Introduction
Phosphatidylethanolamine (PE) is an important
phospholipid in mammalian membranes. PE is present primarily in the
inner leaflet of the membrane bilayer in a viable, typical
mammalian cell (1). PE is
sustained in the bilayer configuration by interacting with other
phospholipids in biological membranes. However, the reorganisation
of membrane phospholipids could lead to the expression of the
non-bilayer nature of PE and the induction of bilayer instability
(2). Accumulating evidence has
demonstrated that the translocation of PE to the cell surface in
many distinct biological events (1). For instance, PE is exposed to the
cell surface, thus providing a hallmark for detection in apoptotic
cells (1). As a viable
alternative, PE-binding probes have been shown to be selective in
detecting dead and dying cells (3). Moreover, findings of a previous
study indicated that exogenous PE induces apoptosis in hepatoma
HepG2 cells (4). However, the
effects of PE on apoptosis in SMMC-7721 cells and the underlying
mechanism of PE remain unclear.
Mitochondria play a critical role in the regulation
of apoptosis. Changes in the mitochondrial membrane potential (ΔΨm)
may switch the committed cells to apoptotic death (5). Findings of previous studies
demonstrated that mitochondrial dysfunction, which occurs during
apoptosis, causes the release of cytochrome c and, thus,
contributes to apoptosis (6,7).
The Bcl-2 family proteins regulate the release of cytochrome
c and other proteins through the outer mitochondrial
membrane (OMM) (8,9). Some members of the Bcl-2 family
inhibit apoptosis, such as Bcl-2, Bcl-xl, and Mcl-1, whereas Bax
and Bak, activate apoptosis. Bax and Bak induced the release of
cytochrome c, whereas anti-apoptotic Bcl-2 family members
inhibited the release of cytochrome c (7). During the apoptotic process, the
cytochrome c that is released from the mitochondria
sequentially triggers a caspase cascade, which is characteristic of
the apoptotic pathway, in which caspase-3 plays a dominant role
(10). Therefore, a balance
between pro-apoptotic (Bax/bad) and anti-apoptotic (Bcl-2/Bcl-xl)
members of the Bcl-2 family proteins and their up- and
downregulation usually determine whether cells undergo apoptosis or
survive (11). However, the
effect of the mitochondrial pathway on the PE-induced apoptosis of
SMMC-7721 cells remains unclear.
As a member of the mitogen-activated protein kinases
(MAPK) family, Erk is crucial in regulating cell growth and
differentiation (12) and has
been shown to act as an important modulator of various
apoptosis-inducing signals in different systems (13,14). It was reported that the MEK/ERK
signalling pathway regulated the expression of Bcl-2 (15). The aim of the present study was to
investigate whether the Erk pathway is also involved in exogenous
PE-induced apoptosis. Stat1 is partially phosphorylated by the Erk
pathway (16), while the
phosphorylation of Stat1 is generally associated with cell cycle
arrest and apoptosis (17,18).
Stat1 is important in the interferon-response, following various
stressful stimuli that induce apoptotic or cell cycle checkpoint
responses (16,19–22). Results of a previous study showed
that the high expression of Stat1 and its activator (IFNγ) reduced
the basal expression of the Bcl-2 promoter (23). To study the underlying mechanisms
of PE-induced apoptosis, we investigated whether Erk and Stat1/2
signalling pathways were involved in PE-induced apoptosis in the
hepatic cancer line SMMC-7721.
Materials and methods
Chemicals and reagents
Chemicals and cell culture reagents (RPMI-1640
medium, penicillin/streptomycin, and FBS) were obtained from Sigma
(St. Louis, MO, USA) and Gibco Laboratories (Grand Island, NY,
USA), respectively. An Annexin V-FITC Apoptosis Detection kit was
obtained from BD Bioscience (Franklin Lakes, NJ, USA).
Antibodies to phosphospecific Erk1/2, Stat1, Stat2
and antibodies against Bax, Bcl-2, caspase-3 were purchased from
Cell Signalling Technology, Inc. (Beverly, MA, USA). All the
reagents were of analytical grade.
Cell culture and treatment
SMMC7721 cells were provided by the Molecular
Biology Centre of the First Affiliated Hospital, Xi’an Jiaotong
University, China. SMMC-7721 cells were grown in RPMI-1640 medium,
which was supplemented with 10% bovine serum albumin (BSA) and 1%
penicillin/streptomycin in a humidified atmosphere of 95% air/5%
CO2 at 37°C. The cells were cultured at different
densities depending on the assay. The treatment was initiated with
PE 24 h after plating. After the cells were treated with 0.125––1.0
mM/l PE for 6–48 h treatment, the cultures were terminated, and
then adherent cells were collected for evaluation. The
morphological change after exposure to PE was observed using a
phase-contrast inverse microscope (DMIRBHC; Leica, Mannheim,
Germany).
Cell viability assay
Cells were cultured at a density of 2×104
cells/well in a 96-well plate in RPMI-1640 medium. Cells were
allowed to adhere and then treated with the indicated concentration
of PE for 24 and 48 h. Subsequently, 20 μl of MTT (5 mg/ml) was
added into each well. After incubation at 37°C for 4 h, the medium
was removed, 100 μl of DMSO was added to each well and absorbance
was read at 490 nm using a microplate reader (BMG Labtech GmbH,
Ortenberg, Germany). The experiments were performed three times,
and the mean absorbance values were calculated. The results are
expressed as the percentage of inhibition that produced a reduction
in the absorbance by PE treatment compared with the control group
(not treated with PE).
Cell cycle analysis
In total, 1×106 cells were synchronised
by exposure to medium with a low concentration of FBS for 24 h to
induce cell cycle arrest. The culture medium was then replaced with
nutrient-rich medium, and the cells were treated with various
concentrations of PE for 24 h. The cells were collected by
trypsinisation and washed twice with cold PBS. The cells were then
fixed with 75% cold ethanol at 4°C for 24 h. Prior to analysis, the
cells (1×105) were labelled with propidium iodide (PI)
(1 mg/ml) in the presence of 1% RNase A for 30 min. The cells were
sorted in a FACS Calibur flow cytometer using BD Cell Quest
software (San Jose, CA, USA).
Apoptotic assay
Cell apoptosis was determined using an Annexin
V-FITC Apoptosis Detection kit I according to the manufacturer’s
protocol. SMMC-7721 cells were cultured at 1×106
cells/ml in 6-well plates for 24 h. The culture medium was replaced
with fresh medium, and the cells were treated with indicated
concentrations of PE for 24 h. After digesting with 0.25%
trypsin-EDTA for 3 min, the cells were collected and centrifuged at
71.55 × g for 8 min. The pellets were then washed twice with cold
PBS. Approximately 1×105–1×106 cells were
resuspended in 100 μl 1X binding buffer and were transferred to a
sterile flow cytometry glass tube. The cells were incubated with 5
μl Annexin V-FITC and 5 μl of 20 μg/ml PI in the dark at room
temperature for 15 min. The apoptotic cells were analysed using a
flow cytometer (FACS Calibur; BD Biosciences). Annexin V-FITC and
PI emissions were detected in the FL 1 and FL 2 channels.
Measurement of ΔΨm
ΔΨm was analysed using a rhodamine 123 assay as
previously described (24).
Rhodamine 123 can be selectively absorbed by mitochondria and is
proportional to the ΔΨm (25).
Briefly, the cells were seeded in 24-well plates at
2×105 cells/well. Subsequent to PE treatment for 24 h,
the cells were washed twice with cold PBS and incubated with cold
PBS containing 1 μl of 10 μg/μl rhodamine 123 at 37°C for 30 min.
The fluorescent intensity of rhodamine 123 in the mitochondria was
detected using a fluorescence microplate reader at an excitation
wavelength of 505 nm and an emission wavelength of 527 nm. The data
were expressed as a percentage of the control.
Confocal microscopy
For immunofluorescence studies, SMMC-7721 cells were
seeded in 6-well plates for 24 h. The cells were washed twice with
ice-cold PBS and fixed with 75% cold ethanol for 30 min at 4°C.
After washing with PBS three times, the cells were incubated with
0.5% Triton X-100 for 25 min. The cells were blocked by incubation
with 10% goat serum in PBS containing 0.3% Triton X-100 and 0.5%
BSA at room temperature for 20 min, followed by incubation with a
mouse monoclonal antibody against Bcl-2, Bax, caspase-3,
phospho-Erk1/2, phospho-Stat1 and phospho-Stat2 (1:100 in PBS) at
4°C overnight. Cells were then incubated with FITC-conjugated goat
anti-mouse IgG (1:100 in PBS) for 2 h at 37°C in a humidified
atmosphere. After washing three times with PBS, the fluorescent
images of cells were collected using a Leica TCS SP2 laser scanning
confocal microscope (Leica).
