Introduction
The bronchial epithelium represents the interface
between the lungs and air and is known to induce and sustain
inflammatory events in respiratory diseases through the production
of inflammatory cytokines. Understanding the regulatory mechanisms
underlying the bronchial epithelial cell inflammatory response is
important for the effective treatment of respiratory diseases,
including asthma (1). The human
bronchial epithelium is continuously exposed to Gram-negative
bacteria of which lipopolysaccharide (LPS) is a glycolipid that
constitutes the major portion of the outer membrane (2). High levels of airborne (up to 1
μg/m3) LPS have been reported in a variety of
environments, and LPS as a contaminant in house dust is a factor
that increases the severity of asthma (3). LPS also induces the expression of
inflammatory cytokines, including interleukin (IL)-8 and IL-6 in
bronchial epithelial cells (4).
Among the known inflammatory response regulators, nuclear factor
(NF)-κB is the most important, as many genes involved in
inflammation have binding sites for NF-κB in their promoter regions
(5).
β-catenin is a member of the WNT/β-catenin pathway
regulating various cellular processes, including proliferation,
differentiation and development (6). In our previous study, we reported
that a promoter polymorphism of β-catenin that affects its mRNA
expression level was significantly associated with the risk of
asthma in human subjects, suggesting that β-catenin may be involved
in the disease mechanism of asthma (7). It is plausible that β-catenin may
modulate the inflammatory response of the bronchial epithelium
stimulated by inflammatory inducers. In this study, we investigated
whether β-catenin is involved in the regulation of inflammatory
cytokine expression, as well as NF-κB activity in BEAS-2B human
bronchial epithelial cells treated with LPS.
Materials and methods
Cell culture
BEAS-2B human bronchial epithelial cells were
purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA), and cultured in Dulbecco’s modified Eagle’s
medium (DMEM) supplemented with 10% fetal bovine serum, 100 μg/ml
streptomycin and 100 U/ml penicillin at 37°C in 5% CO2.
The cells were treated with 0.001 to 10 μg/ml of LPS
(Sigma-Aldrich, St. Louis, MI, USA) to induce an inflammatory
response. Untreated cells were used as controls.
Real-time reverse transcription
polymerase chain reaction (PCR) of inflammatory cytokines
The cells were cultured in 12-well plates and total
RNA was extracted using an RNeasy kit (Qiagen, Hilden, Germany).
Total RNA was reverse transcribed using a cDNA Reverse
Transcription kit (Applied Biosystems Inc., Foster City, CA, USA).
Briefly, the reaction was performed in a final volume of 20 μl that
included 100 mM dNTP, random primers, MultiScribe™ Reverse
Transcriptase, RNase inhibitor and 1 μg total RNA. The reaction
mixtures were heated at 25°C for 10 min, 37°C for 120 min and 85°C
for 5 sec. Real-time PCR was performed using a StepOne PCR System
(Applied Biosystems, Inc.) in triplicate in a final volume of 20 μl
that included TaqMan gene expression master mix, an optimized
concentration of each primer, 250 nM TaqMan probe and 2.0 μl cDNA
reaction mixture. The reaction mixtures were pre-heated at 95°C for
10 min to activate the enzyme, and then subjected to 40 cycles of
melting at 95°C for 15 sec and annealing/extension at 60°C for 1
min. The real-time PCR efficiencies were approximately 100%. The
assay-on-demand gene expression products (Applied Biosystems, Inc.)
were used to evaluate the mRNA expression levels of IL-6
(Hs00174131_m1), IL-8 (Hs99999034_m1), IL1-β (Hs01555410_m1), tumor
necrosis factor (TNF)-α (Hs01113624_g1), monocyte chemoattractant
protein (MCP)-1 (Hs00234140_m1), β-catenin (Hs00170025_m1) and 18S
rRNA (Hs99999901_s1). The 18S rRNA was used as an internal control.
For each sample, the mRNA levels were normalized against the 18S
rRNA level and the ratios of normalized mRNA to the untreated
control sample were determined using the comparative Ct method, as
previously described (8).
Enzyme-linked immunosorbent assay (ELISA)
to measure NF-κB DNA binding activity
Nuclear protein extracts were prepared from the
LPS-treated cells using a nuclear extract kit (Active Motif,
Carlsbad, CA, USA) and analyzed to determine the protein
concentration using a BCA protein assay kit (Pierce, Rockford, IL,
USA). The binding activity of NF-κB to its target DNA sequence
(5′-GGGACTTTCC-3′) was measured using a TransAM NF-κB ELISA kit
(Active Motif). Briefly, 10 μg of protein in the nuclear protein
extracts were added to 96-well plate wells coated with
oligonucleotide containing the target DNA sequence. Following
incubation and washing, an anti-NF-κB antibody was added to the
wells followed by a horseradish peroxidase-conjugated secondary
antibody.
Electrophoretic mobility shift assay
(EMSA) to determine NF-κB activity
Primer sets containing an NF-κB target sequence
consisting of a forward primer, 5′-AGTTGAGGGGA CTTTCCCAGGC-3′ and a
complementary reverse primer, 5′-GCCTGGGAAAGTCCCCTCAACT-3′ were
biotin-labeled at their 5′ end. The forward and reverse primers
were annealed by heating at 95°C for 5 min and cooled slowly to
room temperature. Subsequently, 10 ng of annealed primer, 8 μg of
protein in the nuclear protein extract, and 1 μg of poly d(I-C)
were incubated at 15°C for 30 min in a final volume of 10 μl. For
competition experiments, 660 ng of unlabeled NK-κB probe was added.
The reaction mixtures were separated by 6% non-denaturing
polyacrylamide gel electrophoresis at 120 V in Tris-Borate-EDTA
buffer, and electrotransferred to a nylon membrane at 300 mA for 30
min. The location of the primer-protein complexes was visualized by
incubating the membrane with horseradish peroxidase-conjugated
streptavidin followed by enhanced chemiluminescence detection.
