Introduction
Parkinson’s disease (PD) is a progressive
neurodegenerative disease with clinical symptoms such as tremor,
rigidity and bradykinesia and abnormal postural reflexes (1). The primary pathology of the disease
is degeneration of the nigrostriatal system, which results in the
loss of dopaminergic neurons and depletion of striatal dopamine. As
the disease progresses, a variety of non-motor symptoms emerge due
to the loss of non-dopaminergic pathways (2). The most effective therapy is
replacement of dopamine with levodopa or dopamine receptor
agonists. Despite their benefits, chronic treatment using these
agents is associated with adverse effects including on-off
fluctuations, wearing-off phenomena, or drug-induced dyskinesia
(3). Therefore, it is important
to develop new therapies.
The pathogenic features identified as being
instrumental in the dopaminergic cell death process that occurs in
PD, including oxidative stress, mitochondrial dysfunction,
inflammation and apoptosis, provide targets for the development of
new neuroprotective compounds (4). Erythropoietin (EPO) is a well-known
hematopoietic hormone produced in the fetal liver and adult kidney.
Neuroprotective effects of EPO have been previously demonstrated
using preclinical models of central nervous system (CNS) diseases
including focal and global ischemia, neurotrauma, autoimmune
encephalitis, kainate-induced seizures, subarachnoid hemorrhage and
spinal cord injury. It has been demonstrated that EPO can provide
neuroprotection of neurons against experimental lesions (5). However, the exact mechanisms
involved in EPO neuroprotection remain to be determined. EPO
protein and receptors are detected in brain neurons, astrocytes,
oligodendrocytes, microglia and cerebral endothelial cells
(6). The mechanisms of
EPO-induced neuroprotection include the prevention of
glutamate-induced toxicity, inhibition of apoptosis,
anti-inflammatory effects, antioxidant effects, and stimulation of
angiogenesis. As EPO can cross the blood-brain barrier (BBB)
(7), it has been reported that
systemic administration of EPO may reduce neuron damage in animal
models of ischemic stroke (8),
traumatic brain injury (9), and
spinal cord injury (10).
Neuroprotective effects of EPO infusion into the striatum were
clarified in a previous study (11). The typical PD rat model is the
6-hydroxydopamine (6-OHDA) model, which is currently the most
commonly used procedure for obtaining an experimental nigrostriatal
lesion in the animal. Recent studies (11) have shown that in 6-OHDA-lesioned
rats, the abnormal activation of c-Jun N-terminal kinases (JNK),
extracellular signal-regulated kinase (ERK), and the p38
mitogen-activated protein kinase (MAPK) pathway has been
identified. Additionally, EPO has been shown to prevent
staurosporine-induced apoptosis via STAT5, AKT and MAPK signaling
pathways (39). The aim of the
present study was to clarify whether different doses of EPO exert
neuroprotective and neurogeneic effects on 6-OHDA-treated
dopaminergic neurons via the MAPK pathway in vivo.
Materials and methods
Animals
Adult female Sprague-Dawley rats, weighing 180–220
g, were used in this study. Protocols involving the animals were
approved by the Institutional Review Board of Xinhua Hospital and
were performed according to the guidelines of the National
Institutes of Health for the Care and Use of Laboratory Animals
(NIH publication no. 80-23). The rats were housed in standard
Plexiglas cages with a maximum of 5 animals per cage and had free
access to food and water. Environmental conditions were strictly
controlled, with a 12-h light/dark cycle, temperature of 22°C and
humidity of 44%. The number of animals used was the minimum
required for statistical analysis (n=98).
The rats were divided into 7 groups. In group 1
(n=14, sham), rats were intraperitoneally injected (i.p.) saline
daily for 5 days. In groups 2 (n=14), 3 (n=14) and 4 (n=14), the
rats were i.p. injected doses of 2,500; 5,000 and 10,000 U/kg EPO
daily for 5 days. In group 5 (n=14), rats received saline via
medial forebrain bundle (MFB), while in group 6 (n=14), rats
received 6-OHDA via MFB. In group 7 (n=14), EPO (10,000 U/kg) was
continuously administered i.p. for 5 days to rats prior to
administration of 6-OHDA injection.
EPO injections
For the systemic administration of groups 2–4, EPO
was dissolved in saline, and i.p. injected at doses of 2,500; 5,000
and 10,000 U/kg on a daily basis for 5 days. For group 7, EPO
(10,000 U/kg) was i.p. injected daily for 5 days prior to injection
of 6-OHDA.
6-OHDA lesion
Rats were anaesthetized with ketamine (100 mg/kg,
i.p.) prior to surgery and placed on a stereotaxic frame (SR-9M,
Stereotaxic Instrument; Narishige Scientific Instrument Lab, Tokyo,
Japan). The skull was exposed and a burr hole was drilled to
introduce a syringe for injection of 6-OHDA (Sigma, St. Louis, MO,
USA) solution containing 4.0 μg 6-OHDA/μl in 0.9% saline with 0.02%
ascorbic acid (Sigma), pH 5.0. To minimize variability due to
degradation of the toxin, the 6-OHDA solution was freshly prepared,
kept on ice and protected from exposure to light. Each animal
received two injections of 4 μl of the solution into the right MFB
with a 10 μl Hamilton Syringe at the stereotaxic coordinates (from
bregma and dura) of AP −3.7 mm, ML +1.7 mm, DV −7.8 mm, and AP −4.4
mm, ML +1.2 mm, DV −7.8 mm. The tooth bar was set to −2.4 mm
(12,13). A total volume of 4 μl 6-OHDA was
injected at a flow rate of 1 μl/min. Following the injection, the
needle was left in place for 10 min and then slowly removed. The
skin was sutured and the animals were removed from the stereotaxic
instrument, placed on a heating pad for 30 min and returned to
their cage. In this rat model, 6-OHDA caused a progressive loss of
dopaminergic neurons in the substantia nigra (SN) as it was
absorbed by the neuron’s terminals in the striatum and transported
to the dopaminergic neurons in the SN, leading to damage of the
dopaminergic neurons (14). Rats
undergoing a sham lesion procedure in which only the vehicle [0.9%
saline with 0.02% ascorbic acid (Sigma), pH 5.0] for 6-OHDA was
injected into the MFB seved as the controls.
Behavioral analysis
At 21 days following surgery, the animals underwent
behavioral testing. Rats were injected with apomorphine (0.25 mg/kg
in 0.9% saline, i.p.) and placed in a stainless steel cylindrical
bowl. Net rotations (contralateral turns minus ipsilateral turns)
were counted over a 30-min period beginning 5 min after the
administration of apomorhine. Assessments were carried out by an
observer who was blind to the animal pretreatments (15).
Quantitative analysis of EPO in the
cerebrospinal fluid (CSF)
EPO was administered into rats at concentrations of
2,500; 5,000 and 10,000 U/kg, respectively, via i.p. injection.
Three hours post-injection, the CSF samples were collected via
cisternal puncture as previously described (16). Briefly, the rats were anesthetized
with ketamine (100 mg/kg, i.p.), and the head of each rat was fixed
at a specific forward angle. The back of the neck and base of the
skull were shaved and disinfected with 70% ethyl alcohol. An
incision was made in the skin over the occipital bone, the fascia
was retracted and superficial muscles were dissected. When the
allanto-occipital membrane was exposed, the cisterna magna was
cannulated by placing a 30-gauge needle, and CSF was carefully
withdrawn. CSF was ejected into a 0.5 ml Eppendorf tube and frozen
at −80°C. The volume of CSF ranged between 50 and 150 μl. EPO
concentrations in CSF samples were measured using an enzyme-linked
immunosorbent assay (ELISA) and an immunoblotting assay.
Saline-injected animals were used as controls. EPO concentrations
in CSF were measured by ELISA using the Quantikine IVD EPO kit
(R&D Systems, Minneapolis, MN, USA). ELISA was used to detect
endogenous rat EPO and EPO. A standard curve was performed as
protocol ranging from 0 to 200 mU/ml EPO. The CSF samples were
diluted based on prior experience to fit the ELISA standard range.
The absorbance was assessed using a Bio-Tek μQuant™ Microplate
Spectrophotometer MQX200 reader (BioTek Instruments Inc, Winooski,
VT, USA).
Immunocytochemistry
Seven randomly selected rats per group were
anesthetized with ketamine (100 mg/kg, i.p.) and transcardially
perfused with 100 ml of 0.1 M phosphate-buffered saline (PBS, pH
7.2) followed by 200 ml of 4% paraformaldehyde at 4°C. The brains
were removed, post-fixed for 4 h in 4% paraformaldehyde, embedded
in paraffin, and cut into 3 μm coronal sections on a sliding rotary
microtome (Leica RM2235; Leica Microsystems Nussloch GmbH,
Nussloch, Germany). Seven random sets of serial SN sections of each
rat were collected for immunocytochemistry.
Immunostaining was carried out in sections using a
standard avidin-biotin immunocytochemical protocol as previously
described (17). Endogenous
peroxidase activity was quenched by incubation for 10 min in 0.1 M
PBS containing 0.2% Triton X-100 with 3% hydrogen peroxide.
