell apoptosis (Fig. 1A and B, p<0.05,
Aβ25–35 20 μM vs. control; p<0.01, Aβ25–35
50 μM vs. control, one-way ANOVA with post hoc Newman-Keuls test)
indicating that Aβ25–35 induced apoptosis in a
dose-dependent manner. Microscopic observation also revealed
similar results (Fig. 1C).
Importantly, cleaved caspase-3 and the release of cytochrome
c were increased following treatment with Aβ25–35
in a time-dependent manner (Fig.
1D). As the activation of caspases, in particular that of
caspase-3, plays a prominent role in the initiation of apoptosis,
the augmented cleaved caspase-3 levels and the release of
cytochrome c induced by treatment with Aβ25–35
indicated that Aβ exerts neurotoxic effects on SH-SY5Y cells and
may contribute to the development of AD.
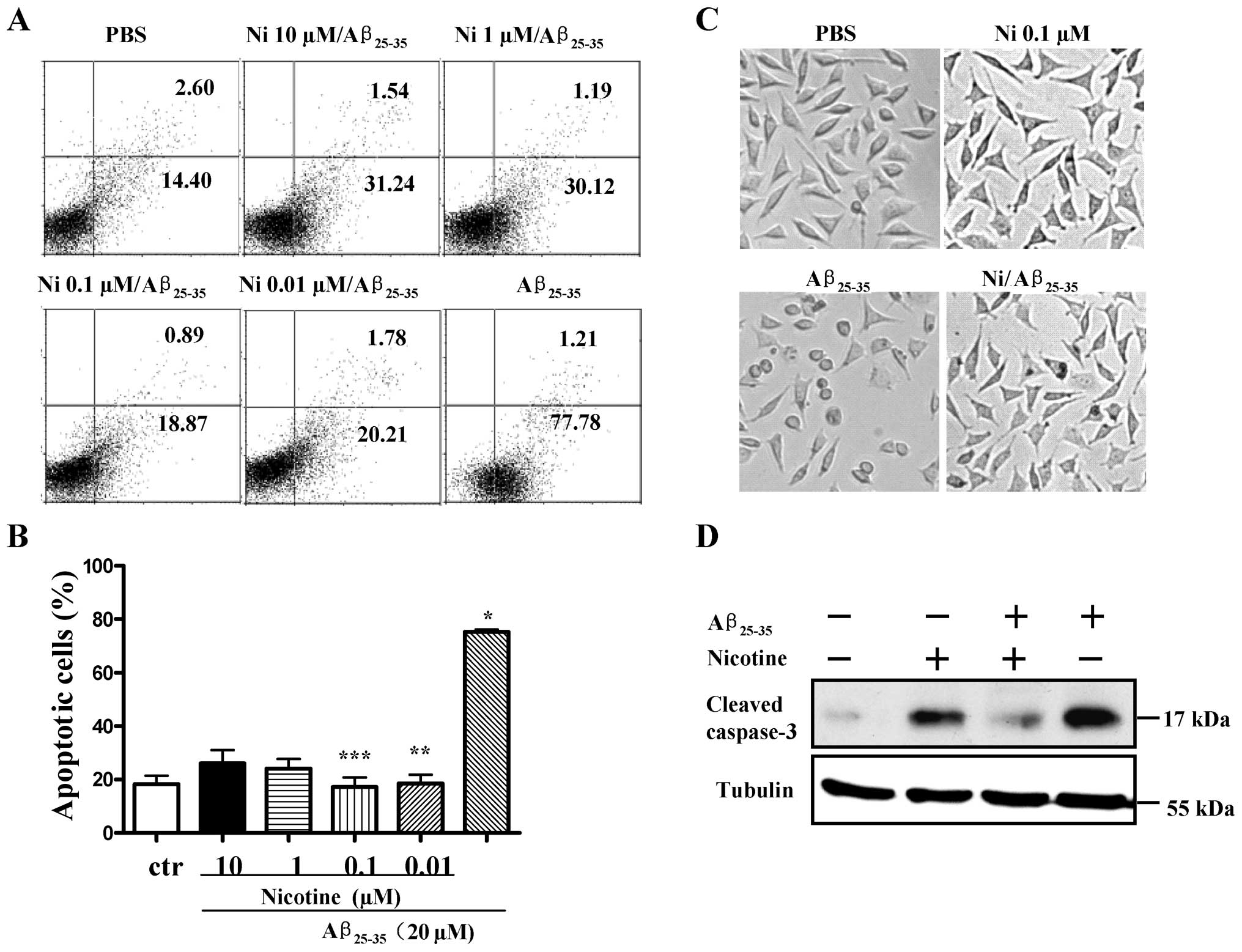
Pre-treatment with nicotine attenuates
Aβ25–35-induced neurotoxicity in SH-SY5Y cells
To explore the potential neuroprotective effects of
nicotine against Aβ-induced neurotoxicity, the SH-SY5Y cells were
treated with nicotine prior to Aβ25–35 stimulation and
cell apoptosis was determined by flow cytometry and microscopic
observation. Western blot analysis was also performed to measure
the levels of cleaved caspase-3. The results revealed that although
treatment with Aβ25–35 induced approximately 79% SH-SY5Y
cell apoptosis, pre-treatment with nicotine markedly abolished the
neurotoxic effects of Aβ25–35 on SH-SY5Y cells (Fig. 2A and B, p<0.001,
Aβ25–35 vs. control; p<0.001, Aβ25–35 vs.