Reporter assay for NF-κB and
β-catenin
For NF-κB reporter assay, the cells were
co-transfected with a pGL 4.32 vector (Promega, Madison, WI, USA)
containing the NF-κB response element linked to a firefly
luciferase reporter gene and a 1:50 ratio of pGL 4.17 vector
(Promega) containing Renilla luciferase reporter gene. Cells
were harvested 24 h after transfection, and luciferase activity was
measured using a Dual-Luciferase Reporter Assay System (Promega).
For each assay, firefly luciferase activity was normalized to the
Renilla luciferase activity to control for variations in
transfection efficiency.
For the β-catenin reporter assay, the cells were
co-transfected with a TOPFlash vector (Millipore, Billerica, MA,
USA) containing the β-catenin response element linked to a firefly
luciferase reporter gene and a 1:50 ratio of pGL 4.17 vector
containing Renilla luciferase reporter gene. Luciferase
activity was measured as described above.
Western blot analysis of total
β-catenin
The cells were lysed with ice-cold RIPA buffer
containing 25 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1% Nonidet P-40,
1% sodium deoxycholate, 0.1% SDS and protease inhibitor cocktail
(Sigma-Aldrich). Total cell lysates were obtained after removing
the insoluble materials by centrifugation at 20,000 × g for 20 min
at 4°C. The protein concentrations were determined using a BCA
protein assay kit (Pierce), and 50 μg protein were separated by 12%
polyacrylamide gel electrophoresis and electrotransferred onto
nitrocellulose membranes at 150 mA for 1.5 h. The membranes were
then blocked for 3 h at room temperature with phosphate-buffered
saline containing 5% skim milk and 0.1% Tween-20 and incubated with
a 1:1,000-dilution of anti-β-catenin antibody (BD Biosciences, San
Jose, CA, USA) overnight at 4°C, and subsequently incubated with a
1:1,000-dilution of horseradish peroxidase-conjugated secondary
antibody (Cell Signaling, Beverly, MA, USA) for 2 h at room
temperature. Peroxidase activity was visualized using an ECL kit
(Bio-Rad Laboratories Inc., Hercules, CA, USA). Anti-β-actin
antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) was
used as the loading control for total cell lysates.
Western blot analysis of nuclear
β-catenin
The cells were harvested and then nuclear fractions