Non-specific binding sites were blocked for 60–90 min with 5–10% of
the appropriate serum in PBS containing 0.2% Triton X-100. To
localize TH-immunoreactivity (TH-IR) neurons a mouse monoclonal
tyrosine hydroxylase (TH) primary antibody (1:1,000; Sigma-Aldrich,
St. Louis, MO, USA) was used. Polyclonal rabbit antibodies to EPO
(1:200; R&D Systems) and erythropoietin receptor (EPOR) (1:50;
Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) were also
used. The primary antibodies were diluted in 0.1 M PBS-TX and 1% of
goat serum thereby yielding the secondary antibody. Following
incubation with the primary antibody overnight at 4°C, the sections
were washed and incubated with the appropriate HRP-conjugated goat
polyclonal secondary antibody to mouse and rabbit IgG 1:500 (all
from Abcam, Cambridge, UK) for 1–2 h at room temperature. The
secondary antibodies were diluted in 0.1 M PBS and 5% BSA. After
washing, the antibody staining was visualized using a DAB kit
(Abcam). After developing the reaction, the stained sections were
mounted, dried, dehydrated and coverslipped with neutral balsam and
examined using a light microscope (Leica Biosystems Nussloch
GmbH).
Double-labeling fluorescent immunohistochemistry of
dopaminergic and EPOR-expressed neurons was performed as previously
described (18). The sections
were incubated with TH antibody (1:1,000; Sigma-Aldrich) and EPOR
antibody (1:50; Santa Cruz Biotechnology, Inc.) at 4°C overnight.
The primary antibodies were diluted in 0.1 M PBS-TX and 1% goat
serum. These proteins were detected using a mixture of Alexa Fluor
488 and Cy3 goat polyclonal secondary antibodies to mouse and
rabbit IgG 1:200 (all from Abcam) for 1.5 h at room temperature in
the dark. The secondary antibodies were diluted in 0.1 M PBS and 5%
BSA. The sections were mounted in fluorescent mounting medium
(Abcam), coverslipped, and kept in the dark at 4°C until they were
examined. The specific immunofluorescence of the Alexa 488 or Cy3
fluorophores was visualized by excitation at 488 or 550 nm,
respectively. Images were captured with an Olympus AX 70
fluorescence microscope using Olympus Fluoview FV1000 LSM and
Fluoview software (v 1.3).
Morphological assessment
Quantification of the TH-positive neurons in the
lesioned and intact SN of each rat was measured in 5–6 coronal
sections per animal using research-grade Image-Pro Plus software
(Media Cybernetics, Inc., Silver Spring, MD, USA). The borders of
the areas of interest were outlined from a live image with a 4X
objective and the entire area of interest was examined using a 20X
objective. Only TH-positive cells with an identifiable unlabelled
nucleus surrounded by TH-immunolabelled cytoplasm or those with
labelled dendrites were counted. Data were expressed as the number
of TH-positive neurons per mm2 of striatum per animal.
Quantification of TH-positive fibres was carried out by optical
density analysis with the aid of an Image-Pro Plus software (Media
Cybernetics, Inc.) using a 20x lens. TH-positive fibre staining
intensity was determined in striatal areas surrounding TH-IR
neurons, through the rostrocaudal extent of the dopaminergic
lesion. Similar measurements were carried out in the unlesioned
striatum in randomly chosen areas. The data from the lesioned side
are presented as a percentage (mean ± SE) of the values from the
unlesioned SN. To estimate the number of EPO-immunopositive cells,
four sections from each of the three levels (rostral, middle and
caudal) of the brain were examined under a 20x lens. Images of four
fields per section per hemisphere were captured and the cell
optical density was measured by Image-Pro Plus software.
Tissue preparation
Animals were sacrificed with an overdose of
pentobarbital (50 mg/kg body weight, i.p.), injected intracardially
with 10 ml of cold saline, and the brains immediately removed.
Brains were transfered to a plastic plate cooled on ice to remove
both sides of the striatums and SN. Tissues were dissected and
frozen in liquid nitrogen and stored at −80°C for immunoblotting.
Seven rats from each group were used for immunoblotting.
Immunoblotting
Samples were homogenized in RIPA buffer [50 mM Tris
(pH 7.5), 150 mM NaCl, 0.1% sodium dodecyl sulfate, 1 mM EDTA
(ethylenediaminetetraacetic acid), 1% Nonidet P-40] containing
protease inhibitors (Roche Diagnostics Corp., Indianapolis, IN,
USA) and 2 mM phenylmethylsulfonyl fluoride (PMSF) by sonication.
The homogenate was centrifuged at 14,000 × g for 10 min at 4°C, and
the pellet containing mainly nuclei and large debris was discarded.
After determining the protein concentration in supernatants using
the Pierce BCA assay kit (Thermo Fisher Scientific, Rockford, IL,
USA), the samples were boiled 5 min in Laemmli Sample Buffer
(Bio-Rad Life Science, Hercules, CA, USA). Samples containing
equivalent amounts of protein were subjected to sodium
dodecylsulfate (SDS)-polyacrylamide gel electrophoresis. Proteins
were electrotransferred to a 0.22 μm Immobilon PVDF membrane
(Bio-Rad, Hercules, CA, USA) in a transfer buffer (25 mM Tris, 192
mM glycine, and 20% methanol) with 250 mA current for 90 min at
4°C.
Subsequent to being transferred, the membrane was
blocked by incubation in blocking buffer (50 mM Tris-HCl, pH 7.5,
150 mM NaCl, 0.1% Tween-20 and 5% w/v non-fat dry milk) for 1 h at
room temperature. The membranes were incubated overnight at 4°C
with mouse monoclonal β-actin antibody (1:5,000), mouse monoclonal
TH antibody (1:5,000; both from Sigma-Aldrich), polyclonal rabbit
EPO antibody (1:500; R&D Systems), polyclonal rabbit p44/42
MAPK (ERK1/2) antibody (1:500), polyclonal rabbit Phospho-p44/42
MAPK (ERK1/2) (Thr202/Tyr204) antibody (1:500), polyclonal rabbit
p38 MAPK antibody (1:500), polyclonal rabbit Phospho-p38 MAPK
(Thr180/Tyr182) antibody (1:500), anti-SAPK/JNK (1:500), polyclonal
rabbit Phospho-SAPK/JNK (Thr183/Tyr185) antibody (1:500),
polyclonal rabbit caspase-3 antibody (1:500), and polyclonal rabbit
cleaved caspase-3 antibody (1:500) (all from Cell Signaling
Technology, Inc., Danvers, MA, USA). The primary antibodies were
diluted in TBST (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1%
Tween-20) and 5% w/v BSA. The membranes were subsequently washed
with TBST (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% Tween-20) and
incubated with HRP-conjugated goat polyclonal secondary antibody to
mouse and rabbit IgG (1:2,000; Cell Signaling Signaling Technology,
Inc.) for 1 h at room temperature. The secondary antibodies were
diluted in blocking buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl,
0.1% Tween-20 and 5% w/v non-fat dry milk). Bound antibodies were
visualized using the enhanced chemiluminescence detection system
(Millipore, Billerica, MA, USA) and analyzed semiquantitatively
using Image Lab Software (Bio-Rad).
Statistical analysis
Data were expressed as the mean ± standard
deviations of the mean. Bars in the figures indicate mean values ±
standard deviations of the mean. A one-factor ANOVA with
Bonferroni’s post-hoc test was used to compare behavioral rotation
rates, cell counts, and optical density between each of the
treatment groups. A paired Student’s t-test was used to observe
differences between the lesion and unlesion side within groups.
Prior to this, homogeneity of variance between the various groups
was ascertained. Differences at P<0.05 were considered
statistically significant. Statistical tests were performed with
SPSS 17.0 software (SPSS Inc., Chicago, IL, USA).
Results
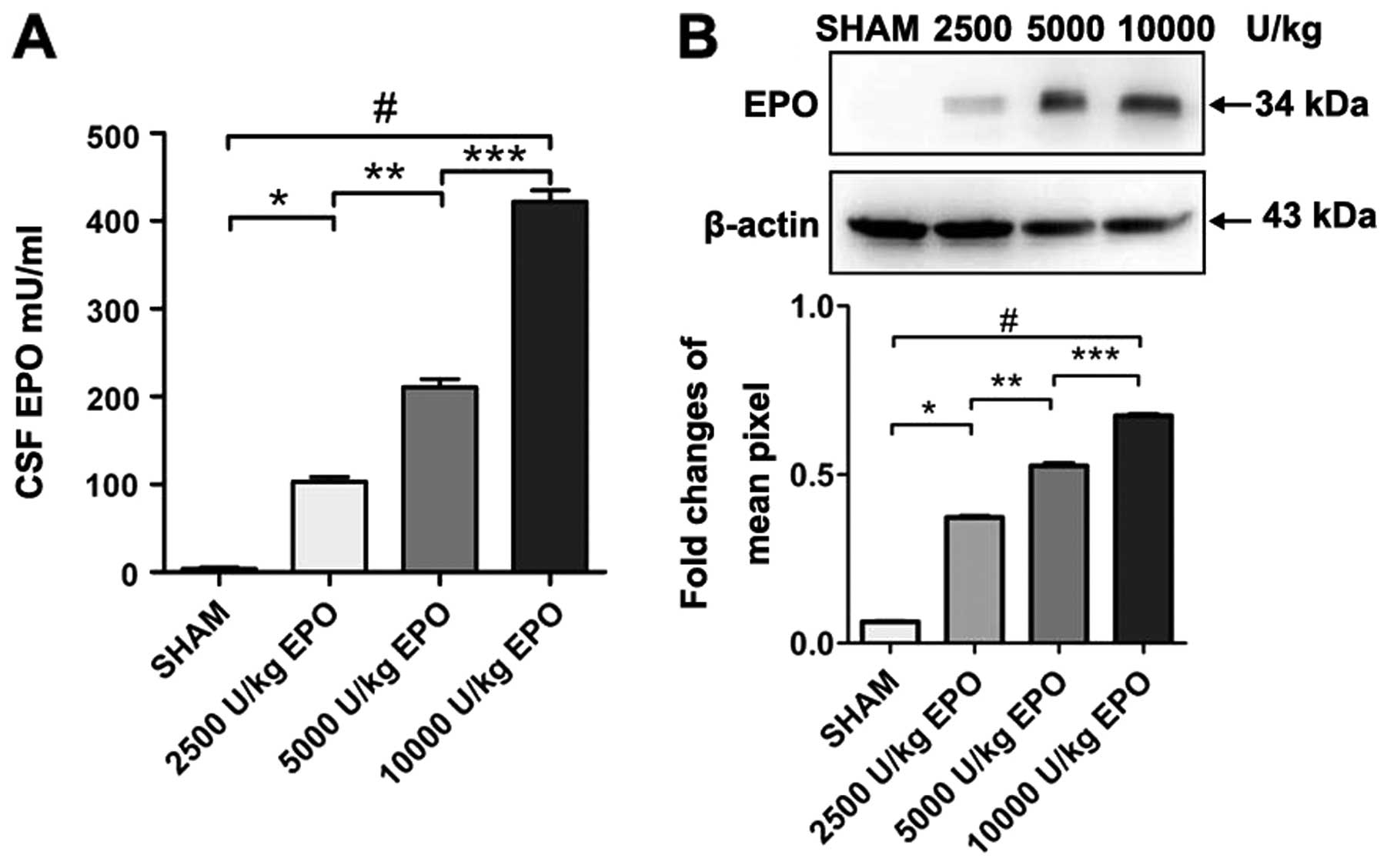
Systemic administration of high-dose EPO
penetrates the BBB and is detectable in brain
EPO was measured in CSF in rats
As expected, we found a dose-dependent increase of
EPO concentration in CSF when comparing 2,500; 5,000 and 10,000
U/kg EPO administered i.p. EPO was undetectable in CSF prior to
injection in groups 1–4. Three hours after the EPO injection,
however, EPO was detected in the CSF of animals administered 2,500;
5,000 and 10,000 U/kg. Western blot analysis revealed a significant
increase in EPO levels in the CSF in the 10,000 U/kg group compared
with the 2,500 and 5,000 U/kg group (P<0.01; Fig. 1).