Western blotting
SMMC-7721 cells (1×106) were incubated in
a 6-well plate for 24 h and treated with different concentrations
of PE for the indicated time periods. Whole-cell lysates were
prepared from PE-treated and untreated cells in RIPA buffer (20 mM
Tris-HCl pH 7.5, 120 mM NaCl, 1.0% Triton X-100, 0.1% SDS, 1%
sodium deoxycholate, 10% glycerol, 1 mM EDTA and 1% protease
inhibitor cocktail). Following centrifugation at 12,879.36 × g at
4°C for 5 min, the protein concentrations in the supernatants were
determined after the cells were centrifuged at 12,879.36 × g at 4°C
for 5 min. Equal amounts of protein (50 μg) in whole-cell lysates
were mixed with reducing sample buffer (0.92 M Tris-HCl pH 8.8,
1.5% SDS, 4% glycerol, and 280 mM 2-ME), and the samples were
boiled at 99°C for 8 min, separated on SDS-polyacrylamide gels and
transferred to polyvinylidene difluoride (PVDF) membranes. The
membranes were blocked in blocking buffer (Tris-buffered saline
containing 3% BSA, 20 mM NaF, 2 mM EDTA, and 0.2% Tween-20) for 2 h
at 37°C. The membrane was then incubated for 2 h with the
appropriate primary antibody at 37°C. The primary antibodies were
used at the following dilutions: 1:800 for phospho-Erk1/2,
phospho-Stat1 or phospho-Stat2, 1:500 for Bcl-2 and 1:3,000 for
GAPDH. The membrane was washed three times with TBST buffer
(Tris-buffered saline containing 20 mM NaF, 2 mM EDTA, and 0.2%
Tween-20), incubated for 60 min with HRP-conjugated secondary
antibodies and washed three times with TBST buffer. Detection was
achieved by chemical fluorescence following an enhanced
chemiluminescence (ECL) western blotting protocol (Amersham
Biosciences, Piscataway, NJ, USA). The image capture and analysis
were performed using a GeneGnome imaging system (Syngene,
Frederick, MD, USA).
Statistical analysis
Data are presented as the means ± SD of at least
three independent experiments that were obtained from different
cultures. All the analyses were performed using the SPSS software
version 15.0. An analysis of variance (ANOVA) was used to test for
significant differences between the groups, which was followed by
the Dunnett’s test for multiple comparisons. Differences were
considered significant when the calculated P-values were
<0.05.
Results
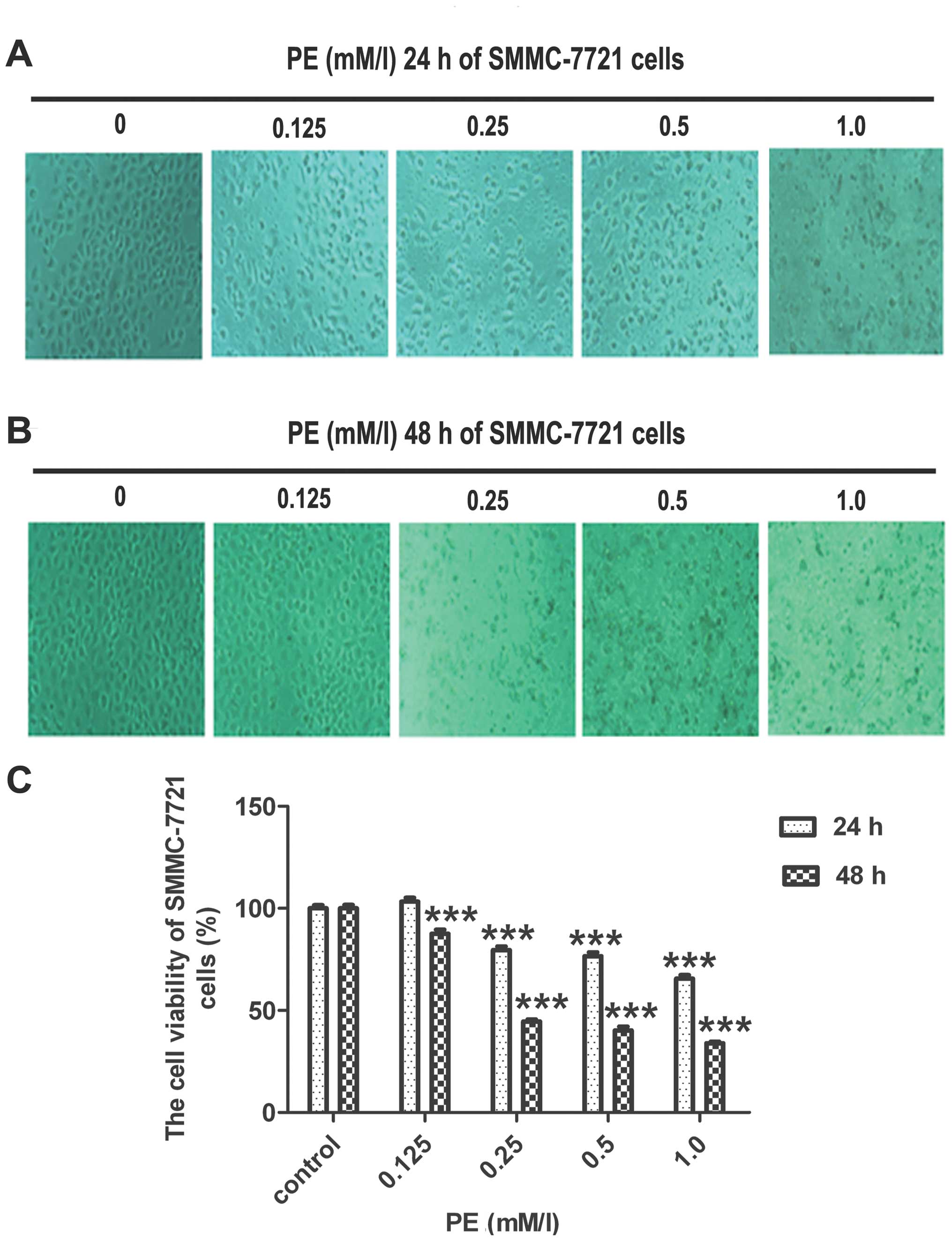
PE treatment decreases viability in
SMMC-7721 cells
The effects of PE on the viability of SMMC-7721
cells were assessed using the MTT uptake method. PE inhibited the
growth of SMMC-7721 cells in a concentration- and time-dependent
manner (Fig. 1). In SMMC-7721
cells, the viability value was 80% after treatment with 0.25 mM/l
PE for 24 h, whereas for treatment with 0.5 and 1.0 mM/l PE after
24 h, the values were 77 and 66%, respectively. After 48 h, in
response to treatment with 0.125, 0.25, 0.5 and 1.0 mM/l PE, the
viability values of SMMC-7721 cells decreased to 88, 45, 40 and
34%, respectively.
Direct observation using an inverted microscope
revealed numerous morphological changes in the cells that were
treated with PE (Fig. 1B and C).
The untreated cells exhibited a regular and healthy shape. After
incubation with PE, the cellular morphology was severely distorted
and polyhedral or irregular and even grew in a tightly connected
manner.