nicotine 0.01 μM/Aβ25–35; p<0.001, Aβ25–35
vs. nicotine 0.1 μM/Aβ25–35, one-way ANOVA with post hoc
Newman-Keuls test). Microscopic observation also revealed similar
results (Fig. 2C). Importantly,
pre-treatment with nicotine markedly attenuated the
Aβ25–35-induced activation of caspase-3 (Fig. 2D). These data indicate that
treatment with nicotine exerts marked neuroprotective effects
against Aβ-induced neurotoxicity.
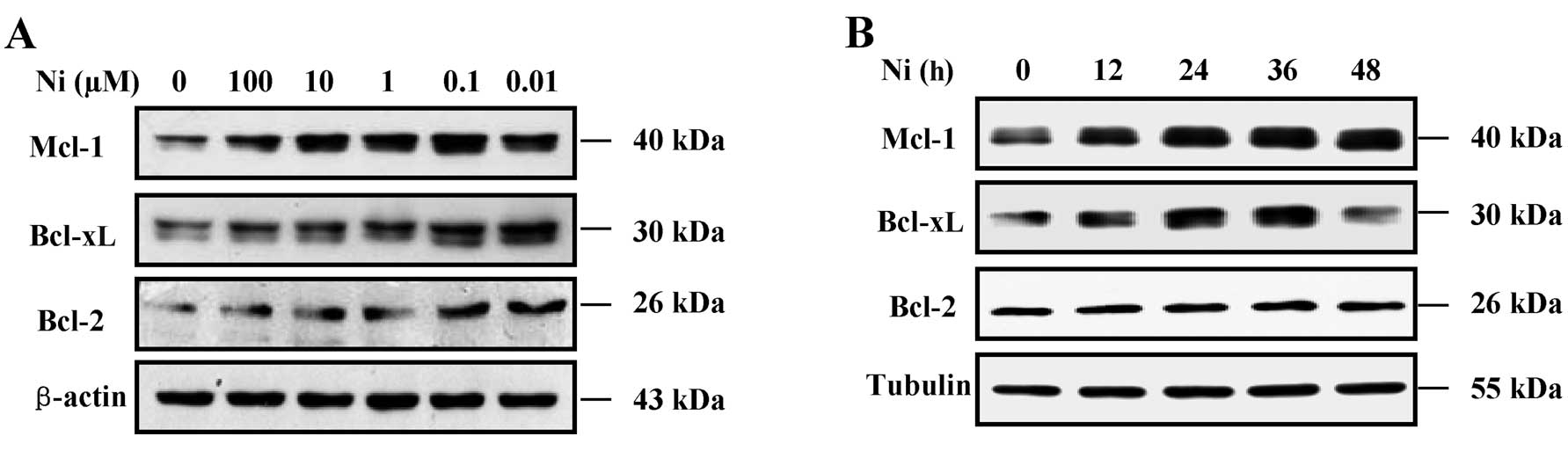
Nicotine increases Bcl-2, Bcl-xL and
Mcl-1 expression in SH-SY5Y cells
Given that Bcl-2 family proteins are important
modulators of cell apoptosis, we determined the effects of nicotine
on Bcl-2, Bcl-xL and Mcl-1 protein expression in the SH-SY5Y cells
treated with Aβ25–35 by western blot analysis. The
results revealed that nicotine markedly increased Bcl-2, Bcl-xL and
Mcl-1 expression in a dose- and time-dependent manner (Fig. 3). The results from western blot
analysis (Fig. 3A) revealed that
pre-treatment with nicotine (0.01–100 μM for 12 h) markedly
increased the Bcl-2, Bcl-xL and Mcl-1 protein levels in the SH-SY5Y
cells. Pre-treatment with 0.1 μM nicotine induced the most marked
upregulation of Bcl-2, Bcl-xL and Mcl-1 expression. Treatment with
0.1 μM nicotine increased Bcl-2, Bcl-xL and Mcl-1 expression in a
time-dependent manner. As the Bcl-2 family play an important role
in regulating apoptosis, as well as the mitochondrial-initiated
release of cytochrome c and activation of caspases (20), the fact that nicotine attenuated
the Aβ-induced activation of caspase-3, indicated that nicotine may
affect Bcl-2 family protein expression (21).