were collected using a nuclear extract kit (Active Motif). Protein
concentrations in each fraction were determined using a BCA protein
assay kit (Pierce). Protein (15 μg) was separated using a 12%
polyacrylamide gel electrophoresis and analyzed by western blot
analysis using an anti-β-catenin antibody followed by horseradish
peroxidase-conjugated anti-mouse secondary antibody.. Anti-TATA box
binding protein (TBP) was used as the loading control for nuclear
protein extracts.
Transfection with small interfering RNA
(siRNA)
The cells were seeded in 12-well plates at a density
of 5×105 cells/well and then transfected with 50 nM of
control siRNA or β-catenin siRNA (Santa Cruz Biotechnology, Inc.)
using Lipofectamine RNAiMAX transfection reagent (Invitrogen,
Carlsbad, CA, USA). After 18 h, the cells were treated with LPS
(0.001 to 10 μg/ml) to induce an inflammatory response. Untreated
cells were used as controls.
Statistical analysis
All data are expressed as the means ± standard
deviation from at least 3 replicate experiments. Statistically
significant differences between the treated and untreated samples
were detected using unpaired t-tests. A P-value of <0.05 was
considered statistically significant. All analyses were performed
using SPSS version 18.0 software (SPSS Inc., Chicago, IL, USA).
Results
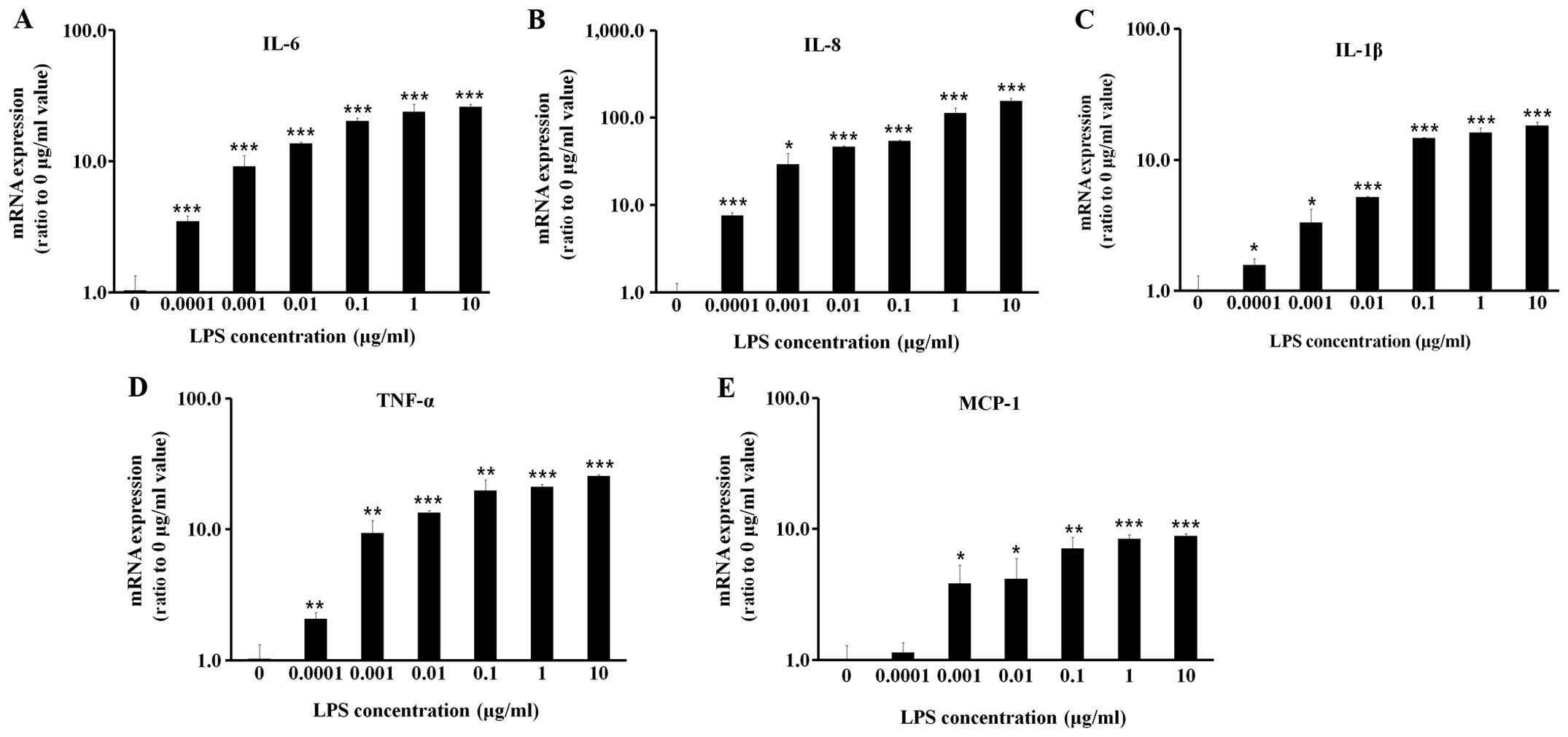
LPS induces inflammatory cytokine
expression in BEAS-2B human bronchial epithelial cells in a dose-
and time-dependent manner
When the BEAS-2B human bronchial epithelial cells
were treated with various concentrations of LPS (0.001 to 10
μg/ml), the expression levels of inflammatory cytokines, including
IL-6, IL-8, IL-1β, TNF-α and MCP-1 increased in a dose-dependent
manner (Fig. 1). It was found
that LPS induced significantly high levels of inflammatory cytokine
expression at a dose of 0.1 μg/ml, and all subsequent experiments
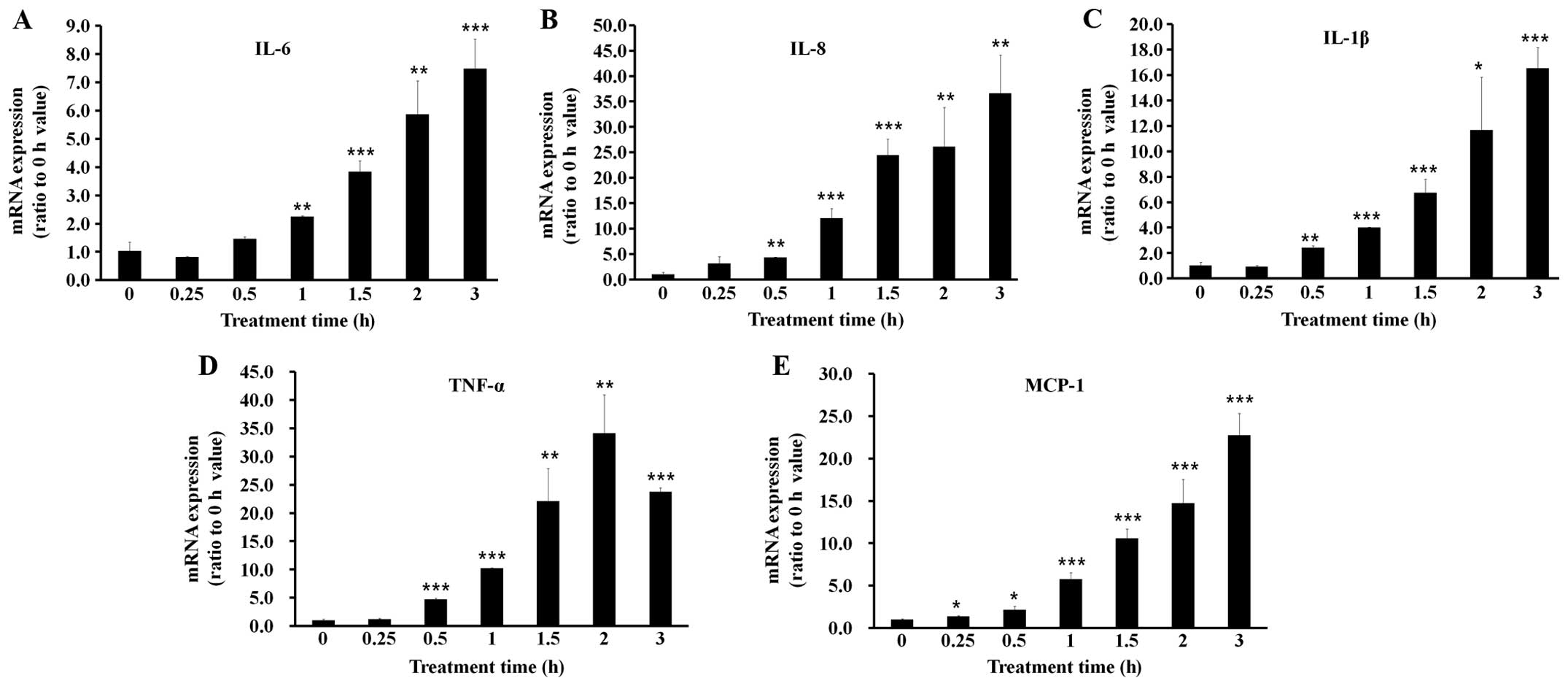
were conducted at this LPS concentration. Subsequently, when the
cells were treated with 0.1 μg/ml LPS for various periods of time,
the inflammatory cytokine expression began to increase 0.5 to 1 h
following treatment with LPS and subsequently increased in a
time-dependent manner for up to 3 h. TNF-α, however, showed its
maximum expression 2 h following treatment with LPS, and decreased
3 h following treatment with LPS (Fig. 2).