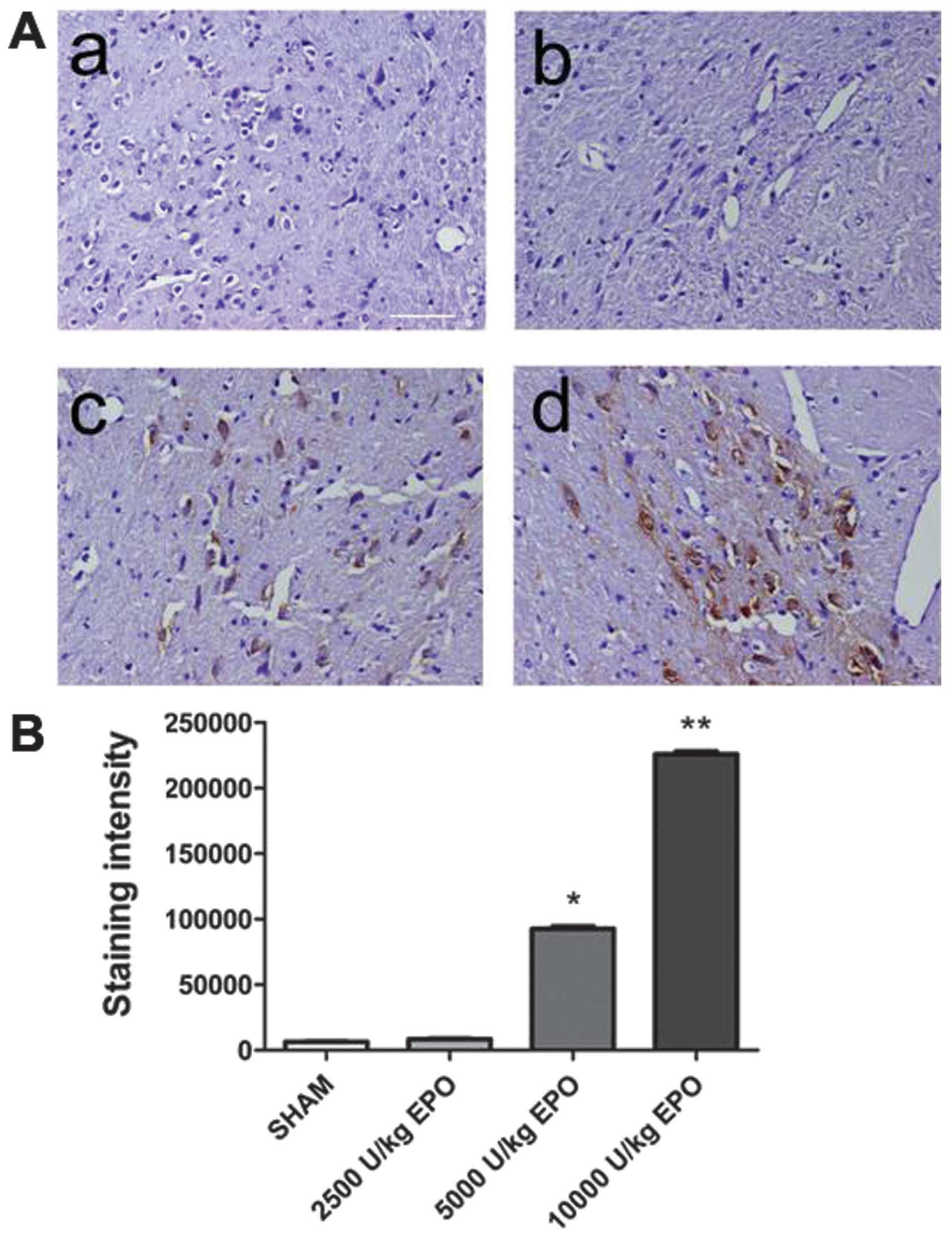
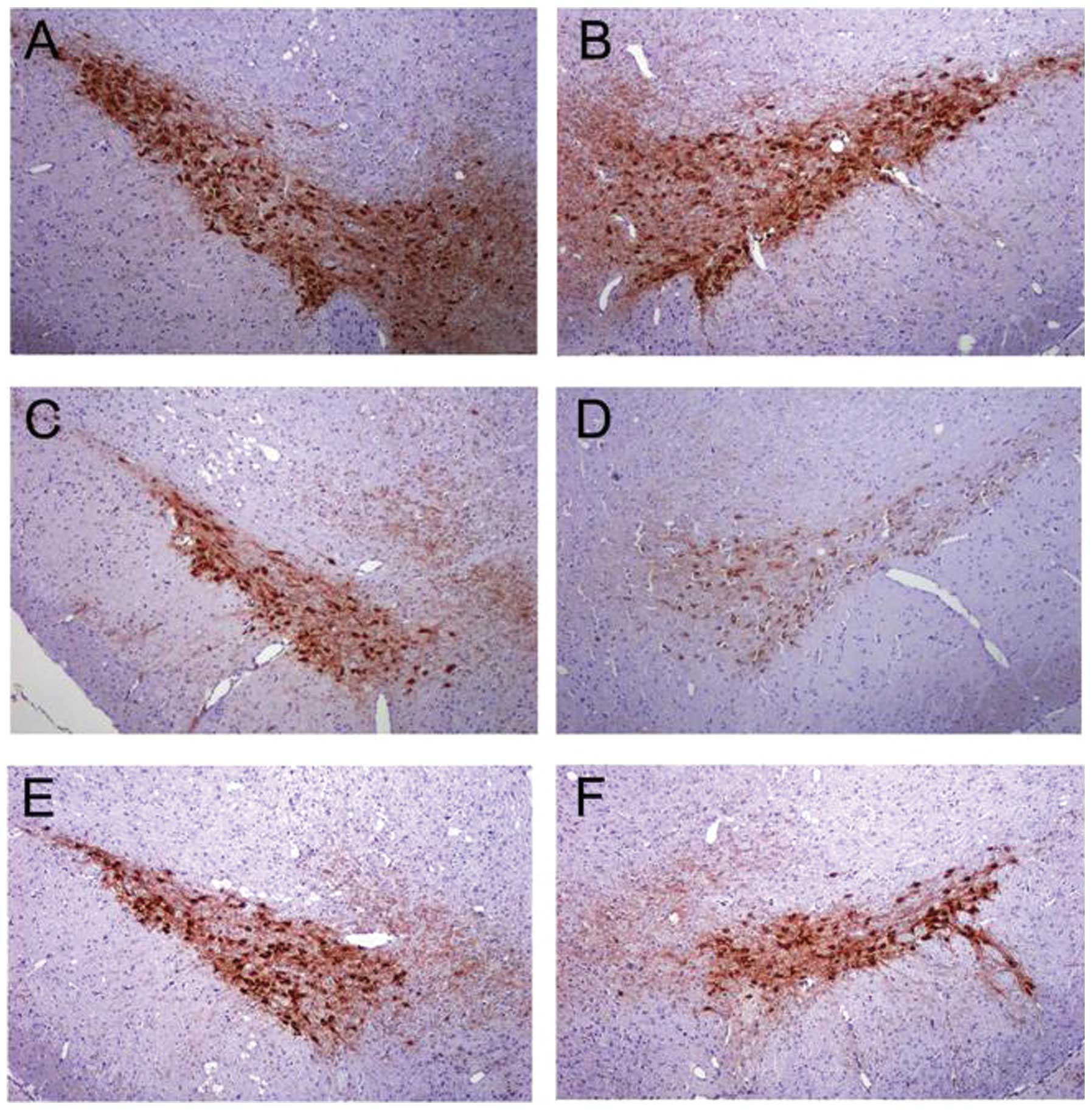
EPO was detected in brain
EPO was undetectable in brain prior to EPO
injection. Three hours after the EPO injection, however, EPO was
detected in the brain of group 1–4 animals administered 5,000 and
10,000 U/kg. A significant increase in the number of EPO-positive
cells in the brain of the 10,000 U/kg group compared with the 2,500
and 5,000 U/kg groups was observed (P<0.01; Fig. 2). A dose of 10,000 U/kg was used
in subsequent experiments.