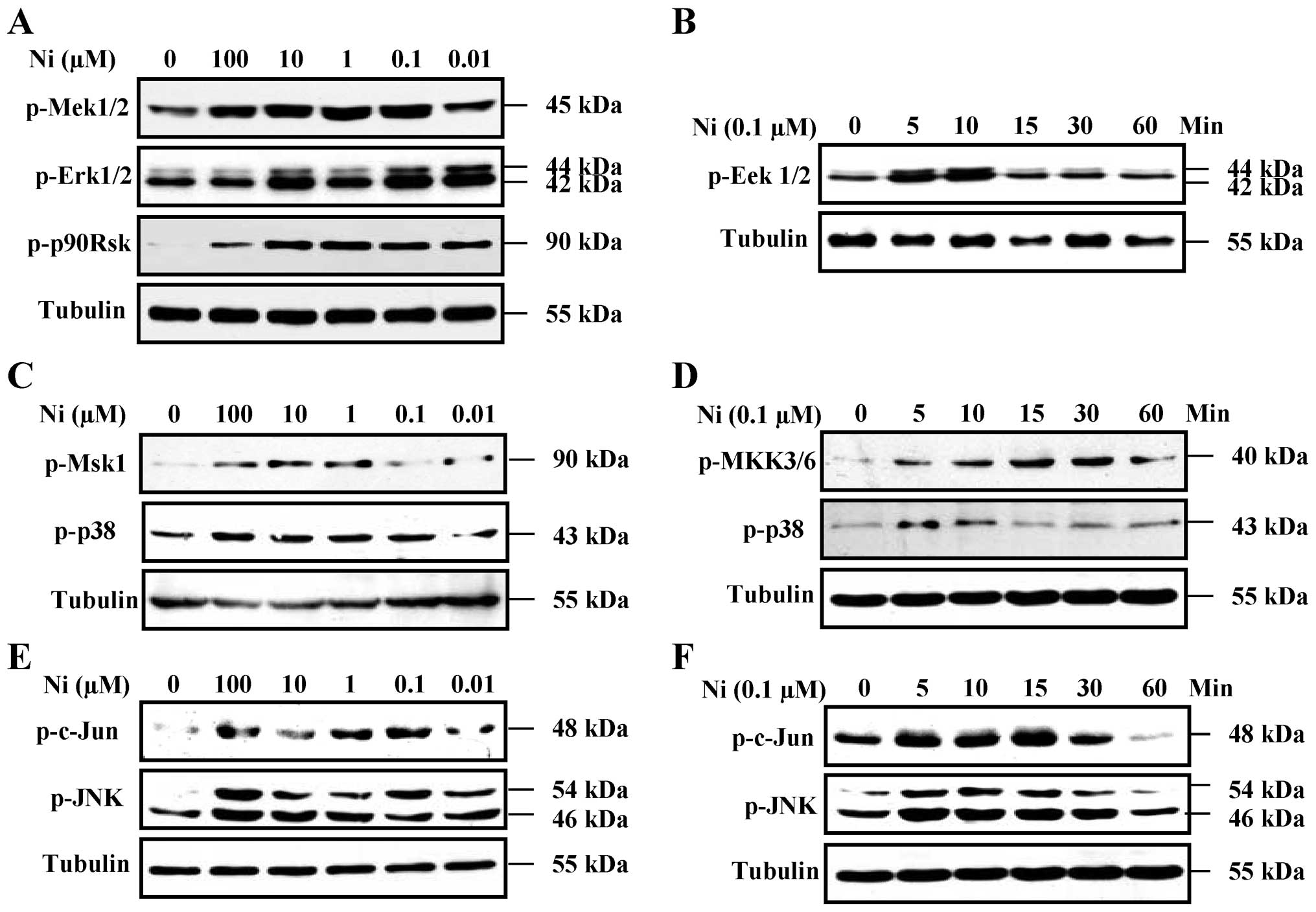
Nicotine increases Erk1/2, p38 and
SAPK/JNK phosphorylation in SH-SY5Y cells
The Erk1/2, p38 and SAPK/JNK pathways are involved
in regulating the expression of anti-apoptotic proteins (22–24). To investigate the role of the
Erk1/2, p38 and SAPK/JNK pathways in the nicotine-induced increase
in Bcl-2, Bcl-xL and Mcl-1 expression, the effects of nicotine on
Erk1/2, p38 and SAPK/JNK MAPK kinase activation were determined. In
initial experiments, SH-SY5Y cells were stimulated with a range of
nicotine concentrations (0.01–100 μM) for 5 min, and the
phosphorylation levels of Erk1/2, p38 and c-Jun MAPKs were
determined by western blot analysis. Nicotine (0.1–100 μM) induced
a significant enhancement of Erk1/2, p38 and c-Jun MAPK
phosphorylation in a dose-dependent manner (Fig. 4A, C and E), approximately 100%
above basal levels. Importantly, Mek1/2, p90Rsk, Msk1, SAPK/JNK and
MKK3/6 were also activated following treatment with nicotine
(Fig. 4A, C and E).