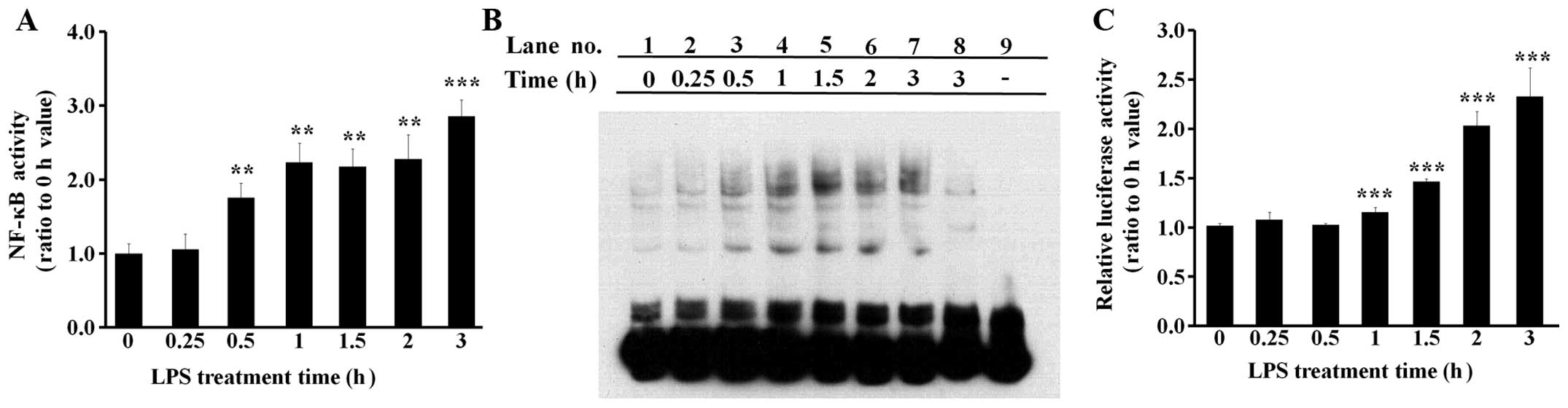
LPS induces NF-κB activity in BEAS-2B
human bronchial epithelial cells
The activity of NF-κB, the major inflammatory
transcription factor, was examined in the cells treated with 0.1
μg/ml LPS for various periods of time. The binding activity of
NF-κB to its target DNA sequence was investigated by both ELISA
(Fig. 3A) and EMSA (Fig. 3B) experiments. The results from
the ELISA and EMSA experiments were similar in that NF-κB was found
to bind its target DNA sequence following treatment with 0.5 h LPS
(Fig. 3A and B). In the EMSA
experiments, the shifted electrophoretic mobility patterns of the
labeled NF-κB probe were inhibited by excess amounts of unlabeled
NF-κB probe, showing that the results were not mediated by
non-specific binding (Fig. 3B,
lanes 3–7 vs. lane 8). The NF-κB reporter assay was conducted using
a pGL4.32 vector, which has a luciferase gene liked to an NF-κB
response element. The results indicated that NF-κB-driven
luciferase expression increased at approximately 1 h following
treatment with LPS (Fig. 3C).
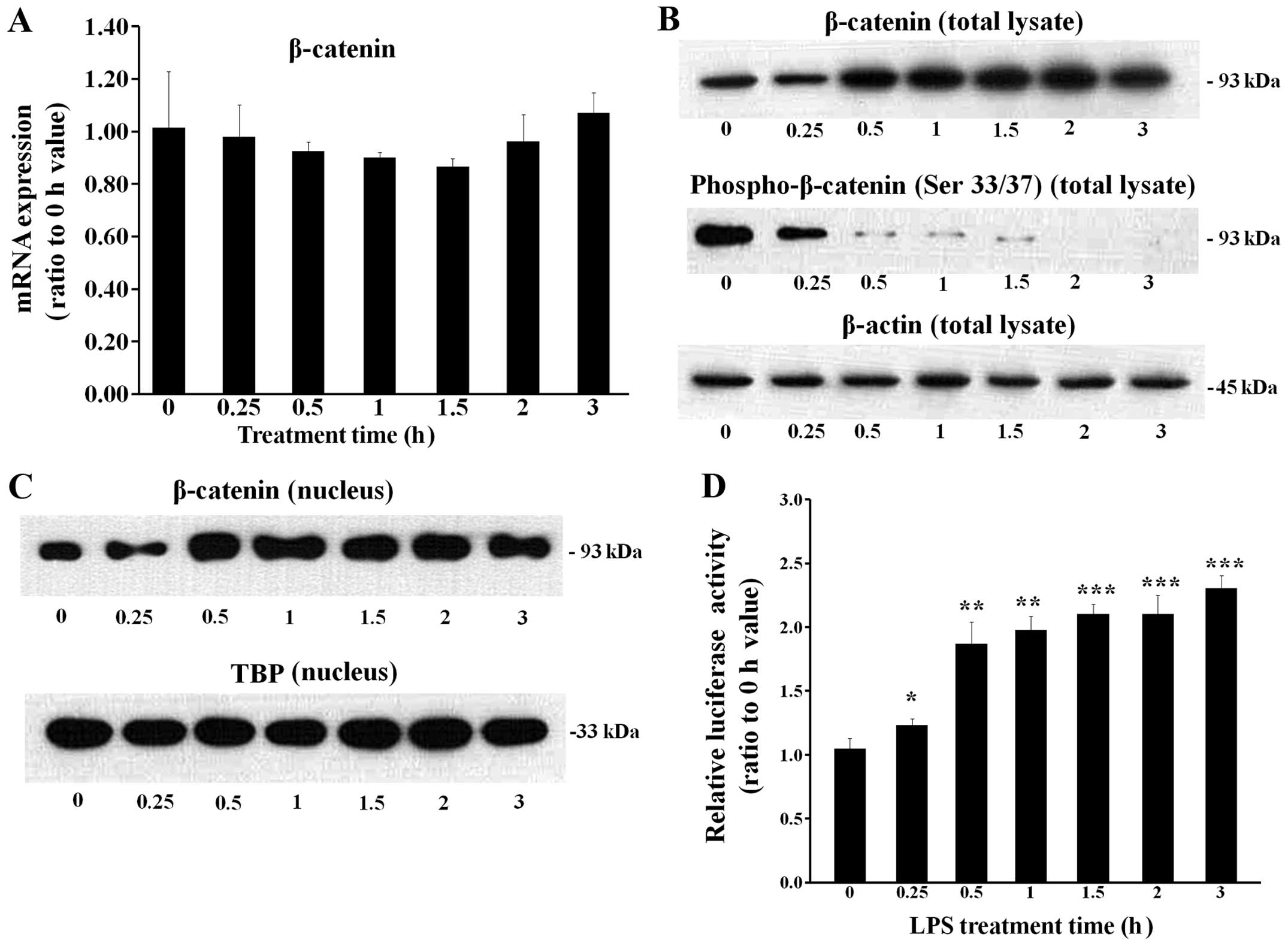
LPS upregulates the level of β-catenin
protein in total cell lysates and nuclear extracts
When the cells were treated with 0.1 μg/ml LPS for
various periods of time, the mRNA level of β-catenin was not
significantly altered (Fig. 4A).