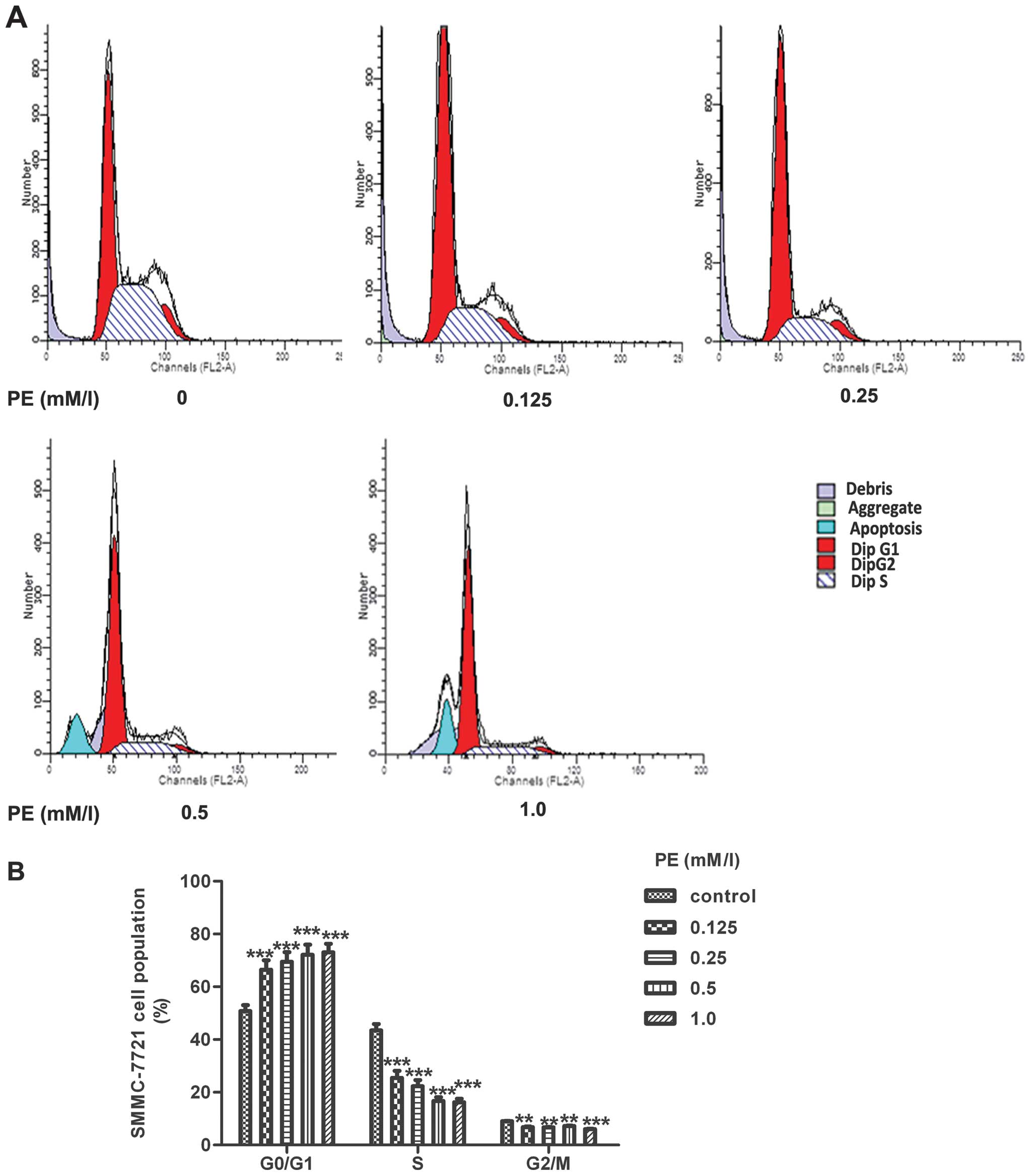
PE induced a G0/G1 cell cycle arrest
To assess whether the PE-induced cell growth
inhibition was due to arrest at a specific point of the cell cycle,
SMMC-7721 cells were synchronised by exposure to medium with a low
FBS concentration for 24 h until starvation. The cells were then
returned to the culture nutrient-rich medium, which stimulated cell
proliferation, with different concentrations of PE for 24 h. The
cells were collected for flow cytometric analysis. As shown in
Fig. 2B, the PE-treated cells
exhibited a dose-dependent accumulation of G0/G1 phase-arrested
cells compared with the corresponding untreated cells. At 1.0 mM/l,
PE significantly increased cell cycle arrest during the G0/G1 phase
of SMMC-7721 cells to 77±0.40%, whereas the percentage of SMMC-7721
cells in the G0/G1 phase in the control group was only 53±0.28%.
This increase in the G1 cell population was mostly at the expense
of the S and G2/M phase cell populations.
PE-induced apoptosis
To examine the degree of apoptosis that was induced
by PE, we used Annexin V-FITC and PI to distinguish apoptotic cells
from necrotic cells. Annexin V is a binding protein with a strong
affinity and selectivity for phosphatidylserine, which appears on
the cell surface as a general indicator of apoptosis. However, the
translocation of phosphatidylserine to the cell surface also occurs
during necrosis (26). Therefore,
measuring Annexin V binding to the cell surface was performed in
conjunction with PI staining. Early apoptotic cells were identified
by positive Annexin V-FITC and negative PI staining, whereas cells
that were in late apoptosis or necrotic cells were positive for
both Annexin V-FITC and PI. Viable cells were negative for both
Annexin V-FITC and PI. The results were interpreted as follows:
cells in the lower left quadrant (Annexin
V−/PI−) were considered to be viable cells,
cells in the lower right quadrant (Annexin
V+/PI−) were considered early apoptotic
cells, cells in the upper right quadrant (Annexin
V+/PI+) were considered late apoptotic cells,
and cells in the upper left quadrant (Annexin
V−/PI+) were considered necrotic cells. The
total apoptotic rate was calculated as the rate of cells in the