The time course of the activation of Erk1/2, p38 and
c-Jun MAPKs by 0.1 μM nicotine in the SH-SY5Y cells indicated that
Erk1/2, p38 and c-Jun MAPK phosphorylation was increased at 5 min
and persisted over 30 min of nicotine stimulation (Fig. 4B, D and F). Importantly, MeK1/2,
p90Rsk, Msk1, SAPK/JNK and MKK3/6 were activated at 5 to 30 min
following treatment with 0.1 μM nicotine (Fig. 4B, D and F). These results indicate
that nicotine activates MAPK pathways and this may play an
important role in the nicotine-mediated induction of anti-apoptotic
proteins.
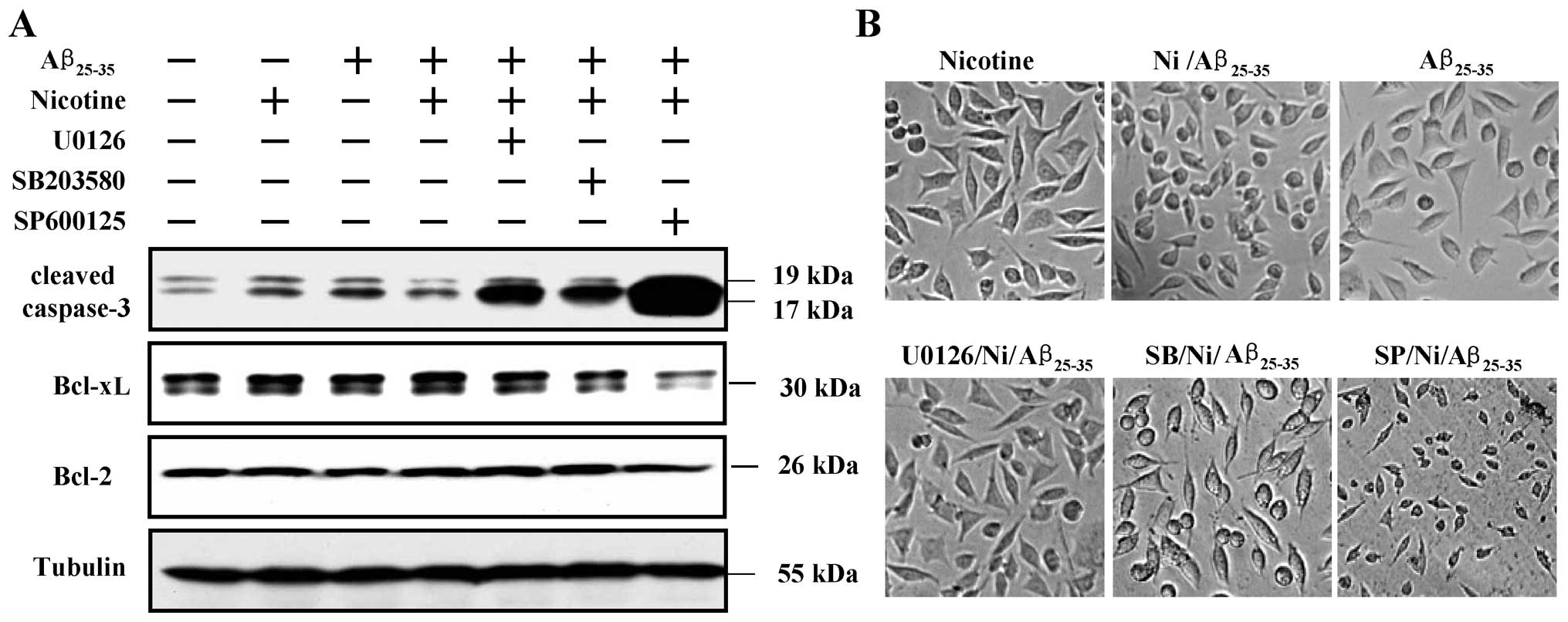
Nicotine upregulates Bcl-xL expression by
activating the Erk1/2, p38 and SAPK/JNK pathways
To explore the role of nicotine-activated MAPK
pathways in the nicotine-induced increase in the expression of
anti-apoptotic proteins, the SH-SY5Y cells were treated with
inhibitors of related kinases and the expression of Bcl-2/Bcl-xL
and the activation of caspase-3 were determined by western blot
analysis. The results revealed that while pre-treatment with
nicotine attenuated the Aβ-induced caspase-3 activation, treatment
with inhibitors of Erk1/2, p38 and SAPK/JNK markedly promoted the
activation of caspase-3. The results also revealed that the
nicotine-induced increase in Bcl-xL expression was dependent on
Erk1/2 (U0126), p38 (SB203580) and SAPK/JNK (SP600125)
phosphorylation (Fig. 5A).