However, the β-catenin protein level in the total cell lysates was
upregulated 0.5 h following treatment with LPS, which was
accompanied by the reduced phosphorylation of β-catenin at serine
33/37 residues (Fig. 4B). The
nuclear β-catenin protein level was also upregulated 0.5 h
following treatment with LPS (Fig.
4C). A reporter assay was conducted using a TOPFlash vector,
which has a luciferase gene liked to a β-catenin response element
(Fig. 4D). The results revealed
that LPS induced β-catenin-driven luciferase expression.
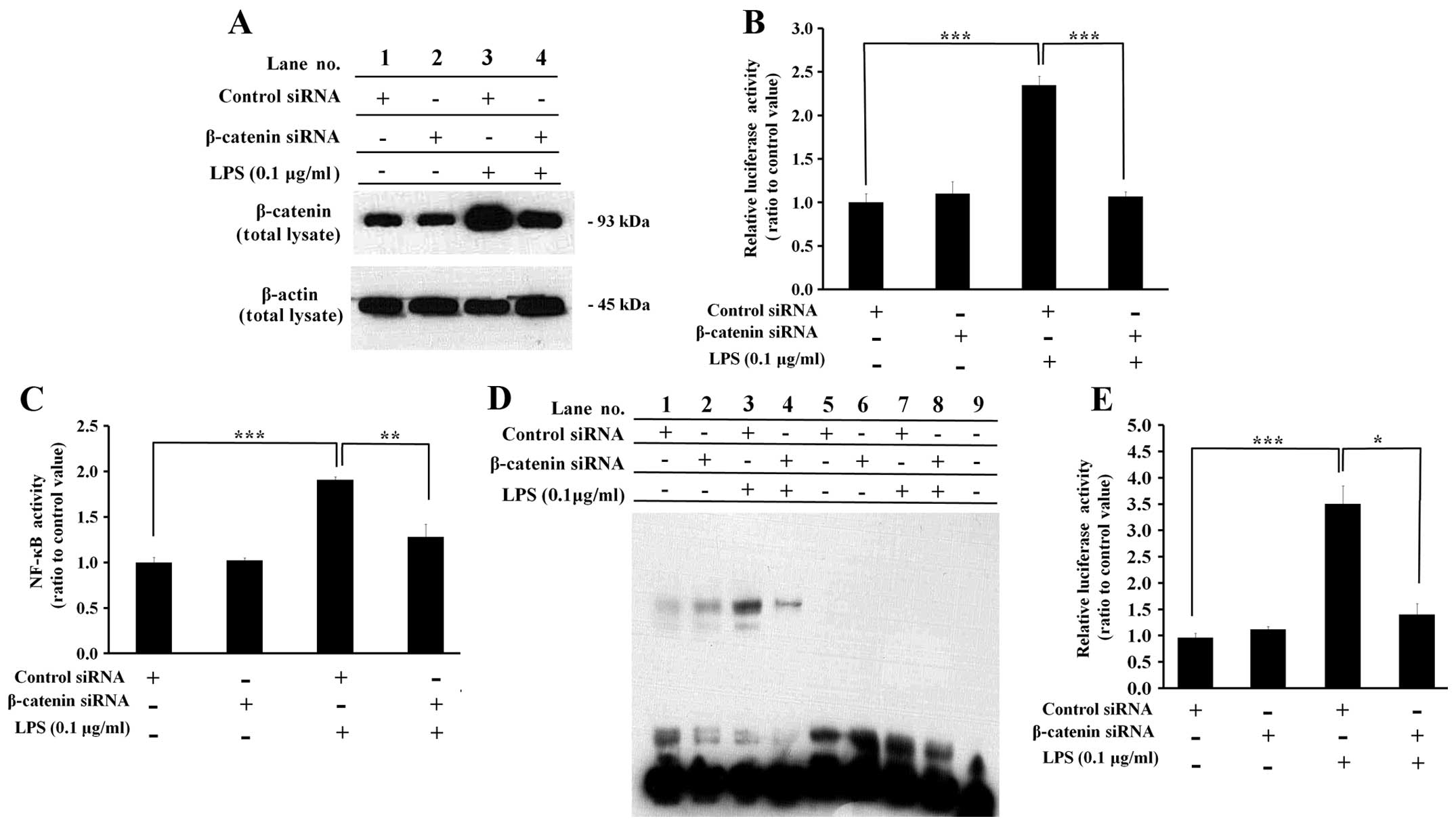
Knockdown of β-catenin results in a
decrease in NF-κB activity in LPS-treated cells
In the knockdown experiments, the cells were
transfected with control siRNA or β-catenin siRNA and treated with
LPS in order to induce NF-κB activity. Both the β-catenin protein
level and luciferase expression by a β-catenin responsive promoter,
which were upregulated following treatment with LPS, were decreased
by β-catenin siRNA transfection compared with control siRNA
transfection [Fig. 5A (lane 3 vs.
lane 4) and B], clearly showing that the siRNA-mediated knockdown
of β-catenin expression had occurred.
