Introduction
Epilepsy is a common, often intractable and
occasionally fatal neurological disease. Many of the sequelae of
chronic seizures result from excitotoxicity and oxidative stress
with ensuing mitochondrial damage and neuronal death (1,2).
Although there are numerous anti-epileptic drugs (AEDs) available
for the management of seizures, they merely treat symptoms, rather
than cure the disease and have side-effects that diminish the
quality of life, including cognitive impairment (3,4).
Oxidative stress associated with seizure activity
can greatly reduce mitochondrial function and is widely accepted as
a contributor to learning and memory deficits resulting from
epileptic seizures (5). Compared
to other organs, the brain is extremely susceptible to damage by
free radicals due to its relatively high levels of oxidative
metabolism and comparatively low levels of both free-radical
scavenging enzymes and antioxidant molecules (6). Glutathione (GSH) plays an important
role in antioxidant defense in mammalian tissues (7,8);
it has been shown to prevent ischemia-induced neuronal death and
improve memory following an ischemic insult (9). The accumulation of malondialdehyde
(MDA), an end product of lipid peroxidation, reflects the extent of
oxidative stress and indirectly that of cellular antioxidant
capacity (10,11). Mitochondrial oxidative stress and
dysfunction not only result from seizures, but may directly
contribute to epileptogenesis (12). High concentrations of reactive
oxygen species (ROS) cause lipid peroxidation and damage to cell
membranes, proteins and mitochondrial DNA (13), which exacerbates mitochondrial
dysfunction, reduces mitochondrial energy production (14) and triggers apoptotic cell death
through the activation of the caspase-3 pathway (3,15).
Hence, exogenous antioxidants may be safe and effective agents for
seizure control and the prevention of cognitive decline.
Dietary antioxidants may preserve mitochondrial
function and energy metabolism, and have other long-term
therapeutic benefits (16). The
natural antioxidant, grape seed proanthocyanidin extract (GSPE), an
extract from red grape seeds containing a variety of phenolic
compounds, is widely marketed in China as a dietary supplement with
a variety of health benefits. It is water-soluble and can cross the
blood-brain barrier more easily than other natural antioxidants,
such as quercetin (17) and
curcumin (18). In addition, GSPE
has demonstrated a wide range of biological effects, including
antioxidant (19,20), anti-inflammatory (21,22), anti-mutagenic (23), anti-carcinogenic (24), cardioprotective (25,26) and neuroprotective (27) activities in various experimental
models; furthermore, it can effectively reduce ischemia/reperfusion
injury (28,29). Moreover, it has been reported to
enhance working memory, ameliorate symptoms of Alzheimer’s disease
(30), and significantly improve
cognitive performance across age groups (31). However, little is known about the
potential neuroprotective efficacy of GSPE against
pentylenetetrazole (PTZ)-induced seizures.
The aim of the present study was to evaluate the
protective effects of GSPE against seizure-induced cognitive
impairment, oxidative stress caused by mitochondrial damage and
neuronal apoptosis mediated by the caspase-3 signaling pathway. Our
results revealed that GSPE effectively suppressed oxidative stress
and neuronal apoptosis induced by epileptic seizures, and improved
cognitive decline in epileptic rats.
Materials and methods
Animals
Ninety male Sprague-Dawley rats (180–200 g) were
obtained from the Experimental Animal Center of Hebei Medical
University. [Animal licenses: SCXK (Hebei 2008-1-003); animal
certification no. 1301020]. The animals were allowed to acclimatize
for 1 week prior to the experiments. The animals were housed in
groups of 4 per cage in a room that was maintained under standard
laboratory conditions (12 h light/dark cycle) and a constant
temperature (25±1°C) and humidity (50–60%). They were allowed free
access to food and water. All experiments conformed to the National
Institutes of Health guidelines for the Care and Use of Laboratory
Animals and with the European Communities Council Directive of 24
November 1986 (86/609/EEC). Every effort was made to minimize the
number of animals used and their suffering.
Materials
GSPE (purity >98%) was kindly provided by Tianjin
Jianfeng Natural Product R&D Co., Ltd. (Tianjin, China). The
GABA(A)-receptor antagonist, PTZ (CAS:54-95-5; Sigma, St. Louis,
MO, USA), was used for epileptic kindling to establish a chronic
epileptic rat model. The levels of MDA and GSH were measured using
ELISA kits (Nanjing Jiancheng Bioengineering Institute, Nanjing,
China). Mitochondrial ROS production was measured by flow cytometry
(BDFACSAria™ II; BD Biosciences, San Jose, CA, USA) using the
oxidation-sensitive fluorescent dye,
2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA, CAS:D6883,
Sigma). The animal model was established by an intraperitoneal
(i.p.) injection of PTZ (35 mg/kg/day) for 36 days. GSPE and 1% PTZ
were dissolved in physiological saline prior to the experiments.
Drugs were administered between 08:30 and 10:30 a.m. to reduce the
effects of circadian rhythms.
Experimental groups and drug
administration
The rats were randomly divided into 5 groups: i) a
control group (n=16) that received normal saline (vehicle) for 60
days (3.5 ml/kg/day, i.p.); ii) a PTZ group (n=22) that received
saline (3.5 ml/kg/day, i.p.) for 24 days followed by PTZ (35
mg/kg/day, i.p.) for 36 days; iii and iv) the PTZ + GSPE groups
(n=18/group) that were treated with GSPE (100 or 200 mg/kg/day, by
gavage) for 24 days then pre-treated with the same doses 30 min
prior to each PTZ administration for 36 days; v) a GSPE alone group
(n=16) that received 200 mg/kg/day GSPE only for 60 consecutive
days. Following drug treatments, the mortality rate and seizure
stages of the animals were recorded. Six rats per treatment group
were tested in the Morris water maze (MWM) and were then
sacrificed. The sections of the hippocampus were divided into 2
parts for the determination of oxidative stress and the measurement
of the expression of pro-apoptotic proteins by western blot
analysis (n=6/group). The remaining rats in each group were
sacrificed and hippocampal tissue was processed for Nissl staining
(n=3/group), the ultrastructural analysis of mitochondria by
transmission electron microscopy (n=3/group) or assays of
mitochondrial ROS generation and the degree of mitochondrial
swelling (n=4/group).
Behavioral assessment
The animals were observed for 1 h after each PTZ
administration for changes in the seizure stage, which was assessed
using the Racine scale: stage 0: no response; stage 1:
hyperactivity, vibrissae twitching; stage 2: head nodding, clonus
and myoclonic jerks of the head; stage 3: unilateral forelimb
clonus; stage 4: rearing with bilateral forelimb clonus; stage 5:
the generalized tonic-clonic seizure with loss of writing
reflex.
Behavioral screening
For behavioral screening, some rats (n=6/group) were
tested in the MWM following the protocols previously described
(37). The MWM consisted of a
circular water tank (180 cm in diameter, 70 cm in height) filled
with 22±1°C water. The pool was divided into 4 equal quadrants
labeled N, W, S and E. A colorless escape platform (10 cm in
diameter) was submerged 2 cm below the surface. Training was
performed in 2 sessions daily for 5 days with an inter-session
interval of 2 h. Each session consisted of 4 trials with an
inter-trial interval of 30 sec. In each trial, the rat was gently
placed in a randomly selected quadrant with its nose pointing
toward the wall and allowed to find the escape platform. If a rat
failed to find the platform within 120 sec, the escape latency was
recorded as 120 sec and the rat was placed on the platform. The
rats remained on the platform for 10 sec before the start of the
next trial. On the 6th day, a probe trial of spatial memory was
conducted by removing the platform and measuring both the time
spent in the target quadrant and the number of crossings over the
former platform location. The MWM was conducted between 9:00 a.m.
and 18:00 p.m. to minimize performance variations due to circadian
rhythmicity.