Notably, SAPK/JNK, but not Erk1/2 or p38, was shown to play a more
important role in the attenuation of Aβ25–35-induced
caspase-3 activation and Bcl-xL expression (Fig. 5A). Microscopic observation also
revealed similar results (Fig.
5B). These data indicate that the Erk1/2, p38 and SAPK/JNK
pathways, particularly SAPK/JNK, play an important role in the
nicotine-mediated anti-apoptotic effects.
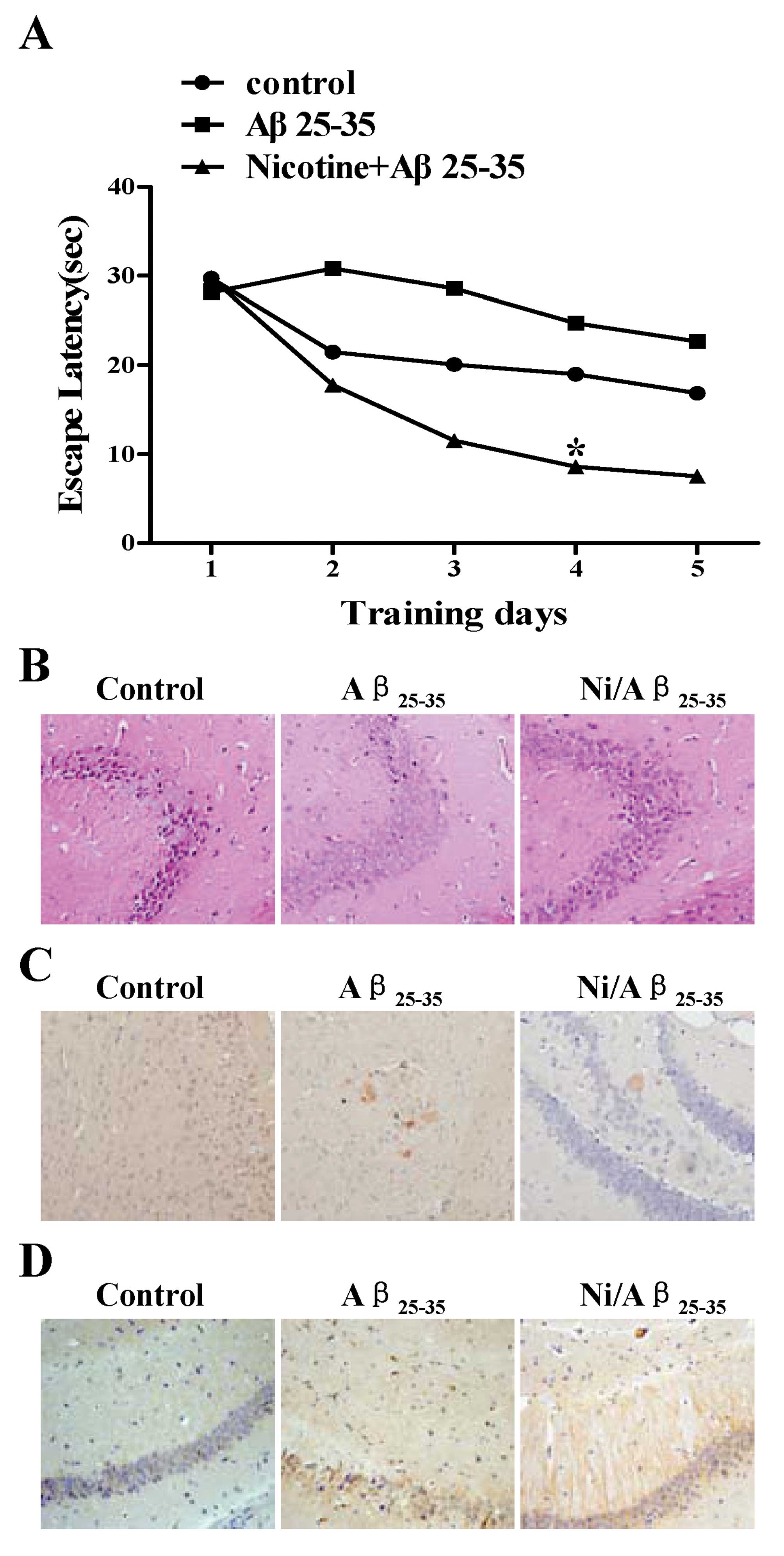
Treatment with nicotine markedly
ameliorates cognitive deficits in mice injected with
Aβ25–35
To investigate the effects of nicotine on spatial
learning deficits induced by Aβ25–35 injection, C57BL/6
mice were administered nicotine prior to the Aβ25–35
injection and the ability of the mice to learn and process spatial
information was examined by a Morris water maze. In the hidden
platform test, the mice became more efficient at finding the
platform on a successive trail (Fig.