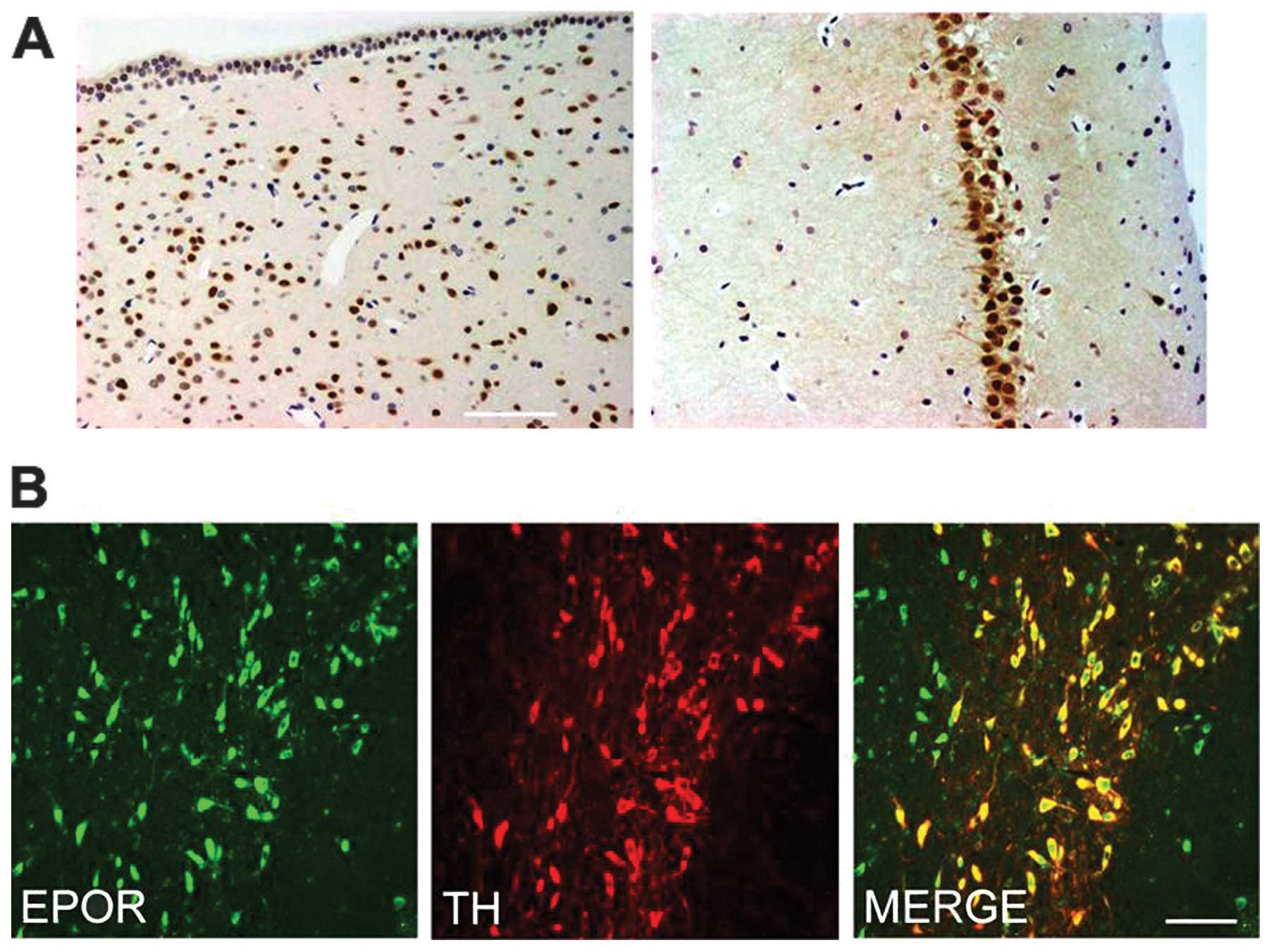
EPOR is expressed in the brain
We first determined the expression of EPOR in the
brain of rat. The periventricular zone and hippocampus exhibited
intense immunoreactivity for EPOR in many medium to large neurons
(Fig. 3A). In the SN pars
compacta (SNpc), we examined the co-expression of EPOR in TH-IR
dopaminergic neurons. Notably, the SNpc TH-IR neurons were strongly
immunoreactive for EPOR (Fig.
3B).
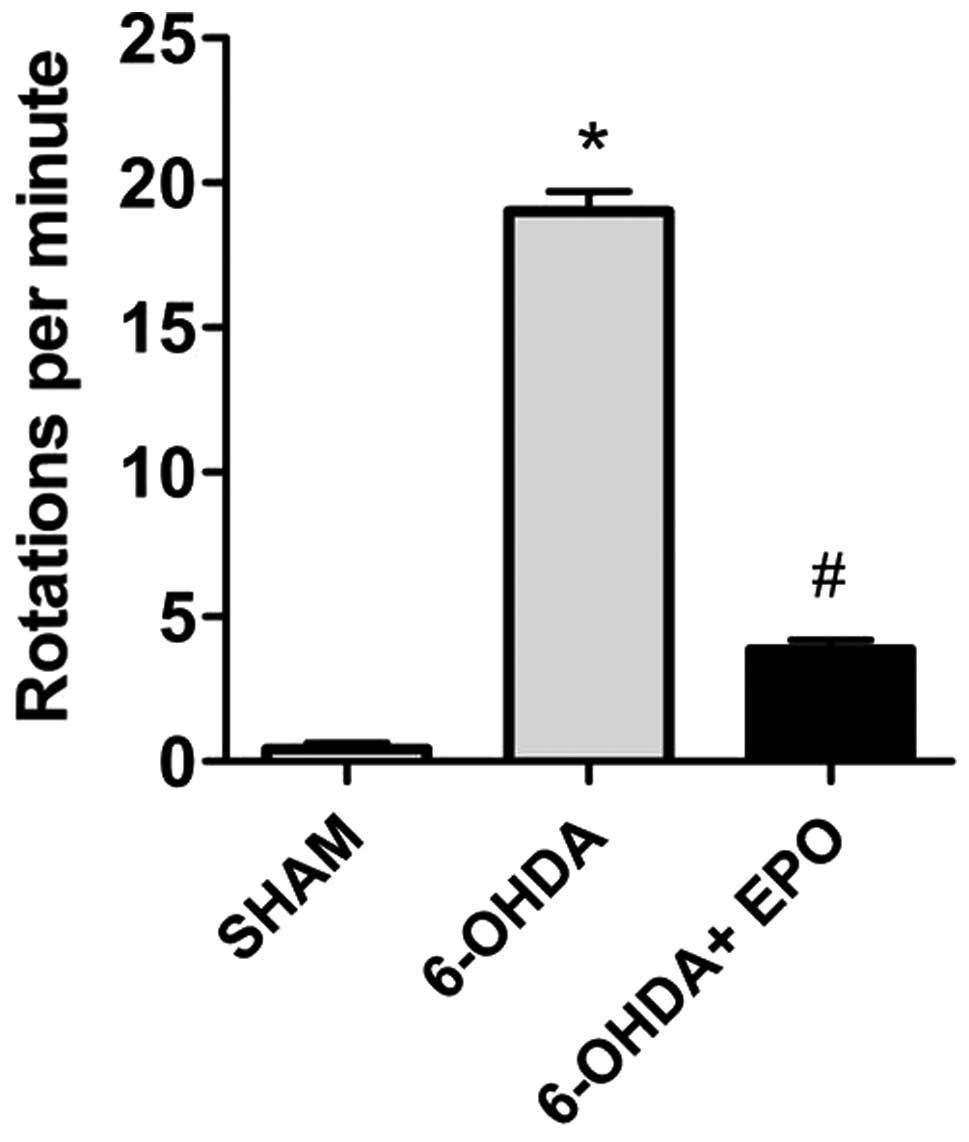
EPO improves behavioral performance
Apomorphine-induced rotations
The effect of pretreatment with EPO on
6-OHDA-induced rotational behavior in response to apomorphine was
assessed. The rats from groups 5–7 were observed at 7 days prior to
the lesion and the mean rotational rate was 0±1, indicating that
there was no variability between animals. The apomorphine-induced
rotation rates are shown in Fig.
4. The data demonstrate that at 21 days after 6-OHDA
administration, rats rotated at mean rates of 19±2 net ipsilateral
turns when administered 6-OHDA alone, indicating a lesion of the
nigrostriatal pathway. Control (sham-lesioned) rats did not rotate.
Intraperitoneal injection of 10,000 U/kg EPO prior to lesion
induction was significantly reduced in rotation rates (4±1 turns,
respectively; P<0.01; Fig
4).
