Introduction
The phosphatase and tensin homolog deleted on
chromosome 10 (PTEN; MMAC1) tumor suppressor was identified as a
novel gene product commonly deleted in various types of cancer
(1–3). PTEN is both a dual-specificity
protein and lipid phosphatase (3,4),
with PTEN suppressing tumorigenesis in its latter form. Mutations
in the PTEN gene have been associated with various types of
cancer (1,2,5–7),
and are the causative factor in Cowden syndrome (8,9).
The homozygous deletion of the PTEN gene is embryonically
lethal in mice (10).
PI3-kinase (PI3K) is an important enzyme in signal
transduction. PI3K relays extracellular growth signals, which are
derived from receptor tyrosine kinases, integrins and
G-protein-coupled receptors, into intracellular signals. This is
achieved by phosphorylating the second messenger lipid,
phosphatidylinositol-4,5-bisphosphate (PIP2) to
phosphatidylinositol-3,4,5-triphosphate (PIP3) (11). PIP3 in turn recruits
AKT to the plasma membrane, resulting in AKT activation. AKT
subsequently regulates a number of downstream processes connected
with cell survival and growth (12). PTEN exhibits lipid phosphatase
activity (3,4), and dephosphorylates PIP3
to PIP2, thereby preventing the recruitment of AKT to
the plasma membrane and downregulating AKT activation (12). PTEN also inhibits focal adhesion
kinase (FAK) (13), a kinase that
is activated by binding to integrins (14) and promotes cell migration,
adhesion, spreading and angiogenesis (13,14). Thus, PTEN impedes various cell
processes, including proliferation, cytoskeleton reorganization and
angiogenesis.
Shank-interacting protein-like 1 (SIPL1; Sharpin) is
a PTEN-negative regulator (PTEN-NR) that reduces PTEN-derived
lipid-phosphatase activity through a direct association with PTEN
(15). This decrease in PTEN
function correlated with increased AKT activation and the promotion
of tumorigenesis in in vivo xenograft models (15). Additionally, SIPL1 expression
correlated with AKT activation and PTEN expression in primary
cervical cancer (15). SIPL1 has
been shown to play a key role in the activation of NF-κB as part of
the linear ubiquitin chain assembly complex (LUBAC) (16–18). As part of the LUBAC, SIPL1
promotes the formation of linear ubiquitin chains on NEMO, leading
to the subsequent activation of NF-κB and the promotion of cell
survival. Additionally, SIPL1 is involved in the inside-out
activation of β1-integrin through direct binding to the α-integrin
subunit (19). Loss of SIPL1
expression is also a causative factor of the chronic proliferative
dermatitis (CPDM) phenotype in mice as a result of abnormal
activation of NF-κB (20–22).
In the present study, we examined the role of SIPL1
in the reduction of PTEN function in CHO-K1 cells with regard to
cell proliferation, adhesion and migration. Ectopic SIPL1 decreased
the PTEN protein, increased cell proliferation, and enhanced cell
attachment and migration. Collectively, our study supports the
hypothesis that SIPL1 plays an important role in inhibiting PTEN
function.
Materials and methods
Cell lines, plasmids and inhibitors
Human embryonic kidney 293T cells (HEK 293T) and
Chinese hamster ovary K1 (CHO-K1; CHO) cells were purchased from
the American Type Culture Collection (ATCC; Manassas, VA, USA), and
cultured in Dulbecco’s modified Eagle’s medium (DMEM) (HEK 293T) or
F-12 media (CHO-K1) supplemented with 10% fetal bovine serum (FBS;
Sigma Aldrich, Oakville, ON, Canada) and 1% penicillin-streptomycin
(Life Technologies, Burlington, ON, Canada). The pLHCX and pLHCX
SIPL1 plasmids were constructed as previously described (13). AKT inhibitor VIII and the
proteasomal inhibitor (MG132) were purchased from Calbiochem (EMD,
Mississauga, ON, Canada) and Sigma Aldrich, respectively.
Retroviral overexpression of SIPL1
The overexpression of SIPL1 was carried out using a
Gag-Pol (GP) and an envelope expressing vector (VSV-G) (Stratagene,
Mississauga, ON, Canada). Briefly, the GP and VSV-G vectors were
transiently co-transfected with pLHCX or pLHCX SIPL1 into HEK 293T
cells using a calcium-phosphate procedure. The virus-containing
medium was harvested 48 h later, filtered through a 0.45 μM filter,
and centrifuged at 20,000 × g for 120 min to concentrate the
retrovirus. Following treatment with the virus, CHO-K1 cells were
selected for stable integration with hygromycin (0.5 mg/ml; Sigma
Aldrich).
Western blot analysis
Cell lysates were prepared in a buffer containing 20
mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton
X-100, 25 mM sodium pyrophosphate, 1 mM NaF, 1 mM
β-glycerophosphate, 0.1 mM sodium orthovanadate, 1 mM PMSF, 2 μg/ml
leupeptin and 10 μg/ml aprotinin (Sigma Aldrich). Cell lysate (50
μg) was separated on SDS-PAGE gels and transferred onto Amersham
Hybond ECL nitrocellulose membranes (Amersham, Baie d’Urfe, QC,
Canada). The membranes were blocked with 5% skim milk and incubated
with the indicated antibodies at 4°C overnight. Appropriate
HRP-conjugated secondary antibodies were incubated for 1 h at room
temperature. Signals were detected using an ECL Western Blotting
kit (Amersham). The primary and secondary antibodies used were:
anti-Sharpin (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA,
USA); anti-Sharpin (1:1,000; Abcam, Toronto, ON, Canada); anti-AKT
(1:1,000; Santa Cruz Biotechnology); anti-AKT Ser473
phosphorylation (1:1,000; Cell Signaling Technology, Danvers, MA,
USA); anti-GAPDH (1:5,000; Cell Signaling Technology), anti-actin
(1:1,000; Santa Cruz Biotechnology), anti-goat (1:3,000; Santa Cruz
Biotechnology), anti-mouse (1:3,000; GE Healthcare; Mississauga,
ON, Canada) and anti-rabbit (1:3,000; GE Healthcare).