6A). Two-way ANOVA (3 groups, 5 days) with repeated measures
rendered that the effect were statistically significant
(p<0.01). Post hoc analysis (Student-Newman-Keuls) revealed that
the Aβ25–35-injected mice had a significantly longer
escape latency (took longer to find the platform) than the
sham-operated mice on days 2–5; a marked decrease in escape latency
was observed following the administration of nicotine as compared
with the Aβ25–35-injected mice (Fig. 6A), indicating that the injection
of Aβ25–35 greatly impaired spatial working memory and
that treatment with nicotine attenuated this impairment in
memory.
To investigate the effects of nicotine on cell
viability in Aβ25–35-injected mice, Aβ accumulation and
Bcl-2 expression, hippocampal tissue sections from the mice were
stained with H&E and Congo red, and immunohistochemitry was
also performed. The results revealed that despite the fact that the
Aβ25–35 injection markedly decreased cell viability
(Fig. 6B) and increased Aβ
deposition (Fig. 6C), nicotine
administration greatly attenuated Aβ accumulation and abolished the
Aβ25–35-induced neurotoxicity (Fig. 6B and C). Bcl-2 immunohistochemitry
indicated that pre-treatment with nicotine markedly upregulated
Bcl-2 expression (Fig. 6D). These
results indicate that the neuroprotective effects of nicotine on
Aβ25–35-induced neurotoxicity may be mediated by the
upregulation in the expression of anti-apoptotic proteins, such as
Bcl-2.
Discussion
In recent studies, we investigated the biological
role of nicotine and found that nicotine activates bone
marrow-derived dendritic cells and that nicotine exerts potential
anti-tumor effects on dendritic cells (13,14,25). Nicotine has also been shown to
mediate synaptic transmission in regions controlling behavior
(5) and to improve memory
function in a transgenic mouse model of AD (6), indicating that nicotine may be a
potential therapeutic agent in AD. In the present study, JNK and α7
nAchR were found to be potential molecules in the treatment of AD.
Firstly, the Aβ-induced increase in cell apoptosis, caspase-3
activation and the release of cytochrome c were markedly
abrogated by treatment with nicotine. Secondly, the upregulation in
Bcl-2, Bcl-xL and Mcl-1 and MAPK kinase phosphorylation was
achieved following treatment with nicotine. Of note, the inhibition
of MAPK activity markedly promoted caspase-3 activation and the
downregulated nicotine-induced increase in Bcl-xL expression;
importantly, nicotine administration improvement memory function
and reduced Aβ accumulation in a mouse model of Aβ-induced AD.
MAPK signaling pathways, which include Erk, SAPK/JNK
and p38, play important roles in cell survival (10–12,22–24). The Erk 1/2 signaling pathway has
been found to be involved in nicotine-mediated neuroprotection in
spinal cord neurons (26). The
p38/JNK MAPK pathway mediates cortical neuronal apoptosis (27). In the present study, using Erk1/2,
p38 and JNK kinase inhibitors, the inhibition of Erk1/2, p38 and
JNK kinases markedly increased caspase-3 activation and abrogated
the nicotine-mediated Bcl-xL upregulation (Fig. 5A), indicating that nicotine
increased Bcl-xL expression and attenuated Aβ-induced neurotoxicity
by activating the Erk1/2, p38 and JNK pathways in SH-SY5Y cells.
The PI3K/Akt and NF-κB pathways are also involved in
nicotine-mediated neuroprotection (28,29). However, the exact role of the
PI3K-Akt and NF-κB pathways in nicotine-mediated neuroprotective
effects against Aβ stimulation remain unknown and require further
investigation.
JNK/SAPK, one of the vital signal transduction
pathways which transmits and converts stress signaling into
apoptotis signaling (27), has
been reported to induce receptor-mediated apoptosis by upregulating
Fas ligand expression in T lymphocytes (30) and is necessary for
irradiation-induced mitochondrial-mediated apoptosis in embryonic
fibroblasts (31). JNK activation
has an anti-apoptotic function in cardiac myocytes (32). In the present studys, the
inhibition of JNK kinase increased the activity of caspase-3 and
downregulated Bcl-xL (Fig. 5A),
indicating that JNK phosphorylation facilitates the survival of
SH-SY5Y cells. The anti-apoptotic function of JNK may be due to the
nicotine-induced JNK activation and may be cell type-dependent, as
JNK has been found to play either a pro-apoptotic or anti-apoptotic
role depending on cell type (31).
Bcl-2, downstream of the SAPK/JNK cascade, plays
important roles in regulating programmed cell death and has been
shown to be associated with neurodegenerative disorders, including
AD (33). In the present study,
both JNK activation (Fig. 4) and
Bcl-2 upregulation augmented by nicotine treatment were observed.