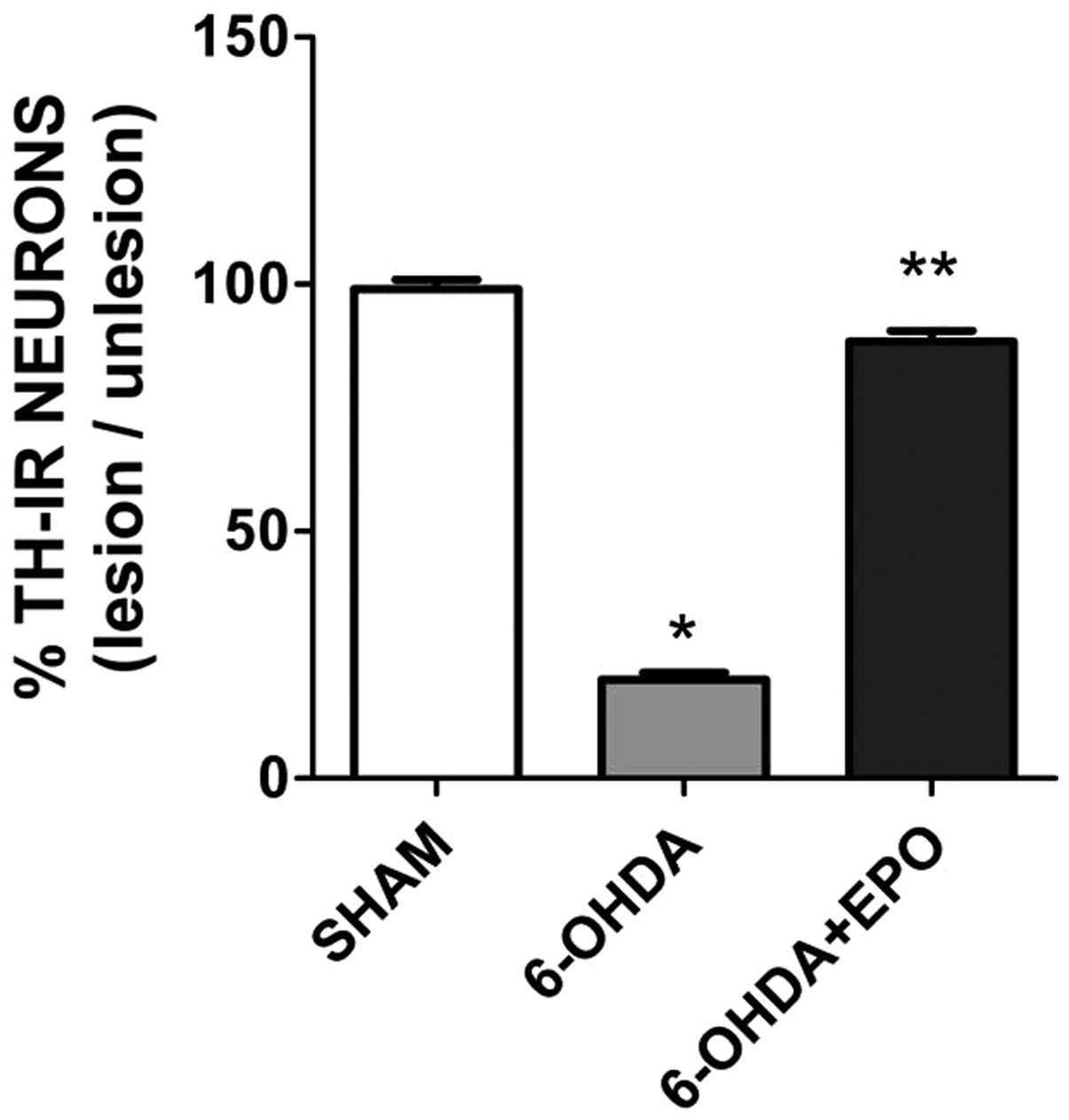
EPO decreases dopaminergic neuron loss in
the SN
TH-positive in the SN
In control rats receiving saline, the dopaminergic
neurons in the SN were intensely immunoreactive to TH (Fig. 5A and B). The number of TH-positive
neurons in the injected SN of control rats did not differ from the
intact SN of control rats (Fig. 5A
and B) or the intact SN of 6-OHDA-lesioned rats and EPO-treated
rats (Fig. 5C and E). However, in
the lesioned hemisphere of 6-OHDA-lesioned rats, the number of
TH-positive neurons in the SN was significantly reduced (Fig. 5D). Numerous TH-positive neurons
were found in the ipsilateral SN in the EPO 10,000 U/kg group
(Fig. 5F).
TH-positive neuron counts in the
SN
The 6-OHDA lesion induced a significant loss of
TH-positive neurons in the ipsilateral SN at 21 days (P<0.01)
post-lesion when compared with rats that underwent sham surgery
(Fig. 6). There were a number of
TH-positive neurons in the lesioned SN in the EPO group 21 days
after the 6-OHDA lesion (Fig. 6).
In the EPO group, the number of TH-positive neurons in the lesioned
SN was significantly increased compared to 6-OHDA (one-factor
ANOVA, P<0.01). No difference was found for the TH-positive
neurons in the unlesioned SN for each group.
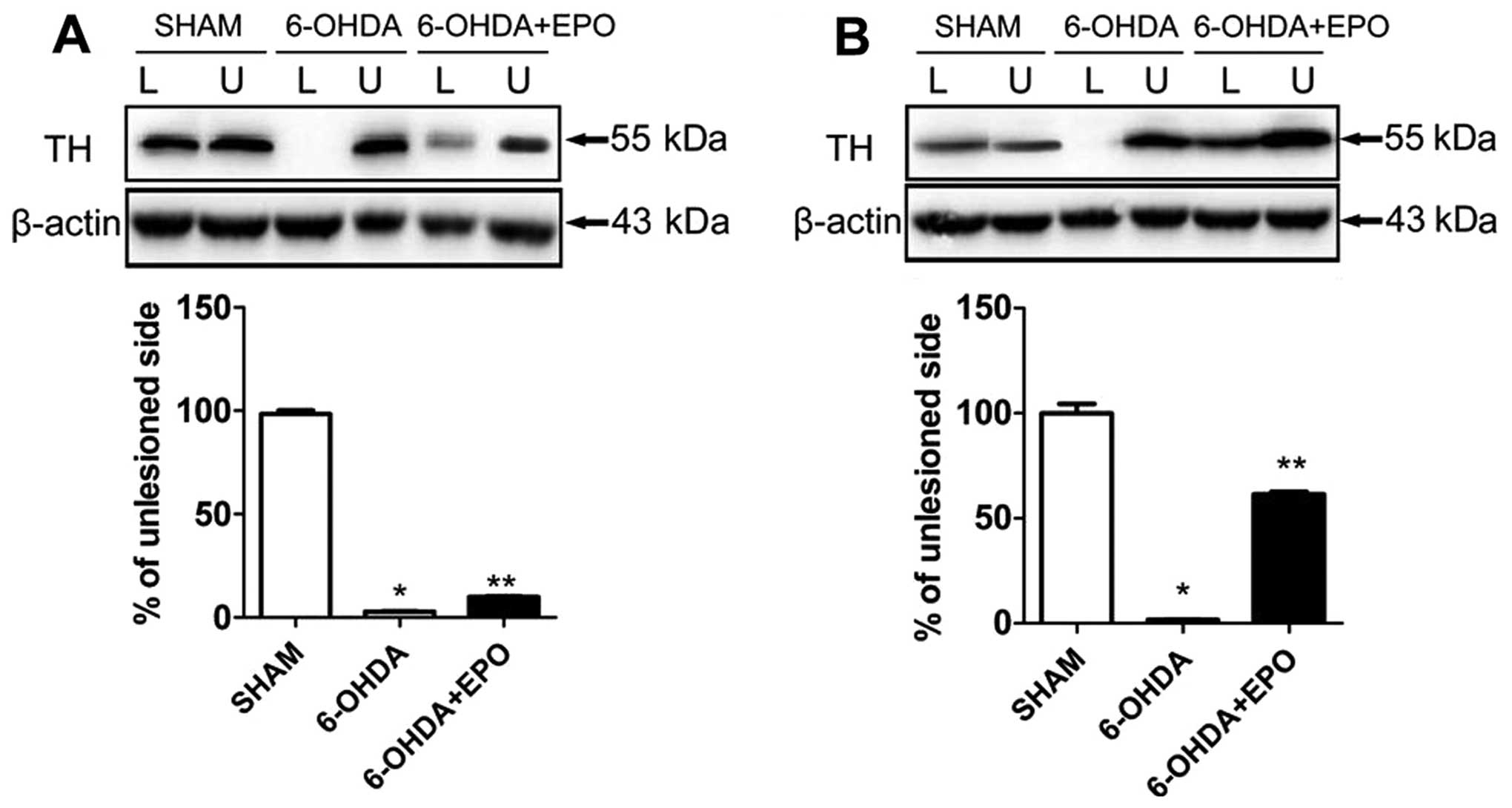
EPO treatment increased TH levels in the
SN and striatum
For each rat, the lesioned and unlesioned striatum
was studied concomitantly on the same gel, and the results were
expressed as a ratio of the lesioned to the unlesioned side. TH
proteins in SN, extracted as described in the experimental
procedure, were investigated. In 6-OHDA-lesioned rats, the
abundance of TH was reduced to 2.4±0.4% in the lesioned SN
(P<0.01, compared with sham rats) relative to the unlesioned SN.
EPO treatment increased TH levels in the SN, and the abundance of
TH was slightly increased to 9.9±0.9% (P<0.01, compared with
6-OHDA-lesioned rats) in the lesioned SN (Fig. 7A). However, an analysis of TH
present in striatum revealed marked alterations. In the EPO
treatment of lesioned rats, the abundance of TH was significantly
increased to 61.5±2.7% (P<0.01, compared with 6-OHDA-lesioned
rats) in the lesioned striatum (Fig.
7B).