Nissl staining
Fifteen animals (n=3/group) were decapitated and a
5-mm-thick coronal section, including the bilateral hippocampus,
was excised from the brains. The sections were fixed in 4%
paraformaldehyde for 24 h, dehydrated in alcohol, cleared with
xylene and embedded in paraffin. The paraffin-embedded brain
sections were sliced at 5 μm thickness and Nissl-stained with 1%
thionin. In every 5th slice (3 slices per animal), we counted the
number of surviving intact pyramidal cells per mm length of
hippocampal CA1 subfield in both hemispheres. Neuronal cells were
counted by 2 observers blinded to the treatment history using high
magnification (×400) light microscopy.
Transmission electron microscopy
Fifteen rats (n=3/group) were deeply anesthetized
and processed for electron microscopy according to the method
described in the study by Xie et al (32). Mitochondrial ultrastructure in the
CA1 subfield was imaged (×40,000) using a transmission electron
microscope (JEM-1230; Jeol Ltd., Tokyo, Japan) from 5 micrographs
per rat.
Measurement of MDA accumulation
The day after the completion of the behavioral
tests, 30 rats (n=6/group) were sacrificed and the hippocampi were
quickly removed and flash-frozen. The samples were thawed and
homogenized in 1:9 w/v ice-cold normal saline. The homogenates were
centrifuged at 3,000 × g for 15 min at 4°C and the supernatants
were used for the determination of the MDA and GSH levels. The MDA
concentration was measured according to the method described in the
study by Ohkawa et al (33). Briefly, the substrate-supernatant
mixture was centrifuged at 3,500 × g for 10 min, and the absorbance
recorded at 532 nm using a spectrophotometer. The results were
expressed in nmol MDA/g protein.
Determination of GSH levels
GSH levels were measured using the method described
in the study by Ellman (34). The
substrate-supernatant mixture was vortexed and the absorbance read
at 412 nm within 15 min. The GSH concentration was expressed in g
GSH/g protein.
Determination of mitochondrial ROS
production and degree of mitochondrial swelling
After the final injection of PTZ, 20 rats
(n=4/group) were sacrificed and the bilateral hippocampus was
quickly removed and divided into 2 sections. Some samples were
homogenized and the fluorescence intensity was immediately
determined by flow cytometry at 488 nm excitation and 530 nm
emission. Data were analyzed using BDFACSAria™ II Cell Sorter
software (Version 7.0; BD Biosciences). Other samples were used to
detect the degree of mitochondrial swelling by measuring the
decrease in optical density at 520 nm, as previously described
(35). The turbidity of the
reaction mixture reflected the degree of mitochondrial swelling.
Freshly prepared rat brain mitochondria (50 μg protein) were
recorded over a period of 10 min at 25°C in 200 μl medium
containing 250 mM sucrose, 5 mM KH2PO4 and 3
mM sodium succinate (pH 7.2).
Western blot analysis
The hippocampi were collected and added to RIPA
buffer with 1% PMSF and then lysed on ice. Total proteins were
extracted (Applygen Technologies, Inc., Beijing, China) following
the manufacturer’s instructions. Brain mitochondria isolation was
conducted as previously described (35). The rat brain homogenate was
centrifuged at 1,000 × g for 10 min, and the resulting supernatant
was subjected to 10,000 × g centrifugation for 10 min. The pellet
was the mitochondrial fraction. The supernatant was recentrifuged
at 100,000 × g for 1 h at 4°C. The resulting supernatant was used
as a cytosolic fraction. After determining protein concentrations,
the proteins in the pellet and supernatant were loaded on 10%
sodium dodecyl sulfate-polyacrylamide gels and transferred onto
PVDF membranes (Millipore Corp., Bedford, MA, USA). The membranes
were blocked for 1 h at room temperature and then incubated
overnight at 4°C with one of the following primary antibodies:
anti-cytochrome c (Cyt c) polyclonal antibody (pAb;
1:800, no. 4272; Cell Signaling Technology, Inc., Danvers, MA,
USA), anti-caspase-9 pAb (1:500; sc-8355, Santa Cruz Biotechnology,
Santa Cruz, CA, USA) and anti-caspase-3 pAb (1:600, bs-6428;
Bioworld Technology, St. Louis Park, MN, USA) and β-actin (1:500,
sc-47778; Santa Cruz Biotechnology). After washing them 3 times,
the membranes were incubated with the appropriate HRP-conjugated
secondary antibodies goat anti-rabbit (1:6,000; Rockland,
Gilbertsville, PA, USA) at room temperature for 1 h. Following
washing three times in TPBS and once in PBS alone, the
immunolabeled marker protein bands on the membranes were scanned
and analyzed (Odyssey; LI-COR, Inc., Lincoln, NE, USA). The
densities of the Cyt c, caspase-9 and caspase-3 protein
bands were measured and expressed as a ratio of the β-actin
density.
Statistical analysis
All data are expressed as the means ± SEM.
Significance of the seizure stage was analyzed using a two-way
analysis of variance (ANOVA). Escape latencies in the MWM were
analyzed with repeated measures and multivariate ANOVA. Other data
were compared by one-way ANOVA followed by the least significant
difference (LSD) or Student-Newman-Keuls (SNK) post-hoc tests for
multiple pair-wise comparisons. All statistical tests were
calculated using SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA)
and P-values <0.05 were considered to indicate statistically
significant differences.
Results
Behavioral assessment
The injection of PTZ alone for 36 consecutive days
caused significant mortality (27.3%), which was reduced by
pre-treatment with 100 mg/kg GSPE (11.1%) and 200 mg/kg GSPE
(5.6%). No fatalities occurred in the control group or the 200
mg/kg GSPE group, indicating that this high dosage has little or no
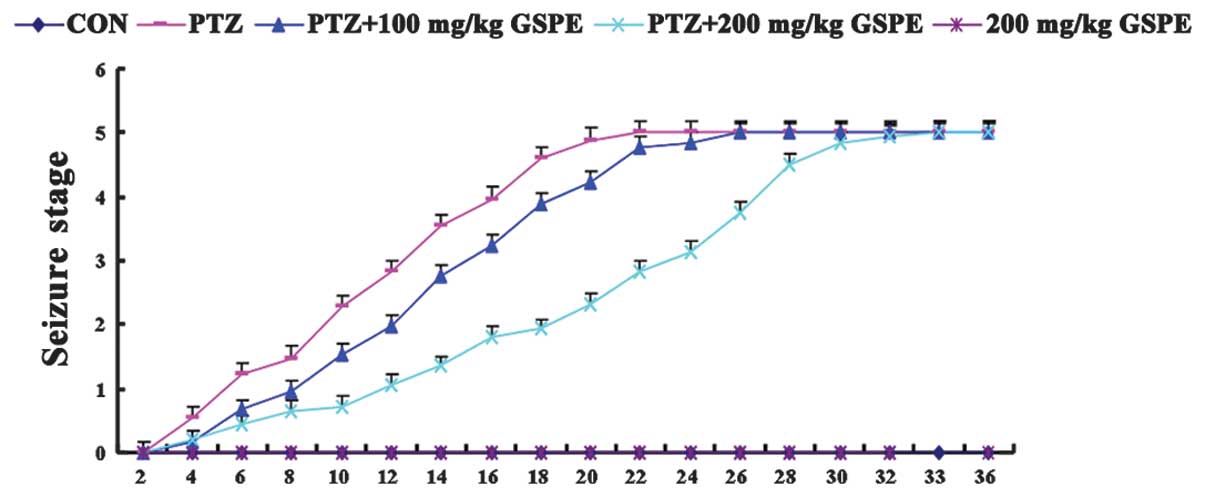
inherent toxicity. As shown in Fig.