As Bcl-2 is located in the outer mitochondrial membrane and
protects cells against a variety of apoptotic stimuli (34), the nicotine-mediated Bcl-2
upregulation (Fig. 3) and
anti-apoptotic effects (Fig. 2)
indicated that the Aβ-induced apoptosis was
mitochondrial-dependent. Hence, it was not surprising to find that
Aβ treatment mrakedly promoted the release of cytochrome c
and mitochondrial-dependent caspase-3 activation (Fig. 1). As nicotine treatment
significantly augmented Bcl-xL and Mcl-1 expression, the exact
roles of these proteins in the nicotine-mediated anti-apoptotic
effects require further investigation.
Taken together, our data reveal that the
anti-apoptotic effects of nicotine may be mediated by the increased
expression of Bcl-2 family proteins and MAPK kinase
phosphorylation, indicating that JNK may be a potential molecule in
the treatment of AD.
Acknowledgements
We thank Jin Hua Su and Fu Chen for providing
excellent animal care. This study was supported by grants from the
National Natural Science Foundation of China (no. 81273203) and the
Natural Science Foundation of Xiamen (no. 3502Z20104002).
References
|
1
|
Chan KY, Wang W, Wu JJ, Liu L, Theodoratou
E, Car J, Middleton L, Russ TC, Deary IJ, Campbell H, Wang W and
Rudan I: Epidemiology of Alzheimer’s disease and other forms of
dementia in China, 1990–2010: a systematic review and analysis.
Lancet. 381:2016–2023. 2013.
|
|
2
|
Rosenberg RN and Lambracht-Washington D:
DNA Aβ42 vaccination as possible alternative immunotherapy for
Alzheimer disease. JAMA Neurol. 70:772–773. 2013.
|
|
3
|
Nalivaeva NN and Turner AJ: The amyloid
precursor protein: a biochemical enigma in brain development,
function and disease. FEBS Lett. 587:2046–2054. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tournier JM and Birembaut P: Nicotinic
acetylcholine receptors and predisposition to lung cancer. Curr
Opin Oncol. 23:83–87. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Albuquerque EX, Pereira EF, Alkondon M and
Rogers SW: Mammalian nicotinic acetylcholine receptors: from
structure to function. Physiol Rev. 89:73–120. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Levin ED, McClernon FJ and Rezvani AH:
Nicotinic effects on cognitive function: behavioral
characterization, pharmacological specification, and anatomic
localization. Psychopharmacology. 184:523–539. 2006. View Article : Google Scholar
|
|
7
|
Cardinale A, Nastrucci C, Cesario A and
Russo P: Nicotine: specific role in angiogenesis, proliferation and
apoptosis. Crit Rev Toxicol. 42:68–89. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kale J, Liu Q, Leber B and Andrews DW:
Shedding light on apoptosis at subcellular membranes. Cell.
151:1179–1184. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ke SZ, Ni XY, Zhang YH, Wang YN, Wu B and
Gao FG: Camptothecin and cisplatin upregulate ABCG2 and MRP2
expression by activating the ATM/NF-κB pathway in lung cancer
cells. Int J Oncol. 42:1289–1296. 2013.PubMed/NCBI
|
|
10
|
Sweatt JD: The neuronal MAP kinase
cascade: a biochemical signal integration system subserving
synaptic plasticity and memory. J Neurochem. 76:1–10. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Thornton TM and Rincon M: Non-classical
p38 map kinase functions: cell cycle checkpoints and survival. Int
J Biol Sci. 5:44–51. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhuang S and Schnellmann RG: A
death-promoting role for extracellular signal-regulated kinase. J
Pharmacol Exp Ther. 319:991–997. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jin HJ, Li HT, Sui HX, Xue MQ, Wang YN,
Wang JX and Gao FG: Nicotine stimulated bone marrow-derived
dendritic cells could augment HBV specific CTL priming by
activating PI3K-Akt pathway. Immunol Lett. 146:40–49. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jin HJ, Sui HX, Wang YN and Gao FG:
Nicotine up-regulated 4-1BBL expression by activating Mek-PI3K
pathway augments the efficacy of bone marrow-derived dendritic cell
vaccination. J Clin Immunol. 33:246–254. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dineley KT, Westerman M, Bui D, Bell K,
Ashe KH and Sweatt JD: Beta-amyloid activates the mitogen-activated
protein kinase cascade via hippocampal alpha7 nicotinic
acetylcholine receptors: In vitro and in vivo mechanisms related to
Alzheimer’s disease. J Neurosci. 21:4125–4133. 2001.PubMed/NCBI
|
|
16
|
Hu SX, Sui HX, Jin HJ, Ni XY, Liu XX, Xue
MQ, Zhang Y and Gao FG: Lipopolysaccharide and dose of nicotine
determine the effects of nicotine on murine bone marrow-derived
dendritic cells. Mol Med Rep. 5:1005–1010. 2012.PubMed/NCBI
|
|
17
|
Oddo S, Caccamo A, Green KN, Liang K, Tran
L, Chen Y, Leslie FM and LaFerla FM: Chronic nicotine
administration exacerbates tau pathology in a transgenic model of
Alzheimer’s disease. Proc Natl Acad Sci USA. 102:3046–3051.