lower right quadrant (Annexin V+/PI−) plus
the rate of cells in the upper right quadrant (Annexin
V+/PI+). As shown in Fig. 3, PE increased the proportion of
SMMC-7721 cells that were stained with both Annexin V and PI in a
concentration- and time-dependent manner. The total percentage of
apoptotic and necrotic SMMC-7721 cells in the untreated cells
increased from 3±0.21 to 25±3.93, 35±3.81, 55±3.46, and 77±1.70%,
when the cells were treated with 0.125, 0.25, 0.5 and 1.0 mM/l PE,
respectively, for 24 h. The early apoptotic rate was 0.76±0.06% in
the control group, whereas this rate markedly increased to 15±0.76,
21±1.08, 37±0.84, and 41±0.52% in the experiment groups of treated
with 0.125, 0.25, 0.5 and 1.0 mM/l PE, respectively for 24 h. These
results showed that PE efficiently induced apoptosis in SMMC-7721
cells.
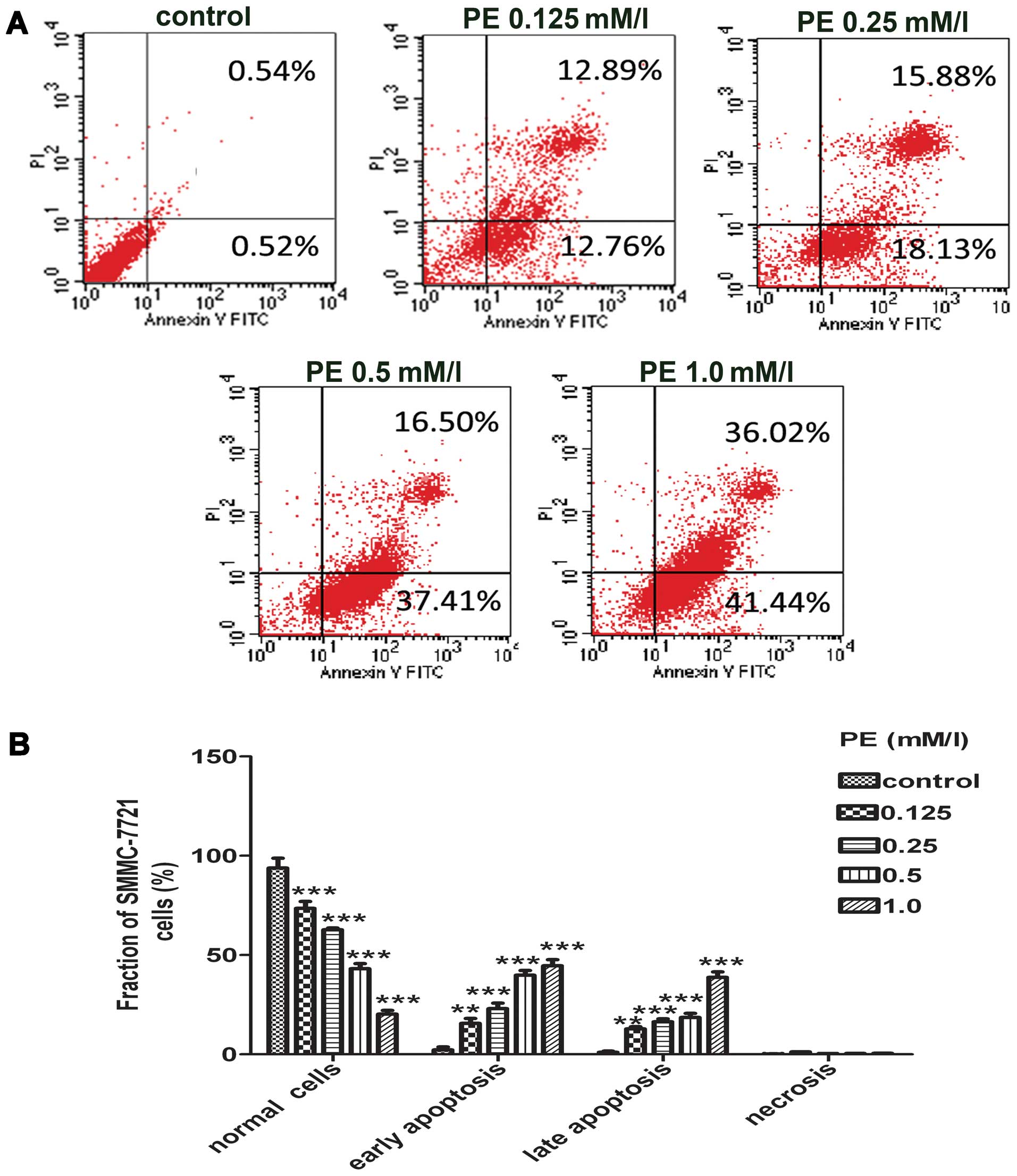 | Figure 3Detection of phosphatidylethanolamine
(PE)-induced apoptosis in SMMC-7721 cells. (A) The representative
images of flow cytometry analysis were shown. After the cells were
treated with PE (0, 0.125, 0.25, 0.5 and 1.0 mM/l) for 24 h, the
cells were stained with FITC-conjugated Annexin V and propidium
iodide (PI), followed by flow cytometric analysis. Cell populations
with Annexin V−/PI−, Annexin
V+/PI−, Annexin V+/PI+,
Annexin V−/PI+ were regarded as living, early
apoptotic, late apoptotic and necrotic cells, respectively. (B)
Statistical analysis of the percentages of the apoptotic cells. The
results were presented as the mean ± SD of three independent
experiments performed. **P<0.01 and
***P<0.001 vs. the control group. |
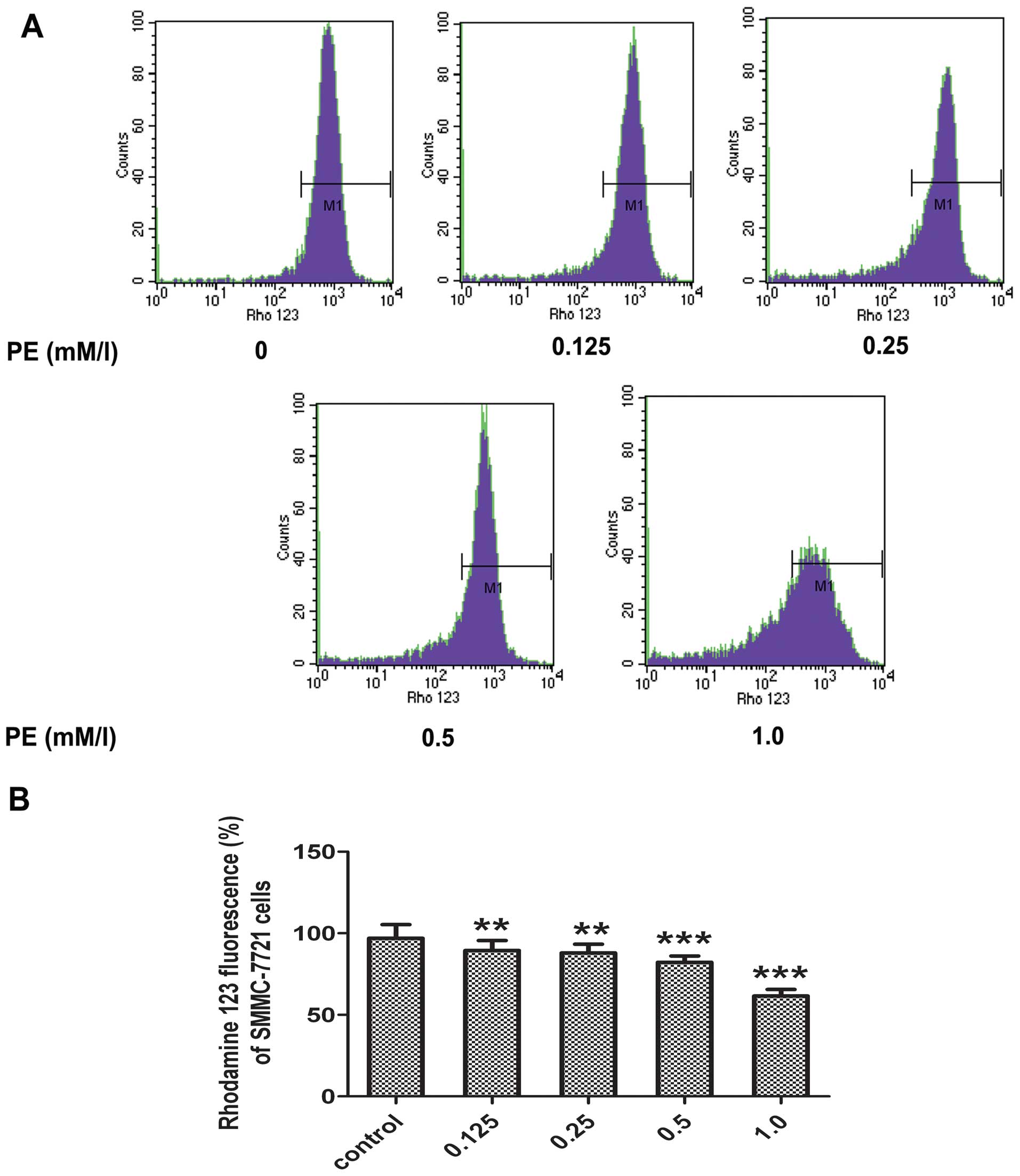
Evaluation of ΔΨm damage
Mitochondrial dysfunction is an important
characteristic of apoptotic cell death. ΔΨm perturbation under PE
treatment was examined. To evaluate the changes in the ΔΨm, a
mitochondria-specific dye rhodamine 123 was used. As shown in
Fig. 4, following the PE
treatment of SMMC-7721 cells for 24 h, the level of the ΔΨm
decreased compared with the control group. A dose-dependent
reduction in ΔΨm was also observed in the PE-treated cells. These
data indicated that PE-induced apoptosis was accompanied by a
collapse in the ΔΨm.