1, there was no seizure behavior in the control group and the
200 mg/kg GSPE group. On the other hand, severe epileptic seizures
were induced by the kindling injections of PTZ for 36 days, and the
PTZ group had epileptic seizures earlier than the other groups,
while high-dose GSPE pre-treatment (200 mg/kg) significantly
delayed the time to seizure. Although the rats in the low-dose
group (100 mg/kg) exhibited mildly delayed seizures, the difference
was not significant compared to the PTZ group (P=0.71), suggesting
that GSPE may partially suppress epileptic seizures.
GSPE reverses cognitive impairment
following seizures
To determine whether GSPE alleviates PTZ-induced
cognitive impairment, we tested spatial learning and memory in the
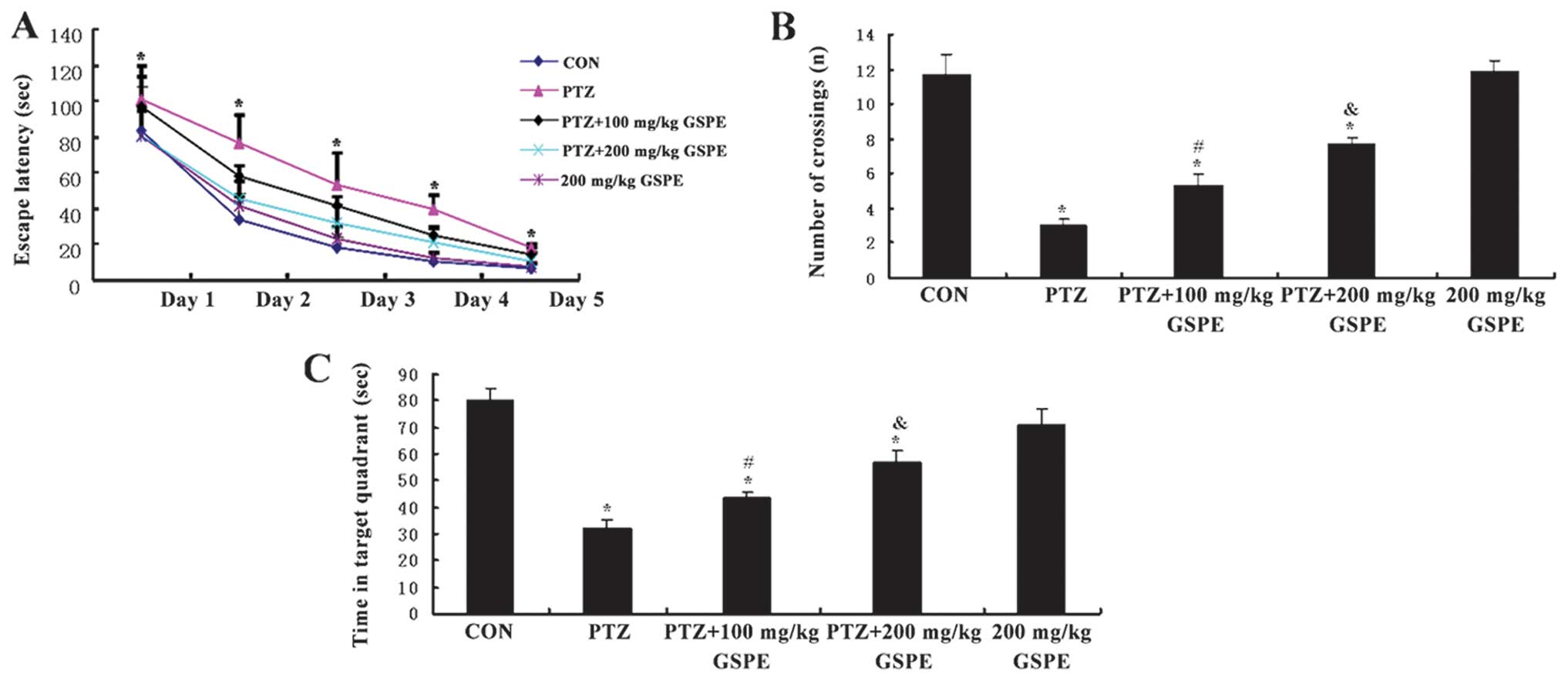
MWM following drug treatments (Fig.
2). The rats from all the groups showed a progressive decline
in the escape latency during training (Fig. 2A), and this decline was more
significant as the training days progressed [F(1,25)=928.84, P<0.001] and drug
treatment group [F(4,25)=13.469, P<0.001]. The rats in the
PTZ alone group (35 mg/kg/d) exhibited significantly prolonged
escape latencies during all sessions compared to the control rats
(P<0.001), while pre-treatment with GSPE dose-dependently
decreased escape latency compared to the PTZ group (P=0.021 at 100
mg/kg and P<0.001 at 200 mg/kg). Escape latencies did not differ
significantly between the high-dose GSPE group and the control
group (P=0.570), indicating the near complete reversal of the
PTZ-induced spatial learning deficit. In the probe trail (Fig. 2B and C), the rats treated with the
vehicle + PTZ crossed the platform location significantly fewer
times than the controls [F(4,25)=30.223, P<0.001] and spent less
time in the target (former platform) quadrant than the control rats
[F(4,25)=23.482, P<0.001]. The PTZ + GSPE
groups also crossed the former platform location more often than
the PTZ group and spent significantly more time in the target
quadrant (P=0.045, P<0.001). Again, there was no significant
difference in mean latency or the time in the target quadrant
between the high-dose GSPE group and the control group (P=0.869,
P=0.128), indicating the near complete reversal of the PTZ-induced
spatial memory deficit.