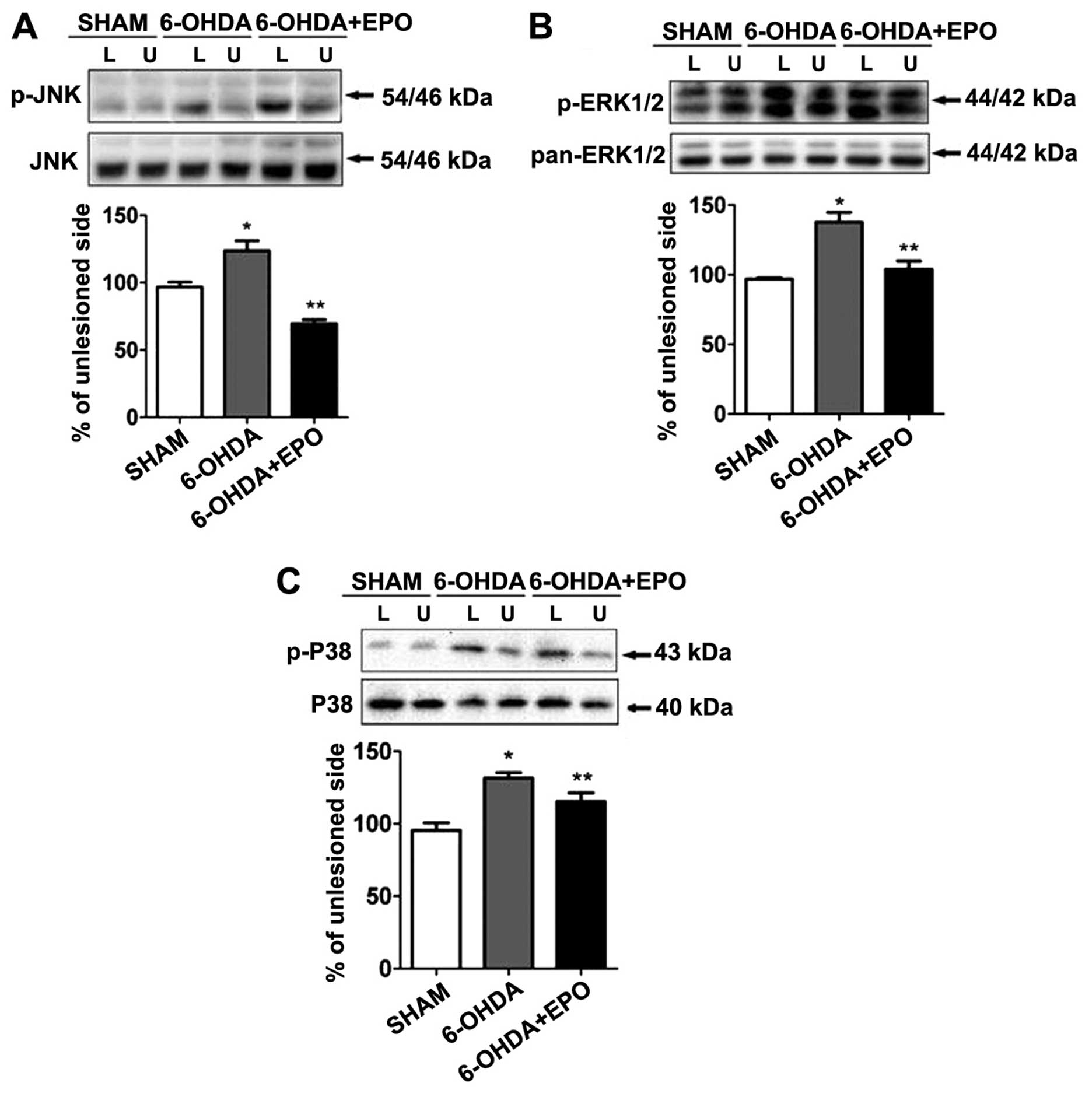
EPO suppresses 6-OHDA-induced activation
of MAPK pathways
MAPKs are a specific class of serine/threonine
kinases that respond to extracellular signals such as growth
factors, mitogens and cellular stress, and mediate proliferation,
differentiation, and cell survival in mammalian cells. Four
distinct groups of MAPKs exist within mammalian cells: the ERKs,
the JNKs, the atypical MAPKs (ERK3, ERK5 and ERK8), and the p38
MAPKs. Since activated MAPK family members, including ERK, JNK,
p38, potentially play a role in inflammation and apoptosis, we
examined the phosphorylation of MAPKs by western blot analysis
using anti-phosphorylated antibodies. As shown in Fig. 8, treatment with 6-OHDA resulted in
the robust phosphorylation of JNK, ERK1/2 and p38. Quantification
revealed that the levels of phosphorylation were significantly
increased for JNK, ERK1/2 and p38 when compared with the sham
(P<0.01) (Fig. 8A–C). By
contrast, the total amount of JNK, ERK and p38 was not altered
during 6-OHDA treatment. We also examined the role of EPO on
6-OHDA-mediated MAPK phosphorylation. Although 6-OHDA treatment
elevated levels of MAPK phosphorylation in rats, EPO treatment
significantly suppressed 6-OHDA-induced MAPK phosphorylation when
compared with 6-OHDA-treated rats (P<0.01) (Fig. 8A–C).
EPO suppresses 6-OHDA-induced activation
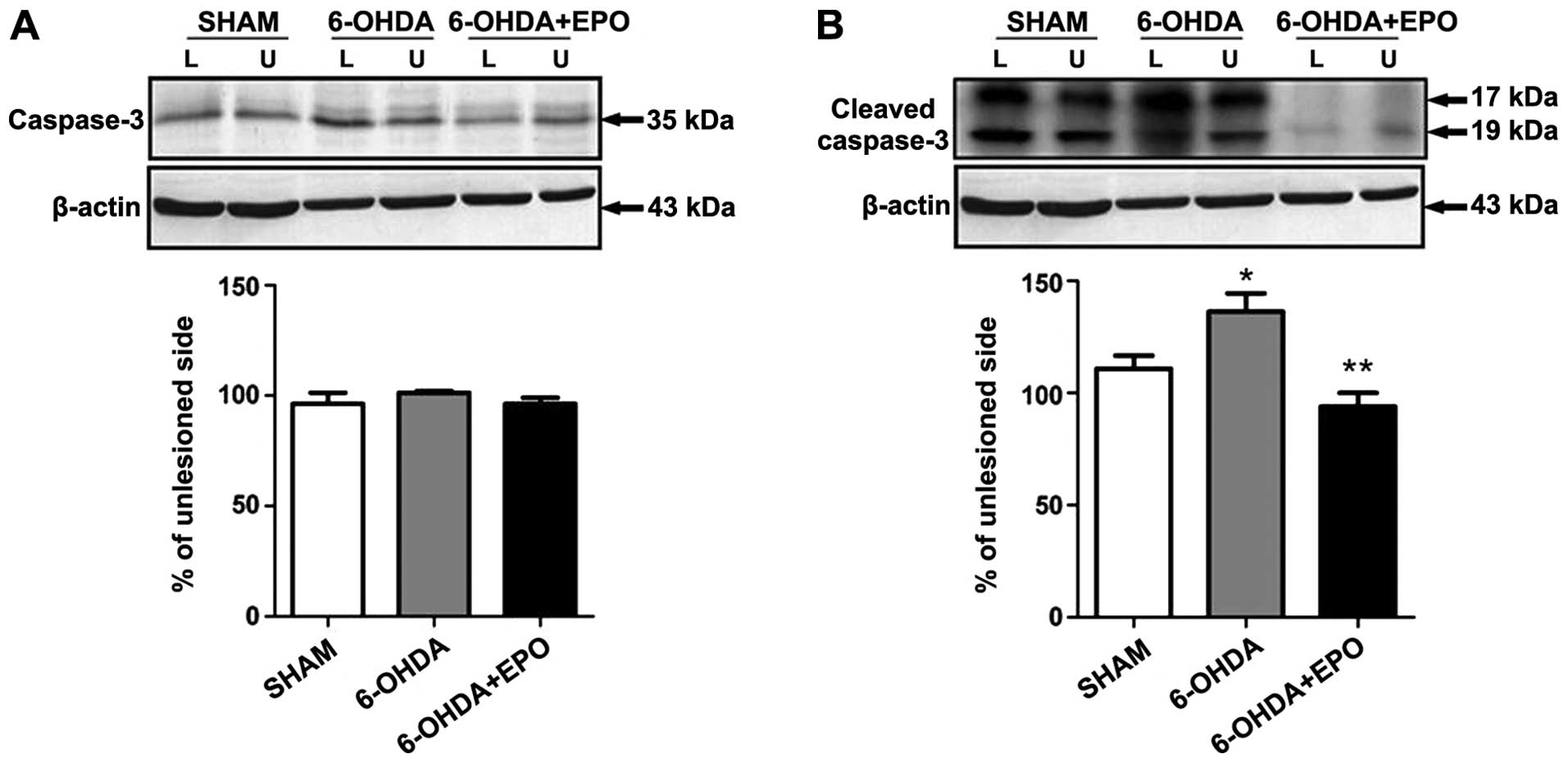
of caspase-3
Western blot analysis revealed that the activated
cleavage product of caspase-3 increased in 6-OHDA treatment as
compared to the control. However, pretreatment with EPO
significantly suppressed caspase-3 activation (Fig. 9). The effect of EPO on
6-OHDA-induced apoptosis may be, at least in part, mediated by
regulating the caspase-3 activation.
Discussion
Results of the present study have demonstrated that
a dose-dependent increase of EPO concentration in CSF as systemic
administration of different doses of EPO for rat via i.p.
injection, and systemic administration of high-dose EPO exerted
neuroprotective effects on a PD model of rats behaviorally and
immunohistochemically. A dose-dependent increase of EPO
concentration in CSF when different doses of EPO were administerd
was found. Systemic administration of EPO prior to injection of
6-OHDA decreased the degeneration of TH-positive neurons in the
lesioned SN. An increase of the TH-positive neurons by EPO leads to
the improvement of apomorphine-induced rotational behavior.