2005.PubMed/NCBI
|
|
18
|
Zhang J, Liu Q, Chen Q, Liu NQ, Li FL, Lu
ZB, Qin C, Zhu H, Huang YY, He W and Zhao BL: Nicotine attenuates
beta-amyloid-induced neurotoxicity by regulating metal homeostasis.
FASEB J. 20:1212–1214. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li J, Wang G, Liu J, Zhou L, Dong M, Wang
R, Li X, Li X, Lin C and Niu Y: Puerarin attenuates
amyloid-beta-induced cognitive impairment through suppression of
apoptosis in rat hippocampus in vivo. Eur J Pharmacol. 649:195–201.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Plotnikov EY, Morosanova MA, Pevzner IB,
Zorova LD, Manskikh VN, Pulkova NV, Galkina SI, Skulachev VP and
Zorov DB: Protective effect of mitochondria-targeted antioxidants
in an acute bacterial infection. Proc Natl Acad Sci USA.
110:E3100–E3108. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cui WY, Wang J, Wei J, Cao J, Chang SL, Gu
J and Li MD: Modulation of innate immune-related pathways in
nicotine-treated SH-SY5Y cells. Amino Acids. 43:1157–1169. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Park HY, Kim GY, Kwon TK, Hwang HJ, Kim
ND, Yoo YH and Choi YH: Apoptosis induction of human leukemia U937
cells by 7,8-dihydroxyflavone hydrate through modulation of the
Bcl-2 family of proteins and the MAPKs signaling pathway. Mutat
Res. 751:101–108. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhu X, Jiang Y, Shan PF, Shen J, Liang QH,
Cui RR, Liu Y, Liu GY, Wu SS, Lu Q, Xie H, Liu YS, Yuan LQ and Liao
EY: Vaspin attenuates the apoptosis of human osteoblasts through
ERK signaling pathway. Amino Acids. 44:961–968. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chi J, Zhu Y, Fu Y, Liu Y, Zhang X, Han L,
Yin X and Zhao D: Cyclosporin A induces apoptosis in H9c2
cardiomyoblast cells through calcium-sensing receptor-mediated
activation of the ERK MAPK and p38 MAPK pathways. Mol Cell Biochem.
367:227–236. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gao FG, Wan DF and Gu JR: Ex vivo nicotine
stimulation augments the efficacy of therapeutic bone
marrow-derived dendritic cell vaccination. Clin Cancer Res.
13:3706–3712. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Toborek M, Son KW, Pudelko A,
King-Pospisil K, Wylegala E and Malecki A: ERK 1/2 signaling
pathway is involved in nicotine-mediated neuroprotection in spinal
cord neurons. J Cell Biochem. 100:279–292. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li Y, Chen G, Zhao J, Nie X, Wan C, Liu J,
Duan Z and Xu G: 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) induces
microglial nitric oxide production and subsequent rat primary
cortical neuron apoptosis through p38/JNK MAPK pathway. Toxicology.
312:132–141. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang X, Cheng Z, Su Q, Zhu X, Wang Q,
Chen R and Wang X: Neuroprotection by nicotine against
colchicine-induced apoptosis is mediated by PI3-kinase - Akt
pathways. Int J Neurosci. 122:324–332. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Barr J, Sharma CS, Sarkar S, Wise K, Dong
L, Periyakaruppan A and Ramesh GT: Nicotine induces oxidative
stress and activates nuclear transcription factor kappa B in rat
mesencephalic cells. Mol Cell Biochem. 297:93–99. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Faris M, Kokot N, Latinis K, Kasibhatla S,
Green DR, Koretzky GA and Nel A: The c-Jun N-terminal kinase
cascade plays a role in stress-induced apoptosis in Jurkat cells by
up-regulating Fas ligand expression. J Immunol. 160:134–144.
1998.PubMed/NCBI
|
|
32
|
Minamino T, Christou H, Hsieh CM, Liu Y,
Dhawan V, Abraham NG, Perrella MA, Minamino T, Christou H, Hsieh
CM, Liu Y, Dhawan V, Abraham NG, Perrella MA, Mitsialis SA and
Kourembanas S: Targeted expression of heme oxygenase-1 prevents the
pulmonary inflammatory and vascular responses to hypoxia. Proc Natl
Acad Sci USA. 98:8798–8803. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Court FA and Coleman MP: Mitochondria as a
central sensor for axonal degenerative stimuli. Trends Neurosci.
35:364–372. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yamaguchi R, Andreyev A, Murphy AN,
Perkins GA, Ellisman MH and Newmeyer DD: Mitochondria frozen with
trehalose retain a number of biological functions and preserve
outer membrane integrity. Cell Death Differ. 14:616–624. 2007.
View Article : Google Scholar : PubMed/NCBI
|