GSPE rescues hippocampal CA1 neurons
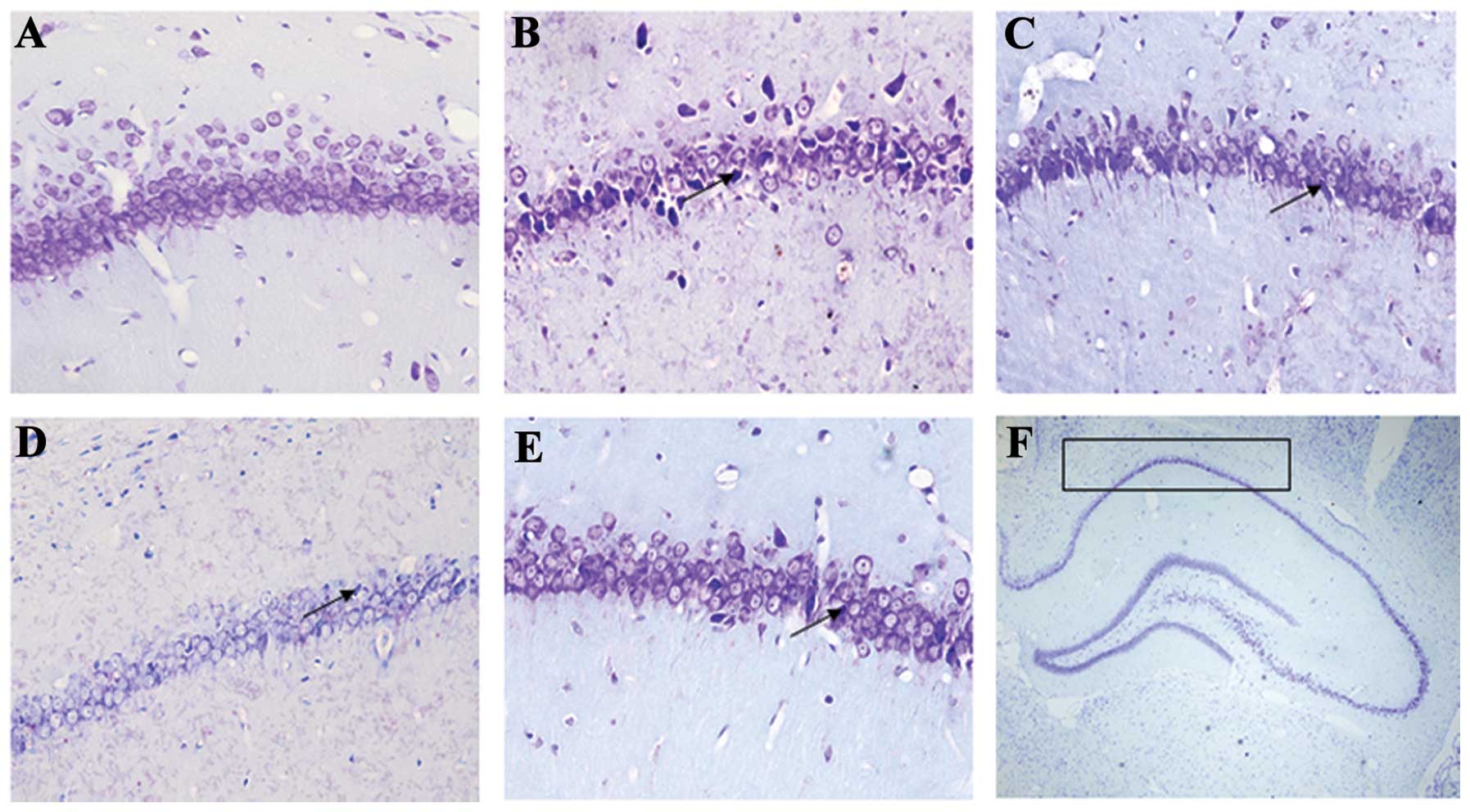
Pyramidal neurons in the Nissl-stained sections from
the control rats were evenly stained light blue and had regularly
shaped cell bodies. By contrast, morphological abnormalities,
including blurred caryotheca and pyknotic nuclei were apparent in
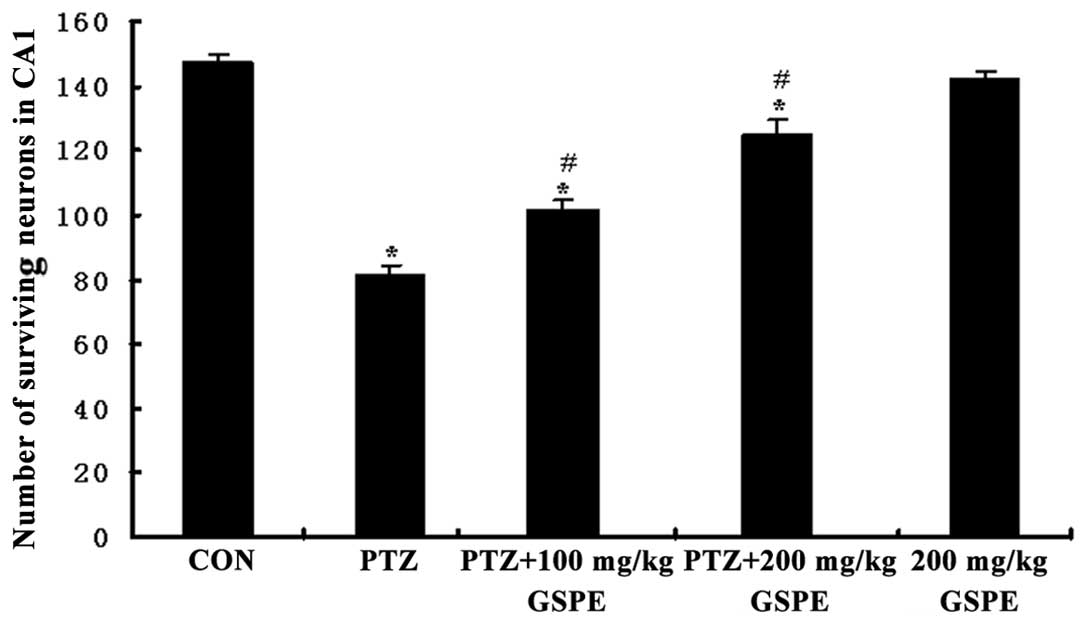
sections from PTZ group rats. The average density of intact
surviving neurons was lower in the PTZ group compared to the
control group, while pre-treatment with GSPE reversed the damage to
cell morphology (Fig. 3) and
reversed the decline in CA1 pyramidal cell density (Fig. 4). Compared to the PTZ group, the
number of damaged neurons was significantly and dose-dependently
reduced by GSPE pre-treatment (P<0.05 for both low and high
doses). The GSPE alone group showed comparable cell density to the
control group, underscoring the low or absent neurotoxicity of the
extract (P=0.254).