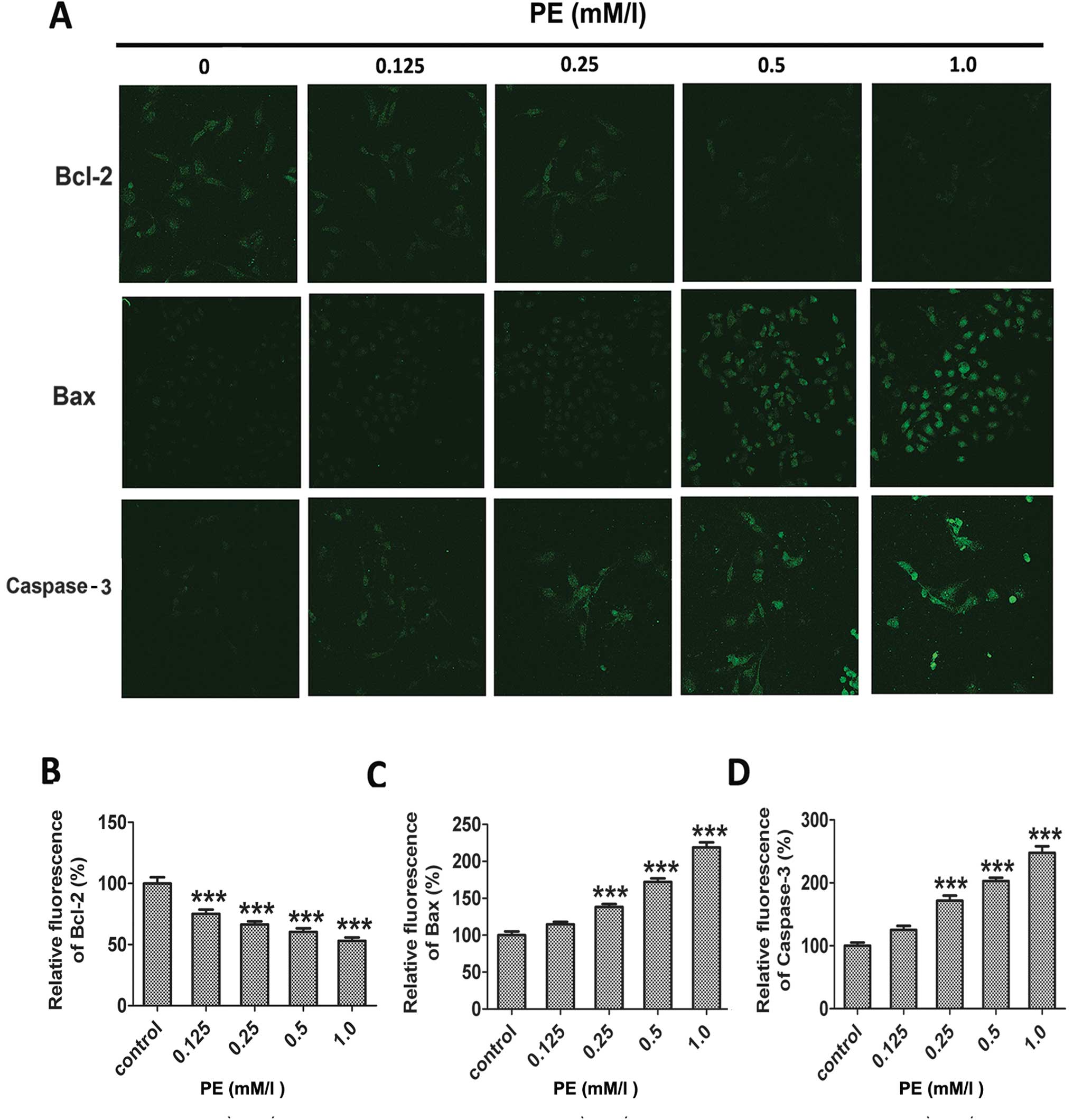
Detection of caspase-3 activity
Since caspase activation is a major step in
apoptosis, we studied the involvement of caspase activation in
PE-stimulated apoptosis in SMMC-7721 cells using immunofluorescence
analysis. As shown in Fig. 6, PE
treatment caused the levels of caspase-3 to increase in a
concentration-dependent manner. Exposure to 0.125, 0.25, 0.5 and
1.0 mM/l PE for 24 h resulted in 1.2-, 1.6-, 2.0- and 2.4-fold
increases in caspase-3 activity.
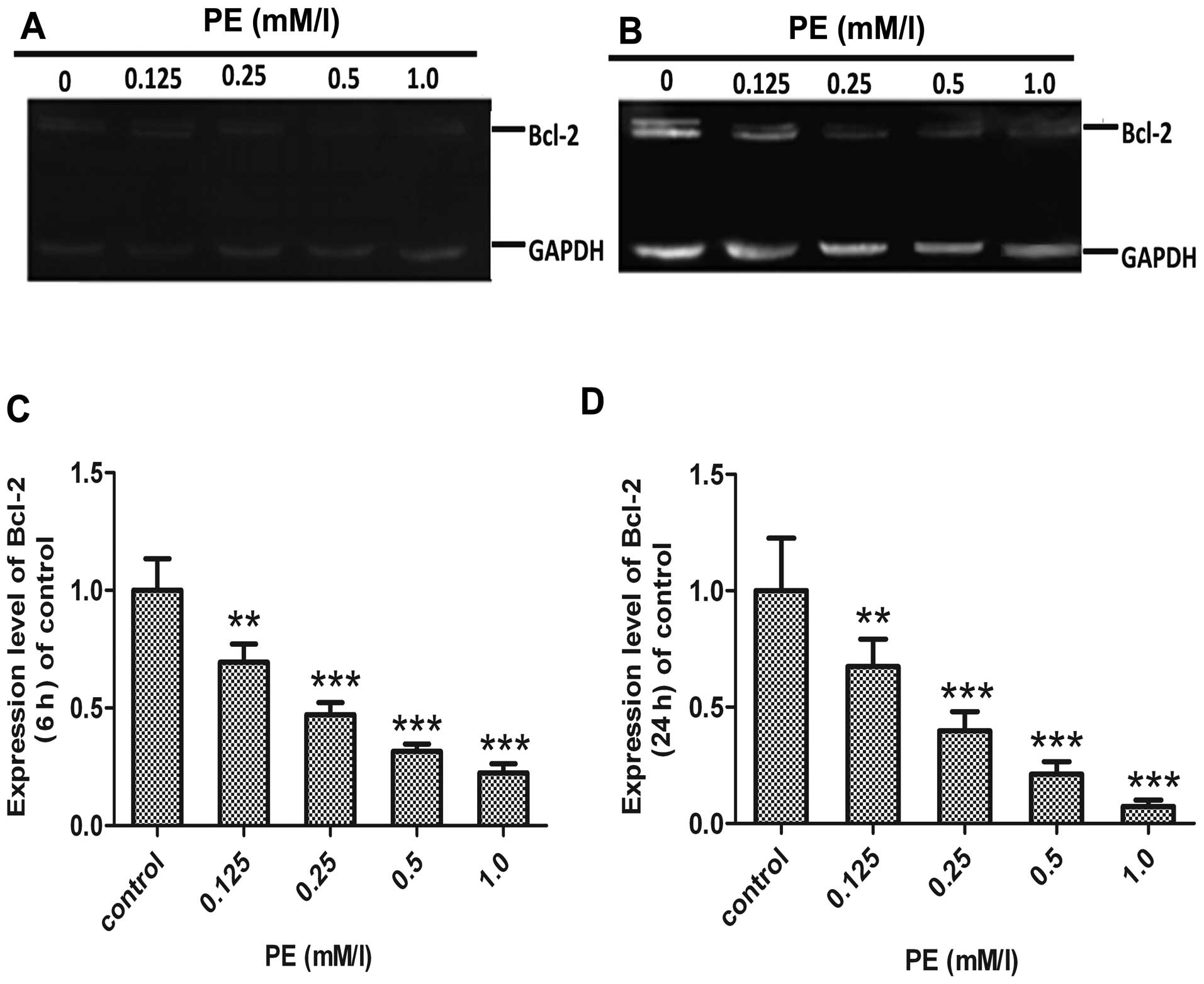
Evaluation of Bax and Bcl-2
The pro-apoptotic protein, Bax, has been reported to
translocate from cytosol to mitochondria following the exposure of
cells to apoptotic stresses. The anti-apoptotic protein Bcl-2 is
known to prevent Bax redistribution to the mitochondria, caspase
activation and apoptosis (27).
In the present study, to detect whether regulation of Bax and Bcl-2
was involved in PE-induced apoptosis, we examined the changes in
Bax and Bcl-2 using immunofluorescence and western blotting in
SMMC-7721 cells. As shown in Figs.
5 and 6, Bax expression was
upregulated, whereas expression of Bcl-2 was downregulated in
SMMC-7721 cells after PE exposure. Therefore, PE treatment induced
apoptosis, which was accompanied by the dose-dependent
downregulation of Bcl-2 and upregulation of Bax.