Moreover, EPO reduces the activation of MAPK pathways in the SN and
striatum, indicating that an anti-apoptotic effect may be a
mechanism of EPO neuroprotection.
The earliest description of EPO in the CNS was that
of Tan et al (19) and
Marti et al (20) who
demonstrated EPO gene expression in human, monkey and murine brain.
In an immunohistochemical study, Juul et al (21) found that EPOR and EPO were
detected broadly in the neuron in postnatal brains. In addition to
neurons, EPO-R is expressed in astrocytes and microglia (22). EPO-R brain expression has been
observed during development and adulthood in non-human primates and
humans (23). Direct binding of
I125-labeled EPO localized EPO binding sites to specific adult
brain regions including the hippocampus, cortex and midbrain in
mouse. EPO-R expression in adult brain was detected in human in
analogous regions and in monkey (24).
The neuroprotective effects of EPO on 6-OHDA-treated
dopaminergic neurons were previously examined (12,25,26). No systemic administration of EPO
was found in those studies. By contrast, Zhang et al
(27) used intrastriatal
injection of EPO. EPO (80 IU/kg) was administered at 30 min prior
to the 6-OHDA lesion. In the present study, 80 IU/kg of EPO was
also intrastriatally injected prior to the 6-OHDA lesion. In a
previous study, 100 IU/day of EPO was daily administered
intraventricularly for 7 days following 6-OHDA lesion (28). In those studies, strong
neuroprotective effects of EPO were demonstrated.
The molecular weight of EPO (30.4 kDa) is greater
than the molecular weight threshold for lipid-mediated transport
across the BBB. Whether EPO is able to cross the BBB is of
significance due to implications for its use as a neuroprotective
agent. Findings of previous studies have shown that, similar to
other large molecule drugs, EPO does not cross the BBB in the
absence of BBB disruption (29).
Intravenous administration of biotinylated-EPO provided evidence
for biotin localization around capillaries in murine brain 5 h
after treatment that was eliminated by an increase of unlabeled
exogenous EPO, suggesting a specific receptor-mediated transport of
EPO into the brain. A pharmacokinetic study directly measured EPO
in homogenized brain tissue obtained from neonatal rats and
confirmed that systemic EPO is only detected in brain after
injection of EPO (5,000 U/kg) and systemic EPO crossed the BBB in a
dose-dependent manner, peaking in brain at 10 h (30). The EPO concentrations in the CSF
increased after a period of slow equilibration. An increase in the
CSF concentration was observed as early as 3 h after intravenous
administration. Peak levels were reached between 9 and 24 h
(31). In the present study, we
found a dose-dependent increase of EPO concentration in CSF. Three
hours after EPO injection, EPO was detected in the CSF of rats.
Western blot analysis revealed a significant increase in EPO levels
in the CSF of the 10,000 U/kg group compared with those of the
2,500 and 5,000 U/kg groups. The dose was determined by a previous
study, and 10,000 U/kg of EPO was administered i.p. on a daily
basis for 7 days prior to the 6-OHDA lesion. TH-positive fibers
were dominantly preserved in the SNc, suggesting that EPO prevented
the neurodegeneration of dopaminergic neurons induced by the 6-OHDA
lesion.
As previously described, we found that the EPO
receptor is co-localized with all TH-positive neurons in the rat
SN. It has been demonstrated that EPO can be taken up by neurons.
EPO elicits effects on nigral TH expression of doparminergic
neurons following 6-OHDA lesion and may prevent neuronal cell
death. The mechanism of action of EPO on the nigral-striatal tract
may be associated with the property of EPO to reduce neuronal
apoptosis (32). In addition, EPO
has an antioxidant effect in brain (33), which may ameliorate the oxidative
stress in the nigra-striatal tract that is caused by the injection
of the 6-OHDA neurotoxin (34).
We found the abundance of TH was significantly increased in the
lesioned striatum. TH is an enzyme that is responsible for a
critical step in the synthesis of dopamine, which occurs in a wide
variety of different tissues serving different functions. A
possible hypothesis of the TH expression in striatal neurons is
that it could rapidly alter in dopamine content. The appearance of
TH-positive neurones in the striatum produced by EPO may be a
consequence of the increase in striatal dopamine levels. The
behavioral performance of rats was also improved.
EPO has trophic effects on dopaminergic neurons.
In vitro evidence has established that EPO promotes the
growth, differentiation, and function of cultured dopaminergic
cells (35). EPO also stimulates
striatal dopamine release (36).
A fundamental mechanism of EPO-induced neuroprotection in cultured
neurons is its ability to inhibit apoptosis, reducing both DNA
damage and cell membrane asymmetry (37). EPO acts by binding to the EPO-R
homodimer. EPO binding activates the phosphorylation activity of
JAK2 and phosphorylation of JAK2, which results in signal
transduction involving STAT5, PI3 kinase, MAPK and other signaling
molecules (38). In a model
study, EPOR signaling resulted in strong activation of STAT5 and
PI3 kinase/AKT, which were required for neuroprotection, as well as
MAPK/ERK1/2 (39).
MAPK pathways play a key role in cell death and
survival. MAPKs are a specific class of serine/threonine kinases
that respond to extracellular signals such as growth factors,
mitogens, and cellular stress and mediate proliferation,
differentiation, and cell survival in mammalian cells. We observed
that 6-OHDA treatment increased the phosphorylation of all three
MAPK members including ERK1/2, JNK, and p38 in a PD rat model as
previously described (40). ERK
signaling is generally considered a pro-survival pathway (41). However, activation of ERK also
contributes to cell death (42).
JNK is a major signaling pathway that is activated by oxidative
stress and is considered an essential molecule in neuronal cell
death. Increased JNK activity has also been reported in the 6-OHDA
model (43). JNK-deficient rats
exhibited resistance to 6-OHDA- or MPTP-induced injury (44). p38 is known to contribute to the
inflammation process as it has been observed in vivo
(45). Results of that study have
also shown that EPO decreased 6-OHDA toxicity by reducing the
activation of MAPK pathways thereby protecting the neuron. Many
signaling pathways convey apoptotic stimuli in cells. Stress
stimulation activates MAPK and various intracellular target
proteins, leading to apoptosis. Caspase-3 activation leads to DNA
breakage, nuclear chromatin condensation and cell apoptosis. In
this study, we found that EPO prevented caspase-3 activation
induced by 6-OHDA. Findings of previous studies have demonstrated
that EPO can cross the BBB (46)
and the systemic administration of EPO improved neurological
function of a rat model of stroke. By contrast, systemic injection
of EPO at a dose of 5,000 U/kg (body weight) did not protect
dopaminergic neurons from 6-OHDA toxicity. Although in this study a
dose of 5,000 U/kg was examined a dose-dependent study to determine
which dose was effective in the brain of a parkinsonian rat was not
conducted (12). Therefore, it
cannot be determined whether a higher dose of EPO could protect
dopaminergic neurons from 6-O HDA toxicity. In the present study,
we performed a dose-dependent study and found that i.p. injection
of EPO at a dose of 10,000 U/kg decreased dopaminergic neuron loss
from 6-OHDA toxicity in parkinsonian rat. Although the precise
mechanisms responsible for EPO need to be determined, the present
results suggest that EPO may be neuroprotective via the anti-MAPK
pathway.
In conclusion, the systemic high dose of EPO exerted
neuroprotective effects by reversing behavioral deficits associated
with PD and prevented loss of the dopaminergic neurons through the
MAPK pathway. As EPO is used in the clinic, recent pharmacological
developments have enabled us to take advantage of EPO without
hematopoietic side-effects. Consequently, EPO serves as a potential
therapeutic candidate for PD patients.
Acknowledgements
This study was supported in part by the Projects of
National Science Foundation of China (nos. 81171203, 81171204,
81200871 and 81373366), and Projects of the Shanghai Committee of
Science and Technology, China (nos. 11nm0503300 and
12XD1403800).
References
|
1
|
Samii A, Nutt JG and Ransom BR:
Parkinson’s disease. Lancet. 363:1783–1793. 2004.
|
|
2
|
Chaudhuri KR, Healy DG and Schapira AH:
Non-motor symptoms of Parkinson’s disease: diagnosis and
management. Lancet Neurol. 5:235–245. 2006.
|
|
3
|
Fahn S, Oakes D, Shoulson I, et al:
Levodopa and the progression of Parkinson’s disease. N Engl J Med.
351:2498–2508. 2004.
|
|
4
|
Olanow CW and Jankovic J: Neuroprotective
therapy in Parkinson’s disease and motor complications: a search
for a pathogenesis-targeted, disease-modifying strategy. Mov
Disord. 20(Suppl 11): 3–10. 2005.
|
|
5
|
Signore AP, Weng Z, Hastings T, et al:
Erythropoietin protects against 6-hydroxydopamine-induced
dopaminergic cell death. J Neurochem. 96:428–443. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Csete M, Rodriguez L, Wilcox M and
Chadalavada S: Erythropoietin receptor is expressed on adult rat
dopaminergic neurons and erythropoietin is neurotrophic in cultured
dopaminergic neuroblasts. Neurosci Lett. 359:124–126. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brines ML, Ghezzi P, Keenan S, et al:
Cerami, Erythropoietin crosses the blood-brain barrier to protect
against experimental brain injury. Proc Natl Acad Sci USA.