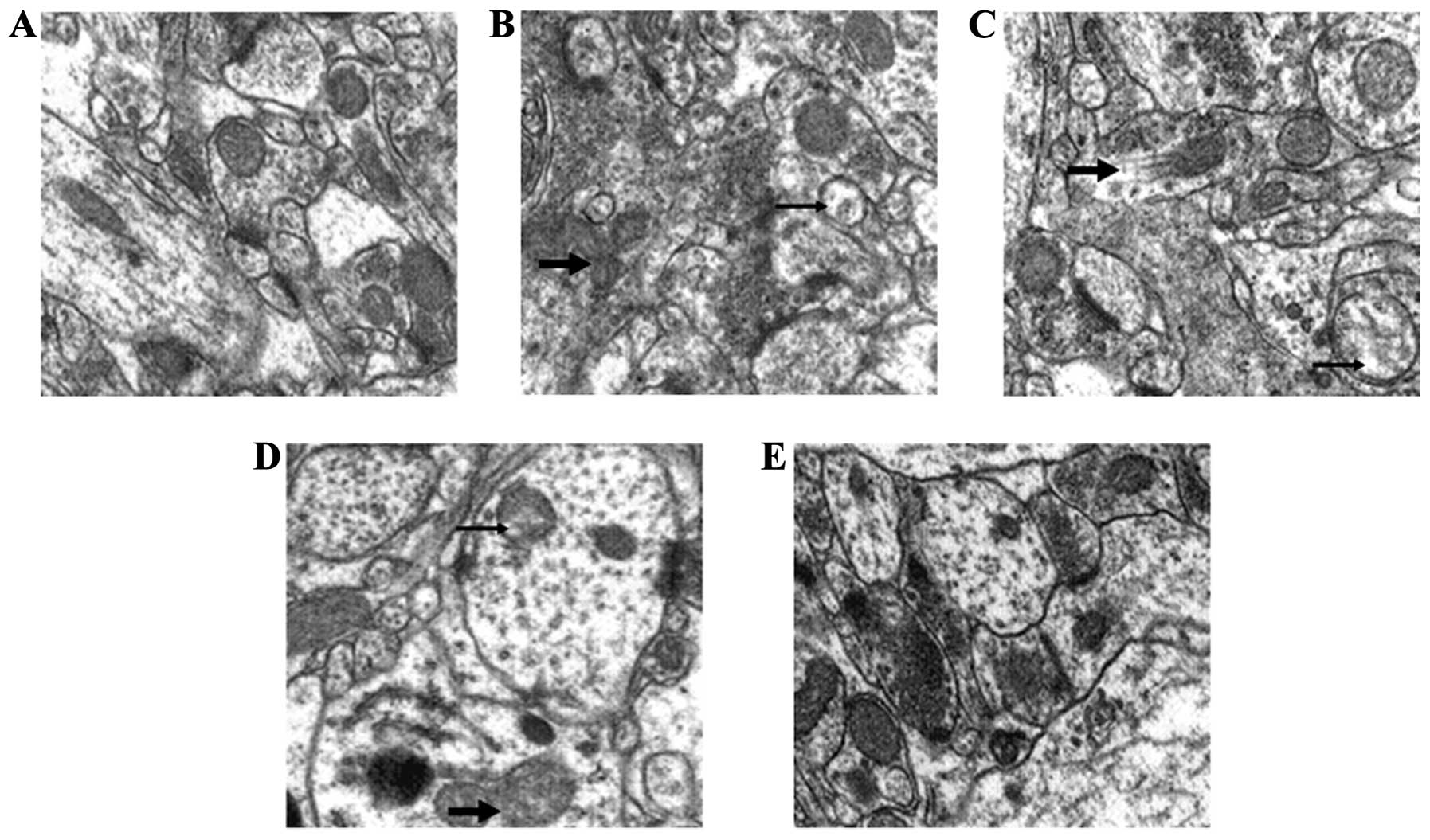
GSPE preserves mitochondrial
ultrastructure during seizures
To demonstrate the effects of GSPE pre-treatment on
the mitochondria, the morphology of the mitochondria was further
observed. Compared to the control group, obvious morphological
changes were observed in the mitochondria from the PTZ group, such
as damaged membranes and obscure boundaries, whereas GSPE
pre-treatment partly reversed these changes (Fig. 5).
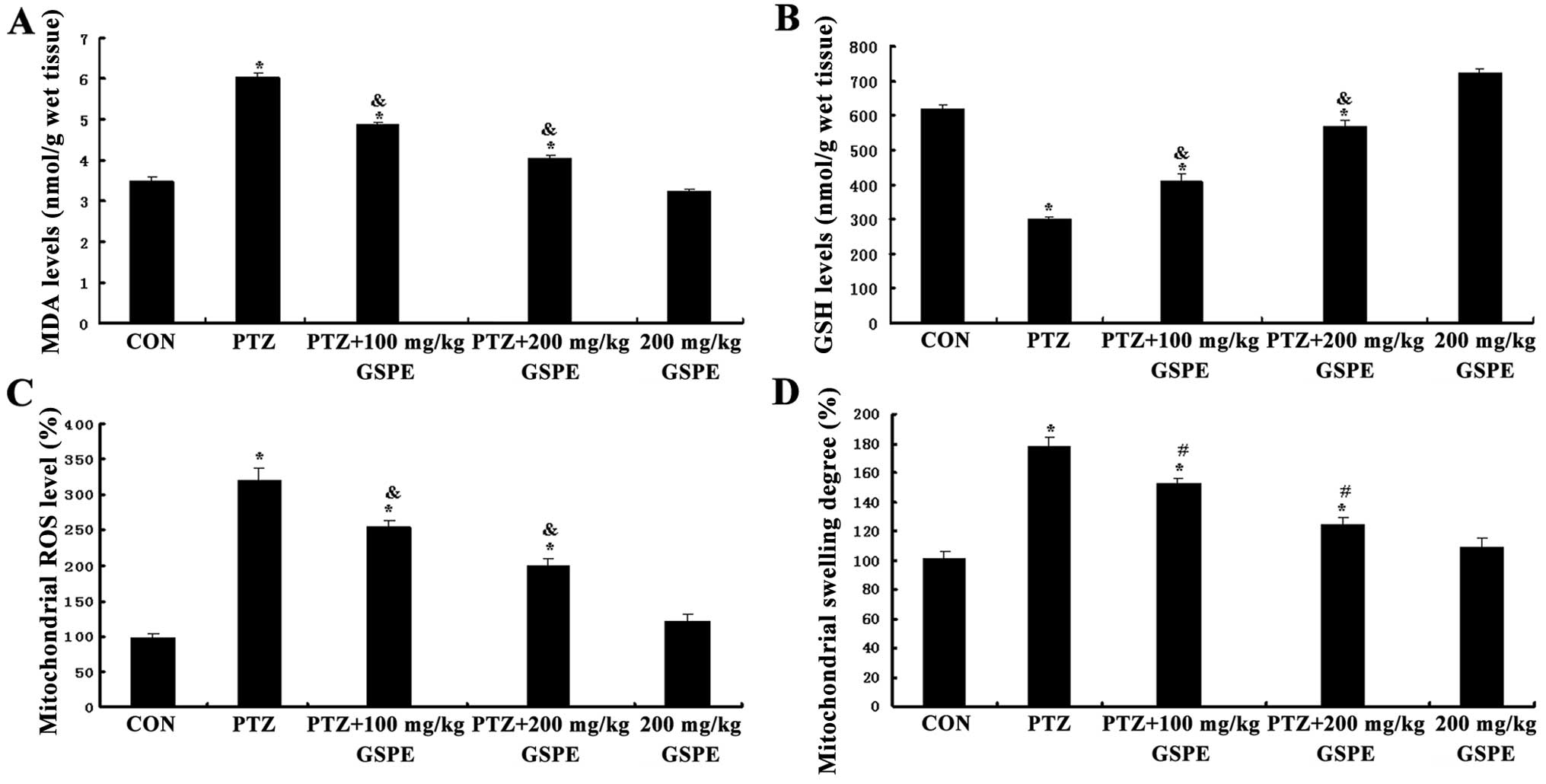
GSPE reduces MDA accumulation and
upregulates GSH levels
As shown in Fig. 6A
and B, there were significant group differences in both
hippocampal MDA accumulation and GSH levels [F(4,25)=178.732, F(4,25)=518.96, P<0.001]. Compared to the
control group, the accumulation of MDA was significantly higher
(P<0.001) and GSH levels were lower in the PTZ group
(P<0.001). Pre-treatment with GSPE dose-dependently reduced MDA
and upregulated GSH levels compared to the PTZ group. Therefore,
GSPE pre-treatment partially alleviated the oxidative stress
induced by PTZ-induced seizures, although MDA levels were still
higher and GSH levels still lower than the control group
(P<0.05, P<0.05). There were no significant differences
between the GSPE alone group and the control group (P=0.035,
P=0.393).