In this experimental condition, NF-κB activity was
measured by ELISA (Fig. 5C), EMSA
(Fig. 5D) and reporter assays
(Fig. 5E). The target DNA binding
activity of NF-κB measured by ELISA was significantly decreased by
β-catenin siRNA compared with control siRNA in the LPS-treated
cells (Fig. 5C), and the EMSA
results showed identical patterns (Fig. 5D, lane 3 vs. lane 4). The shifted
electrophoretic mobility patterns of the labeled NF-κB probe were
inhibited by excess unlabeled NF-κB probe, showing that the results
were not mediated by non-specific binding (Fig. 5D, lanes 5–8). Luciferase
expression mediated by a promoter containing the NF-κB target
sequence was also significantly decreased by β-catenin siRNA
compared with control siRNA in the LPS-treated cells (Fig. 5E).
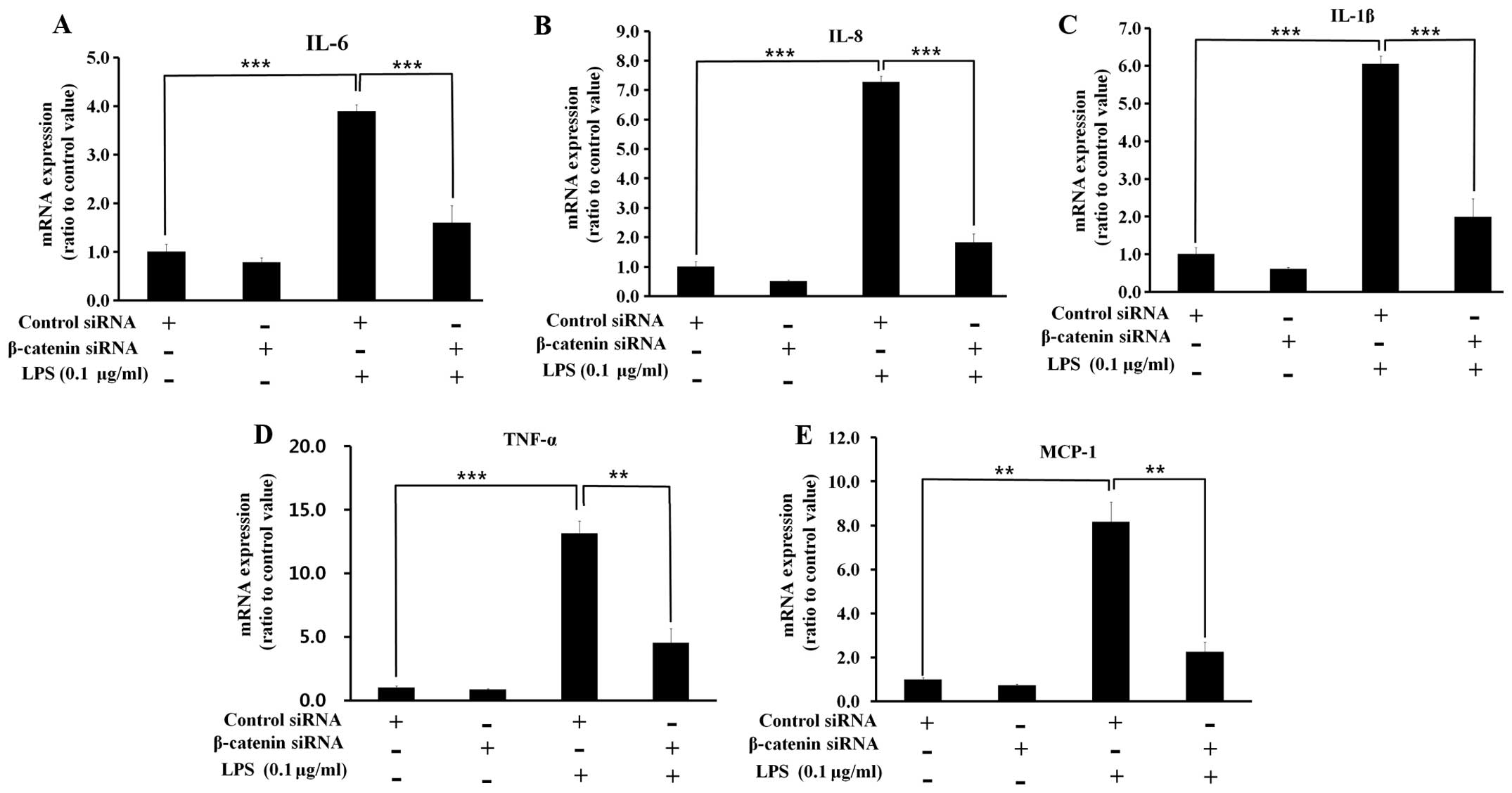
Knockdown of β-catenin results in
decreased inflammatory cytokine expression in LPS-treated
cells
The cells were transfected with control siRNA or
β-catenin siRNA and treated with LPS for the induction of
inflammatory cytokine expression. The experimental results
demonstrated that the expression levels of IL-6, IL-8, IL-1β, TNF-α
and MCP-1, which were induced by treatment with LPS, were all
significantly decreased by β-catenin siRNA transfection compared
with control siRNA transfection in the LPS-treated cells (Fig. 6).
Discussion
Our interest in β-catenin as a modulator of
bronchial inflammation was prompted by a previous study reporting
that a genetic polymorphism of β-catenin was significantly
associated with the risk of asthma in human subjects (7). Until now, however, the role of
β-catenin has not been elucidated in the inflammatory response of
bronchial epithelial cells. In the present study, we demonstrate
that β-catenin plays a role in the regulation of the inflammatory
response of human bronchial epithelial cells treated with LPS.