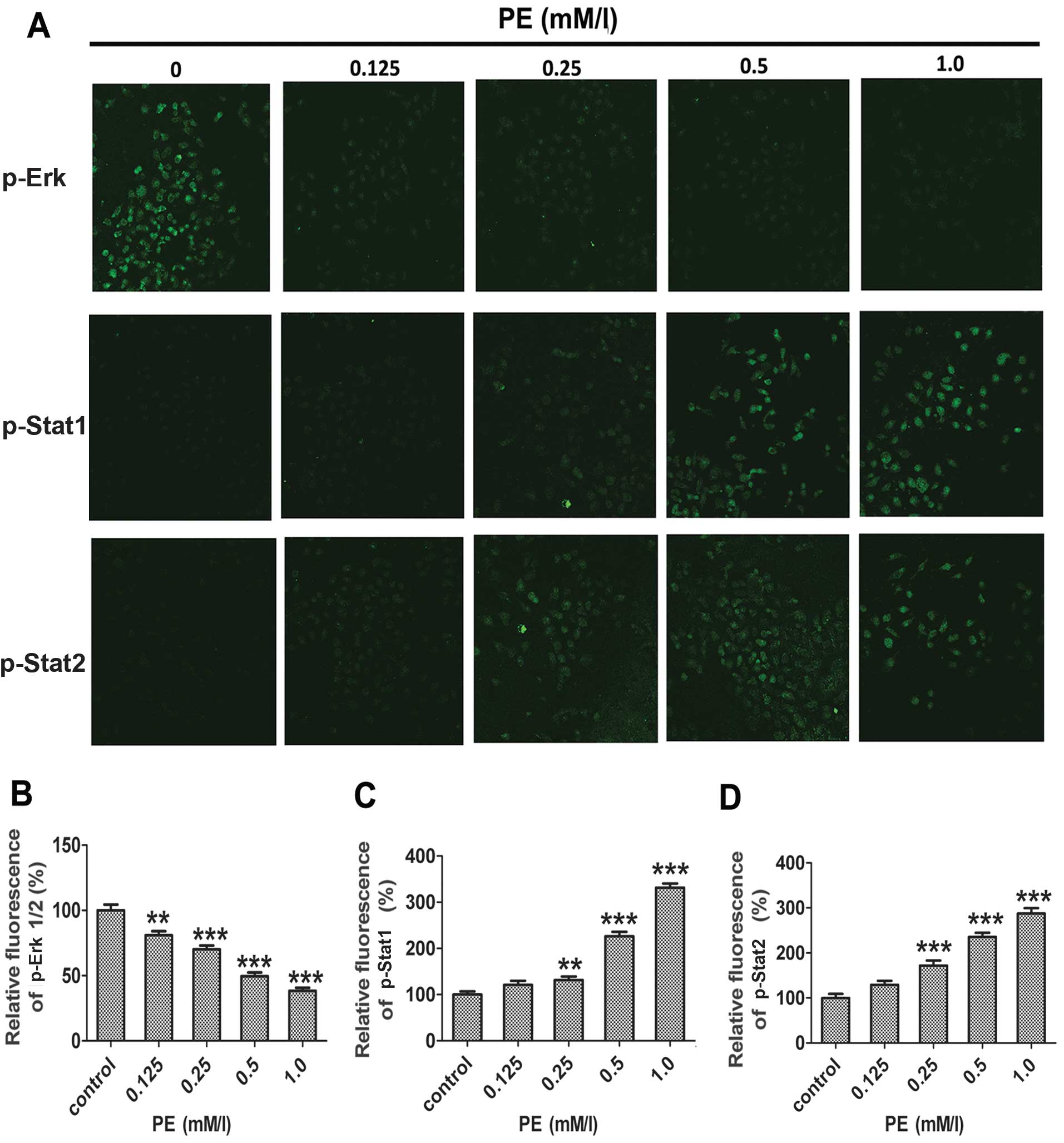
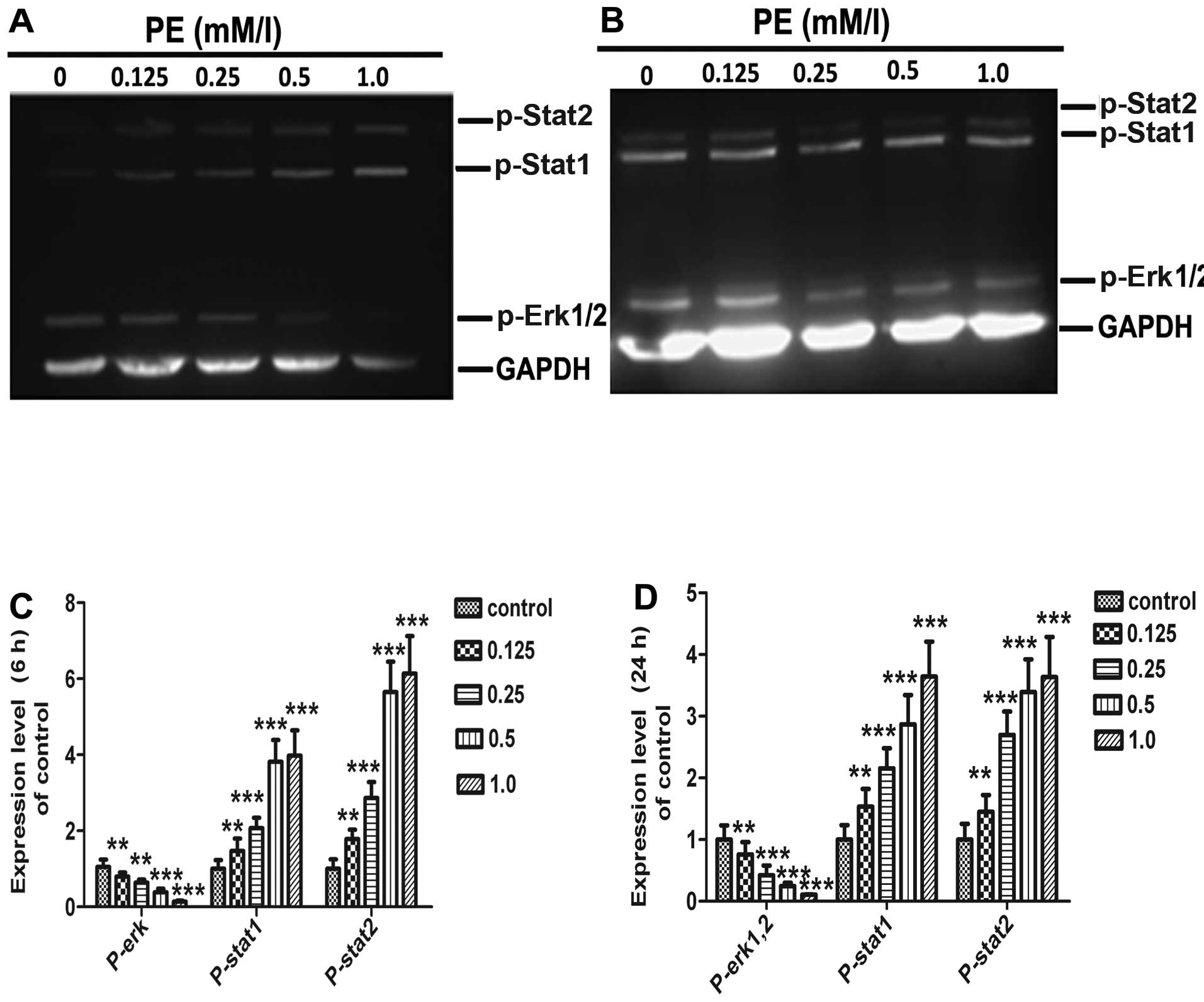
Effects of PE on MAPK phosphorylation and
Stat signalling
MAPKs are known to play an important cellular
regulatory role (12,14). MAPKs and signal transducers and
activators of transcription factor (Stats) signalling pathways
regulate cell proliferation, survival, and differentiation
(28). To investigate the
involvement of MAPKs and Stats in PE-induced apoptosis in SMMC-7721
cells, we examined changes in the phospho-Erk and phospho-Stat
activities of PE-treated cells using western blotting. As shown in
Figs. 7 and 8, Erk phosphorylation decreased
continuously in a time- and dose-dependent manner; however, the
level of Stat1/2 phosphorylation increased.
Discussion
The results of the MTT assay employed in the present
study showed that PE treatment suppressed the viability of the
SMMC-7721 human hepatic cancer line. The effect was gradually
enhanced with increasing PE concentrations (0.125, 0.25, 0.5 and
1.0 mM/l). Moreover, we observed a marked inhibitory effect when
SMMC-7721 cells were treated with PE for 48 h compared with 24 h.
This result suggests that PE inhibited the viability of SMMC-7721
cells in a dose- and time-dependent manner. To determine the
mechanisms that lead to the loss of SMMC-7721 cell proliferation by
PE, the effects of PE treatment on cell cycle arrest were examined.
An analysis of the cell cycle following the treatment of SMMC-7721
cells with different concentrations of PE showed a higher number of
cells in the G0/G1 phase in a dose-dependent manner compared to the
untreated cells. These results suggest that PE inhibited cell
proliferation via a G0/G1 phase arrest. We also found that PE
inhibited cell proliferation via a G0/G1 phase arrest in HEK-293
cells (data not shown). To examine whether apoptosis is involved in
PE-induced cell death in SMMC-7721 and HEK-293 cells, we
investigated the apoptotic effect of PE using flow cytometry for
Annexin V/PI staining. We found that PE treatment directly induced
apoptotic death in SMMC-7721 and HEK-293 cells. Accordingly, we
considered that exogenous PE-induced apoptosis was responsible for
its cytotoxicity in SMMC-7721 and HEK-293 cells. Although PE
externalisation has been identified as a molecular marker of
apoptosis, little is known regarding the effects of exogenous PE on
apoptosis. We hypothesized that PE is stimulated by PE
externalisation of the cytomembrane and that PE externalisation may
be indicative of apoptosis and induce cell apoptosis in
vitro.
Mitochondria are the critical mediators of apoptosis
in the intrinsic pathway (29).