GSPE supresses PTZ-induced mitochondrial
ROS production and degree of mitochondrial swelling
As shown in Fig.
6C, mitochondrial ROS generation was significantly higher in
the PTZ group than the control group [F(4,15)=66.308, P<0.001], whereas
pre-treatment with GSPE dose-dependently decreased mitochondrial
ROS production compared to the PTZ group (P=0.001, P=0.003). Thus,
GSPE partially suppressed mitochondrial ROS production in the
hippocampus, although ROS levels were still higher than those in
the control group. There was no significant difference in
mitochondrial ROS production between the GSPE alone and control
groups (P=0.18). Accordingly, mitochondrial swelling in the
isolated brain mitochondria was monitored by measuring the change
in absorbance of the suspension at 520 nm. As shown in Fig. 6D, the degree of mitochondrial
swelling was significantly increased by 42.98% (P<0.001) in the
PTZ group compared to the control group. Compared with the PTZ
group, GSPE pre-treatment dose-dependently inhibited mitochondrial
swelling by 29.8 and 14.4% (P<0.05 for both doses),
respectively.
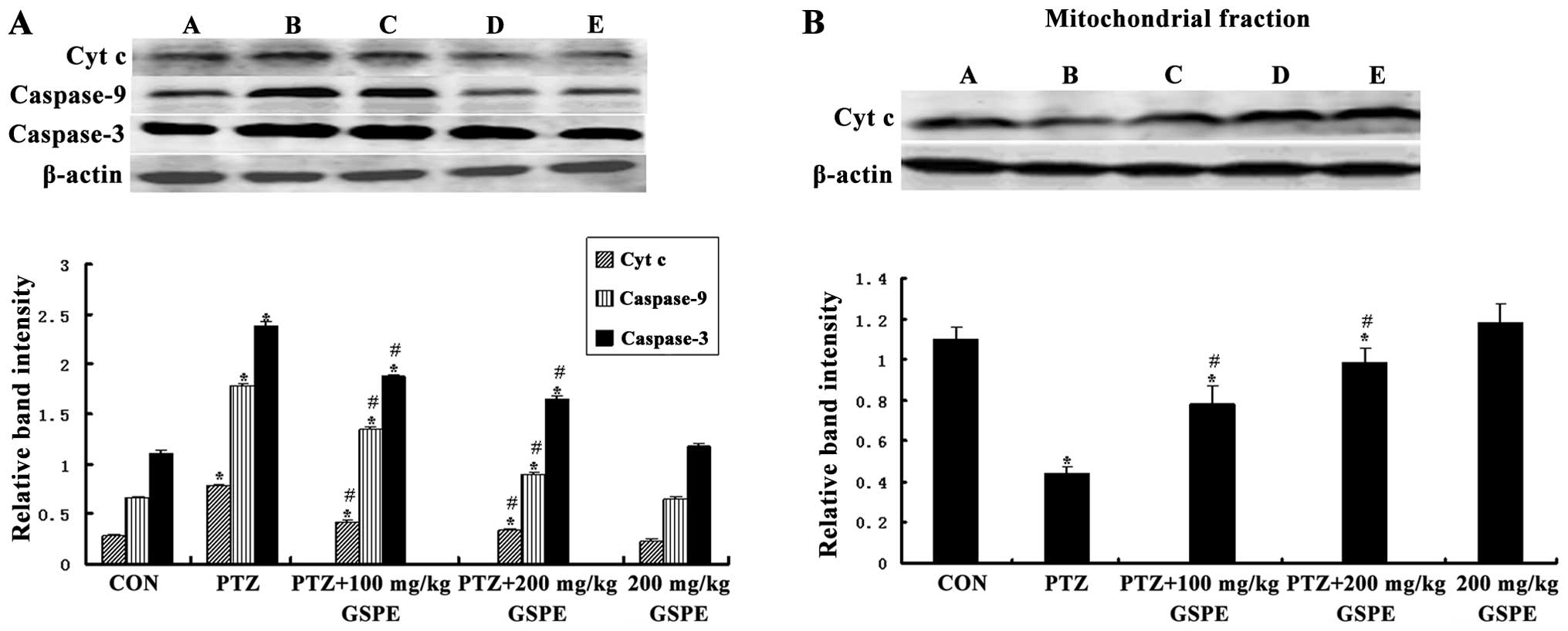
GSPE inhibits mitochondrial Cyt c release
induced by seizures
The translocation of mitochondrial Cyt c to
the cytosol induces the formation of the apoptosomes, leading to
apoptotic death. As shown in Fig.
7, there was a significant group difference in the level of
cytosolic Cyt c [F(4,25)=141.021, P<0.001] and
mitochondrial Cyt c [F(4,25)=234.398, P<0.001]. Compared to
the control group, the cytoplasmic Cyt c concentration was
markedly higher and the mitochondrial Cyt c concentration
was markedly lower in the PTZ group (P<0.001, P<0.001), while
pre-treatment with GSPE dose-dependently reversed those levels
compared to the PTZ group (P<0.05 for both doses). There was no
significant difference between the GSPE alone and control groups
(P=0.096, P=0.632), indicating that high-dose GSPE has no
endogenous pro-apoptotic activity.
GSPE suppresses the PTZ-induced increase
of in caspase-9 and caspase-3 expression
Caspase-3 is the principle effector of the
mitochondrial-dependent apoptotic pathway and is activated during
seizure-induced neuronal death (36). Western blot analysis revealed a
significant group difference in both activated caspase-9 and
caspase-3 expression in the hippocampus [F(4,25)=554.387, P<0.001; F(4,25)=347.38, P<0.001]. Compared to the
control group, activated caspase-9 and caspase-3 levels were
significantly higher in the PTZ group (P<0.001, P<0.001),
while pre-treatment with GSPE significantly decreased these levels
compared to the PTZ group (P<0.001, P<0.001) (Fig. 7).
Discussion
Although numerous advances have been made in the
diagnosis and treatment of epilepsy, the precise pathogenic
mechanism is obscure in the majority of patients, hampering
effective treatment. In our study, GSPE pre-treatment partially
suppressed seizures and reduced the PTZ-induced mortality rate. Of
note, PTZ-induced kindling caused marked cognitive impairment as
measured in the MWM, which is in agreement with our previous report
(37), while GSPE pre-treatment
had significant nootropic and neuroprotective effects, likely
mediated by reduced oxidative stress and the rescue of
mitochondrial function.