97:10526–10531. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wei L, Han BH, Li Y, Keogh CL, Holtzman DM
and Yu SP: Cell death mechanism and protective effect of
erythropoietin after focal ischemia in the whisker-barrel cortex of
neonatal rats. J Pharmacol Exp Ther. 317:109–116. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lu D, Mahmood A, Qu C, Goussev A,
Schallert T and Chopp M: Erythropoietin enhances neurogenesis and
restores spatial memory in rats after traumatic brain injury. J
Neurotrauma. 22:1011–1017. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Celik M, Gökmen N, Erbayraktar S, et al:
Erythropoietin prevents motor neuron apoptosis and neurologic
disability in experimental spinal cord ischemic injury. Proc Natl
Acad Sci USA. 99:2258–2263. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang LJ, Xue YQ, Yang C, Yang WH, Chen L,
Zhang QJ, Qu TY, Huang S, Zhao LR, Wang XM and Duan WM: Human
albumin prevents 6-hydroxydopamine-induced loss of tyrosine
hydroxylase in in vitro and in vivo. PLoS One. 7:e412262012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xue YQ, Zhao LR, Guo WP and Duan WM:
Intrastriatal administration of erythropoietin protects
dopaminergic neurons and improves neurobehavioral outcome in a rat
model of Parkinson’s disease. Neuroscience. 146:1245–1258.
2007.PubMed/NCBI
|
|
13
|
Paxinos G and Watson C: The Rat Brain in
Stereotaxic Coordinates. 6th edition. Amsterdam: Academic
Press/Elsevier; pp. 2212007
|
|
14
|
Woodlee MT, Asseo-García AM, Zhao X, Liu
SJ, Jones TA and Schallert T: Testing forelimb placing ‘across the
midline’ reveals distinct, lesion-dependent patterns of recovery in
rats. Exp Neurol. 191:310–317. 2005.
|
|
15
|
Ziegler M and Szechtman H: Differences in
the behavioral profile of circling under amphetamine and
apomorphine in rats with unilateral lesions of the substantia
nigra. Behav Neurosci. 102:276–288. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pegg CC, He C, Stroink AR, Kattner KA and
Wang CX: Technique for collection of cerebrospinal fluid from the
cisterna magna in rat. J Neurosci Methods. 187:8–12. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Martin AB, Fernandez-Espejo E, Ferrer B,
et al: Expression and function of CB1receptor in the rat
striatum: localization and effects on D1and
D2dopamine receptor-mediated motor behaviors.
Neuropsychopharmacology. 33:1667–1679. 2008.PubMed/NCBI
|
|
18
|
Granado N, Lastres-Becker I, Ares-Santos
S, et al: Nrf2 deficiency potentiates methamphetamine-induced
dopaminergic axonal damage and gliosis in the striatum. Glia.
59:1850–1863. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tan CC, Eckardt KU, Firth JD and Ratcliffe
PJ: Feedback modulation of renal and hepatic erythropoietin mRNA in
response to graded anemia and hypoxia. Am J Physiol. 263:F474–F481.
1992.PubMed/NCBI
|
|
20
|
Marti HH, Wenger RH, Rivas LA, et al:
Erythropoietin gene expression in human, monkey and murine brain.
Eur J Neurosci. 8:666–676. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Juul SE, Yachnis AT, Rojiani AM and
Christensen RD: Immunohistochemical localization of erythropoietin
and its receptor in the developing human brain. Pediatr Dev Pathol.
2:148–158. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nagai A, Nakagawa E, Choi HB, Hatori K,
Kobayashi S and Kim SU: Erythropoietin and erythropoietin receptors
in human CNS neurons, astrocytes, microglia, and oligodendrocytes
grown in culture. J Neuropathol Exp Neurol. 60:386–392.
2001.PubMed/NCBI
|
|
23
|
Knabe W, Knerlich F, Washausen S, et al:
Expression patterns of erythropoietin and its receptor in the
developing midbrain. Anat Embryol (Berl). 207:503–512. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Digicaylioglu M, Bichet S, Marti HH, et
al: Localization of specific erythropoietin binding sites in
defined areas of the mouse brain. Proc Natl Acad Sci USA.
92:3717–3720. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xue YQ, Ma BF, Zhao LR, et al:
AAV9-mediated erythropoietin gene delivery into the brain protects
nigral dopaminergic neurons in a rat model of Parkinson’s disease.
Gene Ther. 17:83–94. 2010.PubMed/NCBI
|
|
26
|
McLeod M, Hong M, Mukhida K, Sadi D,
Ulalia R and Mendez I: Erythropoietin and GDNF enhance ventral
mesencephalic fiber outgrowth and capillary proliferation following
neural transplantation in a rodent model of Parkinson’s disease.
Eur J Neurosci. 24:361–370. 2006.
|
|
27
|
Zhang F, Signore AP, Zhou Z, Wang S, Cao G
and Chen J: Erythropoietin protects CA1 neurons against global
cerebral ischemia in rat: potential signaling mechanisms. J
Neurosci Res. 83:1241–1251. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kadota T, Shingo T, Yasuhara T, et al:
Continuous intraventricular infusion of erythropoietin exerts
neuroprotective/rescue effects upon Parkinson’s disease model of
rats with enhanced neurogenesis. Brain Res. 1254:120–127.
2009.PubMed/NCBI
|
|
29
|
Lieutaud T, Andrews PJ, Rhodes JK and
Williamson R: Characterization of the pharmacokinetics of human
recombinant erythropoietin in blood and brain when administered
immediately after lateral fluid percussion brain injury and its
pharmacodynamic effects on IL-1beta and MIP-2 in rats. J
Neurotrauma. 25:1179–1185. 2008. View Article : Google Scholar
|
|
30
|
Statler PA, McPherson RJ, Bauer LA,
Kellert BA and Juul SE: Pharmacokinetics of high-dose recombinant
erythropoietin in plasma and brain of neonatal rats. Pediatr Res.
61:671–675. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xenocostas A, Cheung WK, Farrell F, et al:
The pharmacokinetics of erythropoietin in the cerebrospinal fluid
after intravenous administration of recombinant human
erythropoietin. Eur J Clin Pharmacol. 61:189–195. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yamada M, Burke C, Colditz P, Johnson DW
and Gobe GC: Erythropoietin protects against apoptosis and
increases expression of non-neuronal cell markers in the
hypoxia-injured developing brain. J Pathol. 224:101–109. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sifringer M, Brait D, Weichelt U, et al:
Erythropoietin attenuates hyperoxia-induced oxidative stress in the
developing rat brain. Brain Behav Immun. 24:792–799. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Aluf Y, Vaya J, Khatib S, Loboda Y,
Kizhner S and Finberg JP: Specific oxidative stress profile
associated with partial striatal dopaminergic depletion by
6-hydroxydopamine as assessed by a novel multifunctional marker
molecule. Free Radic Res. 44:635–644. 2010. View Article : Google Scholar
|
|
35
|
Lee JY, Koh HC, Chang MY, Park CH, Lee YS
and Lee SH: Erythropoietin and bone morphogenetic protein 7 mediate
ascorbate-induced dopaminergic differentiation from embryonic
mesencephalic precursors. Neuroreport. 14:1401–1404. 2003.
|
|
36
|
Yamamoto M, Koshimura K, Kawaguchi M,
Sohmiya M, Murakami Y and Kato Y: Stimulating effect of
erythropoietin on the release of dopamine and acetylcholine from
the rat brain slice. Neurosci Lett. 292:131–133. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Leist M, Ghezzi P, Grasso G, et al:
Derivatives of erythropoietin that are tissue protective but not
erythropoietic. Science. 305:239–242. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhao W, Kitidis C, Fleming MD, Lodish HF
and Ghaffari S: Erythropoietin stimulates phosphorylation and
activation of GATA-1 via the PI3-kinase/AKT signaling pathway.
Blood. 107:907–915. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Um M and Lodish HF: Antiapoptotic effects
of erythropoietin in differentiated neuroblastoma SH-SY5Y cells
require activation of both the STAT5 and AKT signaling pathways. J
Biol Chem. 281:5648–5656. 2006. View Article : Google Scholar
|
|
40
|
Rodriguez-Blanco J, Martin V, Herrera F,
Garcia-Santos G, Antolin I and Rodriguez C: Intracellular signaling
pathways involved in post-mitotic dopaminergic PC12 cell death
induced by 6-hydroxydopamine. J Neurochem. 107:127–140. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Baines CP, Zhang J, Wang GW, et al:
Mitochondrial PKCepsilon and MAPK form signaling modules in the
murine heart: enhanced mitochondrial PKCepsilon-MAPK interactions
and differential MAPK activation in PKCepsilon-induced
cardioprotection. Circ Res. 90:390–397. 2002. View Article : Google Scholar
|
|
42
|
Zhuang S and Schnellmann RG: A
death-promoting role for extracellular signal-regulated kinase. J
Pharmacol Exp Ther. 319:991–997. 2006. View Article : Google Scholar
|
|
43
|
Pan J, Zhao YX, Wang ZQ, Jin L, Sun ZK and
Chen SD: Expression of FasL and its interaction with Fas are
mediated by c-Jun N-terminal kinase (JNK) pathway in 6-OHDA-induced
rat model of Parkinson disease. Neurosci Lett. 428:82–87. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wu SS and Frucht SJ: Treatment of
Parkinson’s disease: what’s on the horizon? CNS Drugs. 19:723–743.
2005.
|
|
45
|
Fyhrquist N, Matikainen S and Lauerma A:
MK2 signaling: lessons on tissue specificity in modulation of
inflammation. J Invest Dermatol. 130:342–344. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lapchak PA: Carbamylated erythropoietin to
treat neuronal injury: new development strategies. Expert Opin
Investig Drugs. 17:1175–1186. 2008. View Article : Google Scholar : PubMed/NCBI
|