The changes in the ΔΨm and mitochondrial dysfunction are considered
an early event in apoptosis (30,31). On receiving a death signal, the
mitochondrial membrane is disrupted, and cytochrome c is
released from mitochondria into the cytosol (30). Bcl-2 family proteins have been
shown to play an important role in the regulation of
mitochondria-mediated apoptosis, and are the upstream regulators of
the ΔΨm (32). The pro-apoptotic
member Bax translocates to the mitochondrion and integrates into
the OMM, where Bax promotes the excretion of cytochrome c
into the cytosol and the disruption of ΔΨm, whereas anti-apoptotic
protein Bcl-2 prevents this process by preserving mitochondrial
integrity (33). Bcl-2 protects
cells against apoptosis and modulates OMM permeability and the
release of cytochrome c (34–36). It has been suggested that Bax
induces a decrease in the membrane potential of mitochondria,
leading to an increase of mitochondrial membrane permeability and
the release of cytochrome c from mitochondria (37–39). Thus, the balance between Bax and
Bcl-2 is crucial in sustaining apoptosis in the intrinsic pathway
(40). Cytochrome c, which
is released from mitochondria leads to the subsequent activation of
downstream caspases, such as caspase-3 (41). Caspase-3, which is a downstream
effector in the caspase cascade, is considered an essential
executor for mitochondrial-dependent apoptotic pathways (42,43). In this study, PE treatment
decreased the ΔΨm in SMMC-7721 and HEK-293 cells (data not shown).
To verify PE-induced apoptosis, we determined the expression of
Bax, Bcl-2 and caspase-3 in SMMC-7721 cells by western blotting. We
found that PE increased caspase-3 expression in a dose-dependent
manner in SMMC-7721 cells. This result suggests that the
mitochondrial pathway is potentially involved in PE-induced cell
apoptosis. Our results also show that PE treatment upregulated the
expression of Bax and downregulated the level of Bcl-2 in a
dose-dependent manner, which eventually leads to an increase in the
ratio of Bax/Bcl-2 protein levels. We demonstrated that Bax/Bcl-2
signalling pathways may be involved in PE-induced apoptosis, which
has been accompanied by conspicuous reduction in the ΔΨm.
MAPKs are key mediators that transduce extracellular
signals from the membrane to the nucleus (44). As a member of the MAPK family, Erk
is important in the regulation of cell growth and mediates a
survival response that counteracts cell death (12). However, other studies have
reported that the activation of Erk is associated with apoptosis
(45,46). Therefore, the role of MAPK
signalling depends on the stimuli and cell type (47). In the present study, whether Erk
activation was involved in the PE-induced apoptotic cell death was
evaluated in SMMC-7721 cells that were treated with PE. The results
showed that PE treatment markedly suppressed Erk activation, which
indicated the involvement of Erk pathways in PE-induced apoptotic
death in SMMC-7721 cells. Tamura et al reported that Erk, as
the responsible kinase for the phosphorylation of Bcl-2, exerts an
effect on the anti-apoptotic function of Bcl-2 in human tumour cell
lines (48). Accordingly,
exogenous PE-induced apoptosis may be related to the downregulation
of Erk and to the repression of Bcl-2. Erk should be an upstream
regulator of Bcl-2. Results of previous studies have shown that the
activation of JNK and P38 is necessary for cancer cell death which
is initiated by a variety of anti-cancer agents and that, notably,
the JNK pathway plays an important role in the activation of the
mitochondrial-dependent apoptotic pathway (49,50). Whether JNK activation is involved
in the mitochondrial apoptotic pathway by PE treatment in SMMC-7721
cells may require further elucidation.
The Stats are a family of latent cytoplasmic
transcription factors that mediate intracellular signalling that is
initiated at cytokine cell-surface receptors and is transmitted to
the nucleus (51). After the
ligation of cytokine receptors, Stats become phosphorylated by
receptor kinases, dimerise and translocate to the nucleus, where
these molecules modulate the expression of Stat-responsive genes
(19). By regulating the target
gene expression, Stat proteins have been shown to play a major role
in mediating extensive biological processes, such as cell
proliferation, survival, apoptosis and differentiation (37–39). Stat1 is partially phosphorylated
by the Erk pathway (16), and
phosphorylation of Stat1 appears to be required for maximal
transcriptional activity (18).
It has been shown that Stat1 may induce apoptotic or cell cycle
checkpoint responses following various stressful stimuli (16,19–22). A previous study also reported that
Stat1 functionally promoted apoptosis and tumour suppression
(52). In this study, we observed
that PE treatment significantly increased the activation of
Stat1/Stat2 in SMMC-7721 cells (Figs.
7 and 8), suggesting that the
Stat1/Stat2 pathway may be involved in PE-induced SMMC-7721 cell
apoptosis. It has been shown that constitutively high activities of
Erk and PI3K-AKT in LU1205 cells inhibit Stat-transcriptional
activities via their effects on JAK2 (53). Another study showed that in
U3A-ST1 cells that constitutively express Stat1, IFNγ, which is a
known activator of Stat1, reduces the basal expression of the Bcl-2
promoter (23). Therefore, PE may
exert its inhibitory effects on Bcl-2 through the inhibition of the
Erk pathway and through the activation of the Stat1/2 pathway,
which would eventually promote SMMC-7721 cell apoptosis. A recent
study proved that testicular lumicrine factors protect the cells of
the initial segment by activating the Erk pathway, repressing Stat
pathways, and preventing apoptosis (54). This result may support our
conclusions from another viewpoint.
In conclusion, our results have demonstrated the
potential apoptotic effect of exogenous PE on SMMC-7721 cells. We
hypothesized that PE may induce a decrease in the membrane
potential of the mitochondria via the upregulation of the ratio of
Bax/Bcl-2 protein levels, which subsequently leads to
caspase-3-dependent apoptosis. In addition, the inhibition of Erk
and the activation of Stat1/2 signalling may be involved in the
PE-induced apoptosis of SMMC-7721 cells.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (81273196). We would like to thank
Tusheng Song and Chen Huang from the Department of Genetics and the
Molecular Biology Department of Xi’an Jiaotong University for their
kind assistance.
References
|
1
|
Zhao M: Lantibiotics as probes for
phosphatidylethanolamine. Amino Acids. 41:1071–1079. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Post JA, Bijvelt JJ and Verkleij AJ:
Phosphatidylethanolamine and sarcolemmal damage during ischemia or
metabolic inhibition of heart myocytes. Am J Physiol.
268:H773–H780. 1995.PubMed/NCBI
|
|
3
|
Zhao M, Li Z and Bugenhagen S:
99mTc-labeled duramycin as a novel phosphatidylethanolamine-binding
molecular probe. J Nucl Med. 49:1345–1352. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yao Y, Huang C, Li ZF, et al: Exogenous
phosphatidylethanolamine induces apoptosis of human hepatoma HepG2
cells via the bcl-2/Bax pathway. World J Gastroenterol.
15:1751–1758. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Keeble JA and Gilmore AP: Apoptosis
commitment-translating survival signals into decisions on
mitochondria. Cell Res. 17:976–984. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Itoh K, Hase H, Kojima H, Saotome K,
Nishioka K and Kobata T: Central role of mitochondria and p53 in
Fas-mediated apoptosis of rheumatoid synovial fibroblasts.
Rheumatology (Oxford). 43:277–285. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Suen DF, Norris KL and Youle RJ:
Mitochondrial dynamics and apoptosis. Genes Dev. 22:1577–1590.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Adams JM and Cory S: The Bcl-2 apoptotic
switch in cancer development and therapy. Oncogene. 26:1324–1337.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chipuk JE and Green DR: How do BCL-2
proteins induce mitochondrial outer membrane permeabilization?
Trends Cell Biol. 18:157–164. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jiang L, Liu Y, Ma MM, Tang YB, Zhou JG
and Guan YY: Mitochondria dependent pathway is involved in the
protective effect of bestrophin-3 on hydrogen peroxide-induced
apoptosis in basilar artery smooth muscle cells. Apoptosis.
18:556–565. 2013. View Article : Google Scholar
|
|
11
|
Brunelle JK and Letai A: Control of
mitochondrial apoptosis by the Bcl-2 family. J Cell Sci.