Chronic kindling by repeated chemical convulsant
administration or electrical stimulation is among the most widely
used models of chronic epilepsy for studies on epileptogenesis,
novel drug targets, and treatment and preventative strategies
(38). The underlying mechanisms
of seizure pathogenesis have been proven to be complex and are not
completely understood; several other molecular mechanisms are
involved, including oxidative stress, inflammation, glutamate
excitotoxicity and calcium overload. This is consistent with our
results that GSPE pre-treatment partially suppressed, but not
completely eliminated epileptic seizures. Moreover, in light of
studies demonstrating that GSPE protects against oxidative stress
(39) and enhances the working
memory and cognitive performance in a rat model of Alzheimer’s
disease (30), we primarily
assessed the effects against epilepsy-associated oxidative stress
and cognitive decline. As expected, PTZ-induced kindling caused an
obvious increase in escape latency in MWM learning sessions and a
marked decrease in time spent in the target quadrant during the
probe (memory) trail. By contrast, pre-treatment with GSPE prior to
PTZ significantly improved spatial learning and memory, as
manifested by shorter escape latencies in learning sessions and
longer time spent in the target quadrant during the probe trail.
These results are consistent with those of a previous study
(30).
Oxidative stress may be a critical pathogenic
mechanism in the development of chronic epilepsy. Oxidative stress
can be self-propagating in that initial oxidative damage creates
additional free radicals, damages antioxidant enzymes, depletes
antioxidant molecules, such as GSH and damages mitochondria,
producing more ROS (11,40). Indeed, PTZ-induced seizures
increased lipid peroxidation (MDA accumulation), decreased GSH in
the hippocampus and enhanced ROS production in the mitochondria
isolated from the epileptic hippocampus, implicating the
exacerbation of oxidative stress in epileptic kindling (41). All these effects were partially or
completely reversed by GSPE pre-treatment.
Mitochondria are the primary site of ROS production.
Free radicals are highly reactive and thus damage biomolecules
close to the site of generation, making mitochondria uniquely
vulnerable to macromolecule dysfunction and DNA damage (12). Mitochondrial dysfunction and the
overproduction of ROS may also enhance neuronal excitability and
increase seizure susceptibility (42). Mitochondrial dysfunction is a
common trigger for apoptosis, as ROS damage can induce Cyt c
release and sequential activation of pro-apoptotic caspase-9 and
caspase-3 (43,44). Therefore, synthetic antioxidants
that protect mitochondrial targets and decrease neuronal death may
be useful supplements for the clinical management of patients with
seizures (45). Our results
revealed that pre-treatment with GSPE decreased PTZ-induced
mitochondrial ROS production and the degree of swelling and
partially suppressed neuronal apoptosis, possibly through the
protection of mitochondrial function and the inhibition of Cyt
c translocation and caspase activation (46,47).
In order to further confirm the molecular change in
the mitochondria that is responsible for the protective effects of
GSPE pre-treatment, we focused on morphological observation. The
findings from Nissl staining were consistent with the molecular
results. Western blot analysis demonstrated that PTZ-induced
seizures significantly increased the hippocampal expression of
active caspase-3 and the release of cytosolic Cyt c; Nissl
staining indicated significant pyramidal cell loss with
ultrastructural signs of apoptosis. Furthermore, electron
microscopy indicated that the mitochondria were damaged in the PTZ
group, while GSPE pre-treatment reversed these morphological
changes. Considering all of the above, targeting mitochondrial
bioenergetics and oxidative stress with GSPE and
non-pharmacological treatments may prove to be useful for the
management of epilepsy (12).
In conclusion, GSPE, a safe and affordable
intervention for clinical use, was effective in attenuating
oxidative stress in the hippocampus of rats with chronic seizures.
More importantly, pre-treatment with GSPE improved cognitive
decline, at least in part, by protecting mitochondrial function and
suppressing caspase-3-dependent apoptosis. However, due to the
complex mechanisms of epilepsy, further studies are required to
define the optimal treatment protocols and identify additional
molecular targets of GSPE in epilepsy.
Acknowledgements
The authors are cordially indebted to the Natural
Science Foundation of Hebei Province, China (C2010000548) for its
financial support. We sincerely thank Ruichun Liu, Dongxia Wu,
Shuyu Wu and Hongran Wu for providing valuable suggestions and
technical assistance.
References
|
1
|
Bertram EH: Temporal lobe epilepsy: where
do the seizures really begin. Epilepsy Behav. 14(Suppl 1): 32–37.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kernan CL, Asarnow R, Siddarth P, et al:
Neurocognitive profiles in children with epilepsy. Epilepsia.
53:2156–2163. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Henshall DC and Simon RP: Epilepsy and
apoptosis pathways. J Cereb Blood Flow Metab. 25:1557–1572. 2005.
View Article : Google Scholar
|
|
4
|
Hermann B: Cognition in epilepsy and its
transient impairment. Epilepsy Behav. 22:4192011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sudha K, Rao AV and Rao A: Oxidative
stress and antioxidants in epilepsy. Clin Chim Acta. 303:19–24.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu H, Zhang X, Du Y, et al: Leonurine
protects brain injury by increased activities of UCP4, SOD, CAT and
Bcl-2, decreased levels of MDA and Bax, and ameliorated
ultrastructure of mitochondria in experimental stroke. Brain Res.
1474:73–81. 2012. View Article : Google Scholar
|
|
7
|
Colle D, Santos DB, Moreira EL, et al:
Probucol increases striatal glutathione peroxidase activity and
protects against 3-nitropropionic acid-induced pro-oxidative damage
in rats. PLoS One. 8:e676582013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sevin G, Ozsarlak-Sozer G, Keles D, et al:
Taurine inhibits increased MMP-2 expression in a model of oxidative
stress induced by glutathione depletion in rabbit heart. Eur J
Pharmacol. 706:98–106. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yabuki Y and Fukunaga K: Oral
administration of glutathione improves memory deficits following
transient brain ischemia by reducing brain oxidative stress.
Neuroscience. 250:394–407. 2013. View Article : Google Scholar
|
|
10
|
Dillioglugil MO, Kir HM, Demir C, et al:
Effect of pentylenetetrazole and sound stimulation induced single
and repeated convulsive seizures on the MDA, GSH and NO levels, and
SOD activities in rat liver and kidney tissues. Brain Res Bull.
83:356–359. 2010. View Article : Google Scholar
|
|
11
|
Mehla J, Reeta KH, Gupta P and Gupta YK:
Protective effect of curcumin against seizures and cognitive
impairment in a pentylenetetrazole-kindled epileptic rat model.
Life Sci. 87:596–603. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Waldbaum S and Patel M: Mitochondria,
oxidative stress, and temporal lobe epilepsy. Epilepsy Res.
88:23–45. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Borniquel S, Valle I, Cadenas S, Lamas S
and Monsalve M: Nitric oxide regulates mitochondrial oxidative
stress protection via the transcriptional coactivator PGC-1alpha.
FASEB J. 20:1889–1891. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fariss MW, Chan CB, Patel M, Van Houten B
and Orrenius S: Role of mitochondria in toxic oxidative stress. Mol
Interv. 5:94–111. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Young C, Roth KA, Klocke BJ, et al: Role
of caspase-3 in ethanol-induced developmental neurodegeneration.
Neurobiol Dis. 20:608–614. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pajuelo D, Díaz S, Quesada H, et al: Acute
administration of grape seed proanthocyanidin extract modulates
energetic metabolism in skeletal muscle and BAT mitochondria. J
Agric Food Chem. 59:4279–4287. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shen Y, Ward NC, Hodgson JM, et al:
Dietary quercetin attenuates oxidant-induced endothelial
dysfunction and atherosclerosis in apolipoprotein E knockout mice
fed a high-fat diet: a critical role for heme oxygenase-1. Free
Radic Biol Med. 65:908–915. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ahmad M: Protective effects of curcumin
against lithium-pilocarpine induced status epilepticus, cognitive
dysfunction and oxidative stress in young rats. Saudi J Biol Sci.