Our results revealed that LPS induced inflammatory
cytokine expression in bronchial epithelial cells in a dose- and
time-dependent manner (Figs. 1
and 2), which was accompanied by
the induction of NF-κB activity. NF-κB activity was measured using
3 different methods, ELISA, EMSA, and a reporter assay (Fig. 3). When the expression of β-catenin
was analyzed in the LPS-treated bronchial epithelial cells, the
mRNA levels were not altered (Fig.
4A). However, the β-catenin protein levels in the total cell
lysates were increased at 0.5 h following treatment with LPS with a
simultaneous reduction in the phosphorylation level at the serine
33/37 residues (Fig. 4B). Nuclear
β-catenin levels were also increased 0.5 h following treatment with
LPS (Fig. 4C). β-catenin is a
member of the WNT/β-catenin pathway that has been reported to
regulate cellular processes, including proliferation,
differentiation and development (6). The level of β-catenin is
post-translationally regulated in the WNT/β-catenin pathway. In its
inactive state, β-catenin protein is degraded by a destruction
complex composed of AXIN, glycogen synthase kinase (GSK)3β and
adenomatous polyposis coli (APC). GSK3β phosphorylates β-catenin at
serine 33 and 37 residues creating a binding site for E3 ubiquitin
for ubiquitination and proteolytic degradation. When the
WNT/β-catenin pathway is activated, the AXIN-GSK3β-APC complex is
disrupted and GSK3β is inactivated, resulting in the
dephosphorylation and stabilization of β-catenin followed by its
nuclear translocation (9,10). The experimental results presented
in this study clearly indicate that LPS induced the
dephosphorylation, stabilization and nuclear translocation of
β-catenin, as well as the reporter activity of the
β-catenin-responsive TOPFlash vector in the bronchial epithelial
cells (Fig. 4).
To elucidate the role of β-catenin in the
LPS-induced inflammatory response of bronchial epithelial cells,
β-catenin was knocked down using siRNA. The results revealed that
the β-catenin protein level, as well as its activity as a
transcriptional activator as measured by the β-catenin-responsive
TOPFlash vector reporter activity, was reduced to basal levels by
β-catenin siRNA transfection (Fig. 5A
and B). In this experimental condition, NF-κB activity was
measured using 3 different methods, ELISA, EMSA and reporter
assays; as shown by all 3 methods, its activity was reduced to
basal levels (Fig. 5C–E).
Similarly, LPS-induced inflammatory cytokine expression was reduced
to almost basal levels by β-catenin siRNA transfection (Fig. 6). These experimental data clearly
demonstrate that β-catenin is involved in the activation of NF-κB,
as well as in the induction of inflammatory cytokine expression in
the inflammatory response of LPS-treated bronchial epithelial
cells.
A number of studies have reported a major role for
NF-κB in the inflammation of bronchial epithelial cells stimulated
by toxic and pathogenic agents, including cigarette smoke extract,
diesel exhaust particles, wood dust, respiratory syncytial virus,
rhinoviruses, Bordetella pertussis and house dust mites
(11–16). The exaggerated activation of NF-κB
has been found in bronchial epithelial cells with a mutation
characteristic of cystic fibrosis, a genetic disease characterized
by chronic airway inflammation (17). Erythromycin, which improves the
clinical symptoms of patients with bronchiolitis, has been reported
to suppress the activation of NF-κB and IL-8 production in human
bronchial epithelial cells (18).
All the above studies have suggested that the regulation of NF-κB
activity has crucial importance for the effective treatment of
respiratory diseases involving bronchial inflammation.
Until now, β-catenin has been mainly associated with
cancer (19), and mutations in
the β-catenin gene have been commonly observed in endometrioid
ovarian cancer, hepatoblastoma, Wilms’ kidney tumors and some
colorectal cancers (20).
Considering the role of β-catenin in various cellular processes of
differentiation and development, however, β-catenin may also be
involved in chronic inflammatory diseases (6). Our previous study reported that
genetic polymorphisms of β-catenin are associated with asthma
(7); however, further studies are
required to elucidate the role of β-catenin in the regulation of
inflammation. A few studies have reported controversial roles of
β-catenin as a regulator of inflammation. Duan et al
reported that β-catenin negatively regulated the inflammatory
response induced by pathogenic Gram-negative bacteria. They
reported that Salmonella typhimurium stimulated the
degradation of β-catenin and increased the expression levels of
IL-6 and TNF-α in a mouse model and in colonic epithelial cells
(21). On the contrary, Kim et
al reported that β-catenin positively regulated the
inflammatory response to LPS. They reported that LPS induced
β-catenin accumulation and nuclear translocation followed by the
induction of NADPH oxidase in RAW 264.7 macrophages and murine
bone-marrow-derived macrophages (22). We have previously demonstrated
that β-catenin positively regulates inflammatory cytokine
expression in THP-1 human monocytic cells stimulated by the Der p 1
house dust mite allergen (23).
Despite discrepancies, these data suggest a role of β-catenin in
the regulation of the inflammatory response.
In this study, we provide clear experimental
evidence that β-catenin positively regulates NF-κB activity, as
well as inflammatory cytokine expression in bronchial epithelial
cells treated with LPS. The results of this study suggest that
β-catenin may be a target for the modulation of bronchial
inflammation, even though further studies are required to elucidate
the molecular mechanisms involved and to provide clinical
evidence.
Acknowledgements
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF-2013R1A1A2A10006146).