122:437–441. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fan Y, Chen H, Qiao B, et al: Opposing
effects of ERK and p38 MAP kinases on HeLa cell apoptosis induced
by dipyrithione. Mol Cells. 23:30–38. 2007.PubMed/NCBI
|
|
13
|
Hetman M, Kanning K, Cavanaugh JE and Xia
Z: Neuroprotection by brain-derived neurotrophic factor is mediated
by extracellular signal-regulated kinase and phosphatidylinositol
3-kinase. J Biol Chem. 274:22569–22580. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tran SE, Holmstrom TH, Ahonen M, Kahari VM
and Eriksson JE: MAPK/ERK overrides the apoptotic signaling from
Fas, TNF, and TRAIL receptors. J Biol Chem. 276:16484–16490. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Boucher MJ, Morisset J, Vachon PH, Reed
JC, Lainé J and Rivard N: MEK/ERK signaling pathway regulates the
expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of
human pancreatic cancer cells. J Cell Biochem. 79:355–369. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kovarik P, Mangold M, Ramsauer K, et al:
Specificity of signaling by STAT1 depends on SH2 and C-terminal
domains that regulate Ser727 phosphorylation, differentially
affecting specific target gene expression. EMBO J. 20:91–100. 2001.
View Article : Google Scholar
|
|
17
|
Battle TE and Frank DA: The role of STATs
in apoptosis. Curr Mol Med. 2:381–392. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wen Z, Zhong Z and Darnell JE Jr: Maximal
activation of transcription by Stat1 and Stat3 requires both
tyrosine and serine phosphorylation. Cell. 82:241–250. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Levy DE and Darnell JE Jr: Stats:
transcriptional control and biological impact. Nat Rev Mol Cell
Biol. 3:651–662. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schindler C, Shuai K, Prezioso VR and
Darnell JE Jr: Pillars article: interferon-dependent tyrosine
phosphorylation of a latent cytoplasmic transcription factor.
Science. 257:809–813. 1992. View Article : Google Scholar
|
|
21
|
Townsend PA, Scarabelli TM, Davidson SM,
Knight RA, Latchman DS and Stephanou A: STAT-1 interacts with p53
to enhance DNA damage-induced apoptosis. J Biol Chem.
279:5811–5820. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Townsend PA, Cragg MS, Davidson SM, et al:
STAT-1 facilitates the ATM activated checkpoint pathway following
DNA damage. J Cell Sci. 118:1629–1639. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Stephanou A, Brar BK, Knight RA and
Latchman DS: Opposing actions of STAT-1 and STAT-3 on the Bcl-2 and
Bcl-x promoters. Cell Death Differ. 7:329–330. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pérez MJ and Cederbaum AI: Antioxidant and
pro-oxidant effects of a manganese porphyrin complex against
CYP2E1-dependent toxicity. Free Radic Biol Med. 33:111–127.
2002.PubMed/NCBI
|
|
25
|
Baracca A, Sgarbi G, Solaini G and Lenaz
G: Rhodamine 123 as a probe of mitochondrial membrane potential:
evaluation of proton flux through F(0) during ATP synthesis.
Biochim Biophys Acta. 1606:137–146. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nera MS, Vanderbeek G, Johnson RO, Ruben
LN and Clothier RH: Phosphatidylserine expression on apoptotic
lymphocytes of Xenopus laevis, the South African clawed
toad, as a signal for macrophage recognition. Dev Comp Immunol.
24:641–652. 2000.PubMed/NCBI
|
|
27
|
Murphy KM, Ranganathan V, Farnsworth ML,
Kavallaris M and Lock RB: Bcl-2 inhibits Bax translocation from
cytosol to mitochondria during drug-induced apoptosis of human
tumor cells. Cell Death Differ. 7:102–111. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Johnson TL, Lai MB, Lai JC and Bhushan A:
Inhibition of cell proliferation and MAP kinase and akt pathways in
oral squamous cell carcinoma by genistein and biochanin A. Evid
Based Complement Alternat Med. 7:351–358. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hellebrand EE and Varbiro G: Development
of mitochondrial permeability transition inhibitory agents: a novel
drug target. Drug Discov Ther. 4:54–61. 2010.PubMed/NCBI
|
|
30
|
Tomasello F, Messina A, Lartigue L, et al:
Outer membrane VDAC1 controls permeability transition of the inner
mitochondrial membrane in cellulo during stress-induced apoptosis.
Cell Res. 19:1363–1376. 2009. View Article : Google Scholar
|
|
31
|
Orrenius S: Mitochondrial regulation of
apoptotic cell death. Toxicol Lett. 149:19–23. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ling YH, Lin R and Perez-Soler R:
Erlotinib induces mitochondrial-mediated apoptosis in human H3255
non-small-cell lung cancer cells with epidermal growth factor
receptorL858R mutation through mitochondrial oxidative
phosphorylation-dependent activation of BAX and BAK. Mol Pharmacol.
74:793–806. 2008. View Article : Google Scholar
|
|
33
|
Kirkin V, Joos S and Zörnig M: The role of
Bcl-2 family members in tumorigenesis. Biochim Biophys Acta.
1644:229–249. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Adams JM and Cory S: Life-or-death
decisions by the Bcl-2 protein family. Trends Biochem Sci.
26:61–66. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Newmeyer DD and Ferguson-Miller S:
Mitochondria: releasing power for life and unleashing the
machineries of death. Cell. 112:481–490. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tsujimoto Y: Cell death regulation by the
Bcl-2 protein family in the mitochondria. J Cell Physiol.
195:158–167. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Baek D, Nam J, Koo YD, et al: Bax-induced
cell death of Arabidopsis is meditated through reactive
oxygen-dependent and -independent processes. Plant Mol Biol.
56:15–27. 2004. View Article : Google Scholar
|
|
38
|
Kondo K, Obitsu S, Ohta S, Matsunami K,
Otsuka H and Teshima R: Poly(ADP-ribose) polymerase
(PARP)-1-independent apoptosis-inducing factor (AIF) release and
cell death are induced by eleostearic acid and blocked by
alpha-tocopherol and MEK inhibition. J Biol Chem. 285:13079–13091.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gogvadze V, Norberg E, Orrenius S and
Zhivotovsky B: Involvement of Ca2+and ROS in
alpha-tocopheryl succinate- induced mitochondrial permeabilization.
Int J Cancer. 127:1823–1832. 2010.
|
|
40
|
Yip KW and Reed JC: Bcl-2 family proteins
and cancer. Oncogene. 27:6398–6406. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kroemer G and Reed JC: Mitochondrial
control of cell death. Nat Med. 6:513–519. 2000. View Article : Google Scholar
|
|
42
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Paris C, Bertoglio J and Bréard J:
Lysosomal and mitochondrial pathways in miltefosine-induced
apoptosis in U937 cells. Apoptosis. 12:1257–1267. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang X, Martindale JL and Holbrook NJ:
Requirement for ERK activation in cisplatin-induced apoptosis. J
Biol Chem. 275:39435–39443. 2000. View Article : Google Scholar
|
|
46
|
Bacus SS, Gudkov AV, Lowe M, et al:
Taxol-induced apoptosis depends on MAP kinase pathways (ERK and
p38) and is independent of p53. Oncogene. 20:147–155. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chang L and Karin M: Mammalian MAP kinase
signalling cascades. Nature. 410:37–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tamura Y, Simizu S and Osada H: The
phosphorylation status and anti-apoptotic activity of Bcl-2 are
regulated by ERK and protein phosphatase 2A on the mitochondria.
FEBS Lett. 569:249–255. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Olson JM and Hallahan AR: p38 MAP kinase:
a convergence point in cancer therapy. Trends Mol Med. 10:125–129.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dhanasekaran DN and Reddy EP: JNK
signaling in apoptosis. Oncogene. 27:6245–6251. 2008. View Article : Google Scholar
|
|
51
|
Yang J and Stark GR: Roles of
unphosphorylated STATs in signaling. Cell Res. 18:443–451. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lee HJ, Oh YK, Rhee M, et al: The role of
STAT1/IRF-1 on synergistic ROS production and loss of mitochondrial
transmembrane potential during hepatic cell death induced by
LPS/d-GalN. J Mol Biol. 369:967–984. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Krasilnikov M, Ivanov VN, Dong J and Ronai
Z: ERK and PI3K negatively regulate STAT-transcriptional activities
in human melanoma cells: implications towards sensitization to
apoptosis. Oncogene. 22:4092–4101. 2003. View Article : Google Scholar
|
|
54
|
Xu B, Abdel-Fattah R, Yang L, Crenshaw SA,
Black MB and Hinton BT: Testicular lumicrine factors regulate ERK,
STAT, and NFKB pathways in the initial segment of the rat
epididymis to prevent apoptosis. Biol Reprod. 84:1282–1291. 2011.
View Article : Google Scholar : PubMed/NCBI
|