20:155–162. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mansouri E, Panahi M, Ghaffari MA and
Ghorbani A: Effects of grape seed proanthocyanidin extract on
oxidative stress induced by diabetes in rat kidney. Iran Biomed J.
15:100–106. 2011.PubMed/NCBI
|
|
20
|
Ding Y, Dai X, Jiang Y, et al: Grape seed
proanthocyanidin extracts alleviate oxidative stress and ER stress
in skeletal muscle of low-dose streptozotocin- and
high-carbohydrate/high-fat diet-induced diabetic rats. Mol Nutr
Food Res. 57:365–369. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou DY, Du Q, Li RR, Huang M, Zhang Q and
Wei GZ: Grape seed proanthocyanidin extract attenuates airway
inflammation and hyperresponsiveness in a murine model of asthma by
downregulating inducible nitric oxide synthase. Planta Med.
77:1575–1581. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Park JS, Park MK, Oh HJ, et al: Grape-seed
proanthocyanidin extract as suppressors of bone destruction in
inflammatory autoimmune arthritis. PLoS One. 7:e513772012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sharma SD and Katiyar SK: Dietary grape
seed proanthocyanidins inhibit UVB-induced cyclooxygenase-2
expression and other inflammatory mediators in UVB-exposed skin and
skin tumors of SKH-1 hairless mice. Pharm Res. 27:1092–1102. 2010.
View Article : Google Scholar
|
|
24
|
Uchino R, Madhyastha R, Madhyastha H, et
al: NFkappaB-dependent regulation of urokinase plasminogen
activator by proanthocyanidin-rich grape seed extract: effect on
invasion by prostate cancer cells. Blood Coagul Fibrinolysis.
21:528–533. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bagchi D, Sen CK, Ray SD, et al: Molecular
mechanisms of cardioprotection by a novel grape seed
proanthocyanidin extract. Mutat Res. 523–524:87–97. 2003.
|
|
26
|
Demirkaya E, Avci A, Kesik V, et al:
Cardioprotective roles of aged garlic extract, grape seed
proanthocyanidin, and hazelnut on doxorubicin-induced
cardiotoxicity. Can J Physiol Pharmacol. 87:633–640. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ahn SH, Kim HJ, Jeong I, et al: Grape seed
proanthocyanidin extract inhibits glutamate-induced cell death
through inhibition of calcium signals and nitric oxide formation in
cultured rat hippocampal neurons. BMC Neurosci. 12:782011.
View Article : Google Scholar
|
|
28
|
Song X, Xu H, Feng Y, Li X, Lin M and Cao
L: Protective effect of grape seed proanthocyanidins against liver
ischemic reperfusion injury: particularly in diet-induced obese
mice. Int J Biol Sci. 8:1345–1362. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wei R, Ding R, Wang Y and Tang L: Grape
seed proanthocyanidin extract reduces renal ischemia/reperfusion
injuries in rats. Am J Med Sci. 343:452–457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu P, Kemper LJ, Wang J, Zahs KR, Ashe KH
and Pasinetti GM: Grape seed polyphenolic extract specifically
decreases abeta*56 in the brains of Tg2576 mice. J
Alzheimers Dis. 26:657–666. 2011.PubMed/NCBI
|
|
31
|
Asha Devi S, Sagar Chandrasekar BK,
Manjula KR and Ishii N: Grape seed proanthocyanidin lowers brain
oxidative stress in adult and middle-aged rats. Exp Gerontol.
46:958–964. 2011.PubMed/NCBI
|
|
32
|
Xie T, Wang WP, Jia LJ, et al:
Environmental enrichment restores cognitive deficits induced by
prenatal maternal seizure. Brain Res. 1470:80–88. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ohkawa H, Ohishi N and Yagi K: Assay for
lipid peroxides in animal tissues by thiobarbituric acid reaction.
Anal Biochem. 95:351–358. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ellman GL: Tissue sulfhydryl groups. Arch
Biochem Biophys. 82:70–77. 1959. View Article : Google Scholar
|
|
35
|
He XL, Wang YH, Gao M, Li XX, Zhang TT and
Du GH: Baicalein protects rat brain mitochondria against chronic
cerebral hypoperfusion-induced oxidative damage. Brain Res.
1249:212–221. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rupinder SK, Gurpreet AK and Manjeet S:
Cell suicide and caspases. Vascul Pharmacol. 46:383–393. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xie T, Wang WP, Mao ZF, et al: Effects of
epigallocatechin-3-gallate on pentylenetetrazole-induced kindling,
cognitive impairment and oxidative stress in rats. Neurosci Lett.
516:237–241. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tourov A, Ferri R, Del Gracco S, Elia M,
Musumeci SA and Stefanini MC: Spike morphology in PTZ-induced
generalized and cobalt-induced partial experimental epilepsy. Funct
Neurol. 11:237–245. 1996.PubMed/NCBI
|
|
39
|
Al-Malki AL, Sayed AA and El Rabey HA:
Proanthocyanidin attenuation of oxidative stress and NF-κB protects
apolipoprotein E-deficient mice against diabetic nephropathy. Evid
Based Complement Alternat Med. 2013:7694092013.
|
|
40
|
Sayre LM, Perry G and Smith MA: Oxidative
stress and neurotoxicity. Chem Res Toxicol. 21:172–188. 2008.
View Article : Google Scholar
|
|
41
|
Freitas RM: Investigation of oxidative
stress involvement in hippocampus in epilepsy model induced by
pilocarpine. Neurosci Lett. 462:225–229. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Heinemann U, Buchheim K, Gabriel S, Kann
O, Kovacs R and Schuchmann S: Cell death and metabolic activity
during epileptiform discharges and status epilepticus in the
hippocampus. Prog Brain Res. 135:197–210. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wu CY, Tang ZH, Jiang L, Li XF, Jiang ZS
and Liu LS: PCSK9 siRNA inhibits HUVEC apoptosis induced by ox-LDL
via Bcl/Bax-caspase9-caspase3 pathway. Mol Cell Biochem.
359:347–358. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hong GL, Liu JM, Zhao GJ, et al: The
reversal of paraquat-induced mitochondria-mediated apoptosis by
cycloartenyl ferulate, the important role of Nrf2 pathway. Exp Cell
Res. 319:2845–2855. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ullah N, Ullah I, Lee HY, et al:
Protective function of nicotinamide against ketamine-induced
apoptotic neurodegeneration in the infant rat brain. J Mol
Neurosci. 47:67–75. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cheng M, Gao HQ, Xu L, Li BY, Zhang H and
Li XH: Cardioprotective effects of grape seed proanthocyanidins
extracts in streptozocin induced diabetic rats. J Cardiovasc
Pharmacol. 50:503–509. 2007.PubMed/NCBI
|
|
47
|
Li J, Liu H, Ramachandran S, et al: Grape
seed proanthocyanidins ameliorate Doxorubicin-induced
cardiotoxicity. Am J Chin Med. 38:569–584. 2010. View Article : Google Scholar : PubMed/NCBI
|