Abbreviations:
|
IL
|
interleukin
|
|
GSK
|
glycogen synthase kinase
|
|
LPS
|
lipopolysaccharide
|
|
MCP
|
monocyte chemoattractant protein
|
|
PCR
|
polymerase chain reaction
|
|
siRNA
|
small interfering RNA
|
|
TBP
|
TATA box binding protein
|
|
TNF
|
tumor necrosis factor
|
References
|
1
|
Montefort S, Herbert CA, Robinson C and
Holgate ST: The bronchial epithelium as a target for inflammatory
attack in asthma. Clin Exp Allergy. 22:511–520. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rietschel ET, Kirikae T, Schade FU, Mamat
U, Schmidt G, Loppnow H, Ulmer AJ, Zahringer U, Seydel U, Padova
FD, Schreier M and Brade H: Bacterial endotoxin: molecular
relationships of structure to activity and function. FASEB J.
8:217–225. 1994.PubMed/NCBI
|
|
3
|
Michel O: Role of lipopolysaccharide (LPS)
in asthma and other pulmonary conditions. J Endotoxin Res.
9:293–300. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Striz I, Mio T, Adachi Y, Bazil V and
Rennard S: The CD14 molecule participates in regulation of IL-8 and
IL-6 release by bronchial epithelial cells. Immunol Lett.
62:177–181. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Matsusaka T, Fujikawa K, Nishio Y, Mukaida
N, Matsushima K, Kishimoto T and Akira S: Transcription factors
NF-IL6 and NF-kappa B synergistically activate transcription of the
inflammatory cytokines, interleukin 6 and interleukin 8. Proc Natl
Acad Sci USA. 90:10193–10197. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moon RT, Bowerman B, Boutros M and
Perrimon N: The promise and perils of Wnt signaling through
beta-catenin. Science. 296:1644–1646. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bae S, Lee H, Choi BW, Lee HK, Chung SI,
Kim W, Kim K, Seo SJ, Kim DS, Kim SM and Yoon Y: Beta-catenin
promoter polymorphism is associated with asthma risk in Korean
subjects. Clin Biochem. 45:1187–1191. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
|
|
9
|
Cadigan KM and Liu YI: Wnt signaling:
complexity at the surface. J Cell Sci. 119:395–402. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Willert K and Nusse R: Beta-catenin: a key
mediator of Wnt signaling. Curr Opin Genet Dev. 8:95–102. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Profita M, Bonanno A, Montalbano AM,
Ferraro M, Siena L, Bruno A, Girbino S, Albano GD, Casarosa P,
Pieper MP and Gjomarkaj M: Cigarette smoke extract activates human
bronchial epithelial cells affecting non-neuronal cholinergic
system signalling in vitro. Life Sci. 89:36–43. 2011. View Article : Google Scholar
|
|
12
|
Tal TL, Simmons SO, Silbajoris R, Dailey
L, Cho SH, Ramabhadran R, Linak W, Reed W, Bromberg PA and Samet
JM: Differential transcriptional regulation of IL-8 expression by
human airway epithelial cells exposed to diesel exhaust particles.
Toxicol Appl Pharmacol. 243:46–54. 2010. View Article : Google Scholar
|
|
13
|
Pylkkanen L, Stockmann-Juvala H, Alenius
H, Husgafvel-Pursiainen K and Savolainen K: Wood dusts induce the
production of reactive oxygen species and caspase-3 activity in
human bronchial epithelial cells. Toxicology. 262:265–270. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bossios A, Gourgiotis D, Skevaki CL,
Saxoni-Papageorgiou P, Lotvall J, Psarras S, Karpathios T,
Constandopoulos AG, Johnston SL and Papadopoulos NG: Rhinovirus
infection and house dust mite exposure synergize in inducing
bronchial epithelial cell interleukin-8 release. Clin Exp Allergy.
38:1615–1626. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ishibashi Y and Nishikawa A: Role of
nuclear factor-kappa B in the regulation of intercellular adhesion
molecule 1 after infection of human bronchial epithelial cells by
Bordetella pertussis. Microb Pathog. 35:169–177. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Thomas LH, Friedland JS, Sharland M and
Becker S: Respiratory syncytial virus-induced RANTES production
from human bronchial epithelial cells is dependent on nuclear
factor-kappa B nuclear binding and is inhibited by
adenovirus-mediated expression of inhibitor of kappa B alpha. J
Immunol. 161:1007–1016. 1998.
|
|
17
|
Venkatakrishnan A, Stecenko AA, King G,
Blackwell TR, Brigham KL, Christman JW and Blackwell TS:
Exaggerated activation of nuclear factor-kappaB and altered
IkappaB-beta processing in cystic fibrosis bronchial epithelial
cells. Am J Respir Cell Mol Biol. 23:396–403. 2000. View Article : Google Scholar
|
|
18
|
Desaki M, Okazaki H, Sunazuka T, Omura S,
Yamamoto K and Takizawa H: Molecular mechanisms of
anti-inflammatory action of erythromycin in human bronchial
epithelial cells: possible role in the signaling pathway that
regulates nuclear factor-kappaB activation. Antimicrob Agents
Chemother. 48:1581–1585. 2004. View Article : Google Scholar
|
|
19
|
Clevers H and Nusse R: Wnt/beta-catenin
signaling and disease. Cell. 149:1192–1205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Polakis P: The many ways of Wnt in cancer.
Curr Opin Genet Dev. 17:45–51. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Duan Y, Liao AP, Kuppireddi S, Ye Z,
Ciancio MJ and Sun J: beta-Catenin activity negatively regulates
bacteria-induced inflammation. Lab Invest. 87:613–624.
2007.PubMed/NCBI
|
|
22
|
Kim JS, Yeo S, Shin DG, Bae YS, Lee JJ,
Chin BR, Lee CH and Baek SH: Glycogen synthase kinase 3beta and
beta-catenin pathway is involved in toll-like receptor 4-mediated
NADPH oxidase 1 expression in macrophages. FEBS J. 277:2830–2837.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jang J, Ha JH, Kim SM, Kim W, Kim K, Chung
SI and Yoon Y: β-catenin mediates the inflammatory cytokine
expression induced by the Der p 1 house dust mite allergen. Mol Med
Rep. 9:633–638. 2014.
|