Real-time PCR analysis (RT-qPCR)
Total RNA was isolated using TRIzol (Life
Technologies). Reverse transcription was carried out using
SuperScript III (Life Technologies) according to the manufacturer’s
instructions. Briefly, 2 μg of RNA was converted to cDNA at 65°C
for 6 min followed by 1-min incubation on ice, 25°C for 11 min,
50°C for 60 min and 70°C for 15 min. qPCR primers used were: actin,
forward: 5′-ACC GAG CGC GGC TAC AG-3′ and reverse: 5′-CTT AAT GTC
ACG CAC GAT TTC C-3′; PTEN, forward: 5′-TGT GGT CTG CCA GCT AAA
GG-3′ and reverse: 5′-CGG CTG AGG GAA CTC AAA GT-3′ and SIPL1,
forward: 5′-GCT ATT GCA GGT GGA GAC GA-3′ and reverse: 5′-GCC TCC
TGA AGC TGA ACA CT-3′. RT-qPCR was performed using the ABI 7500
Fast Real-Time PCR system in the presence of SYBR-Green according
to the manufacturer’s instructions (Applied Biosystems, Burlington,
ON, Canada). Briefly, each reaction consisted of 1 μl cDNA, 0.25 μl
forward primer (10 μM), 0.25 μl reverse primer (10 μM), 4.75 μl
H2O and 6.25 μl of SYBR-Green Master Mix. The PCR
reaction was carried out in a 96-well plate at 50°C for 2 min, 95°C
for 10 min, followed by 40 cycles at 95°C for 15 sec and 60°C for 1
min. The samples were run in triplicate and repeated three
times.
Serum stimulation
CHO-K1 cells (5×105) were seeded in a
6-well plate and incubated at 37°C overnight. The following day the
cells were washed with PBS and incubated with serum-free F-12 media
at 37°C overnight. The media were removed and replaced with
complete F-12 media for 15, 30, 60, 120 or 180 min, followed by
cell lysate collection and western blotting for AKT activation. It
was determined that the parental CHO-K1 cells achieved maximum AKT
activation at 30 min. CHO-K1 empty vector (EV) pLHCX or SIPL1 cells
were seeded and serum-starved as described above. The media were
then replaced with media containing 0, 2, 4, 6, 8 or 10% FBS and
incubated at 37°C for 30 min followed by cell lysate
collection.
Cell proliferation assay
A total of 500 CHO-K1 EV and SIPL1 cells were seeded
in a 96-well plate and incubated at 37°C for 5 days. Proliferation
was measured using the WST-1 cell proliferation assay kit
(Millipore, Mississauga, ON, Canada) according to the
manufacturer’s instructions. Absorbance readings were measured with
a plate reader (μQuant; BioTek Instruments, Inc., Winooski, VT,
USA) at 420 nm. For the treatment groups, the cells were treated
with AKT inhibitor VIII to a final concentration of 2 μM in the
media or an equal volume of DMSO (Sigma Aldrich).
Wound healing assay
CHO-K1 EV of SIPL1 cells (1×105) were
seeded in 6-well plates and incubated overnight at 37°C. Each well
of the plate was scratched using a sterile pipette tip, both in the
vertical and horizontal directions to generate the wound. The cells
were washed with PBS to remove any dislodged cells and incubated at
37°C overnight. The plates were examined daily to observe the
migration of the cell across the wound using a light microscope
(Axiovert 200; Carl Zeiss, Jena, Germany).
Cell attachment
CHO-K1 EV of SIPL1 cells (2×105) were
seeded in 6-well plates and incubated overnight at 37°C for 30, 60,
120 or 180 min, followed by removal of the unattached cells. The
attached cells were released with trypsin (Life Technologies), and
an aliquot of cells was removed and mixed with Trypan blue (Sigma
Aldrich) to stain non-viable cells. The viable cells were counted
under a light microscope (Axiovert 200; Carl Zeiss). The experiment
was repeated in triplicate.
Immunofluorescence
Immunofluorescence staining was performed by fixing
cells with 4% paraformaldehyde for 20 min and permeabilized with
0.05% Triton X-100 for 15 min. One unit of Rodamine phalloidin was
added to each slide at room temperature for 15 min according to the
manufacturer’s instructions. After washing, the slide was
subsequently covered with VECTASHIELD mounting medium with DAPI
(Vector Laboratories, Burlingam, CA, USA). Images were captured
with a fluorescence microscope (Axiovert 200; Carl Zeiss).
Statistical analysis
Data are presented as means ± standard error.
Statistical analysis was performed using a Studen’t t-test or a
two-way ANOVA. P<0.05 was considered to indicate statistical
significance.
Results
SIPL1 expression facilitates AKT
activation
The human SIPL1 cDNA was stably expressed into
CHO-K1 cells using a retrovirus (Fig.
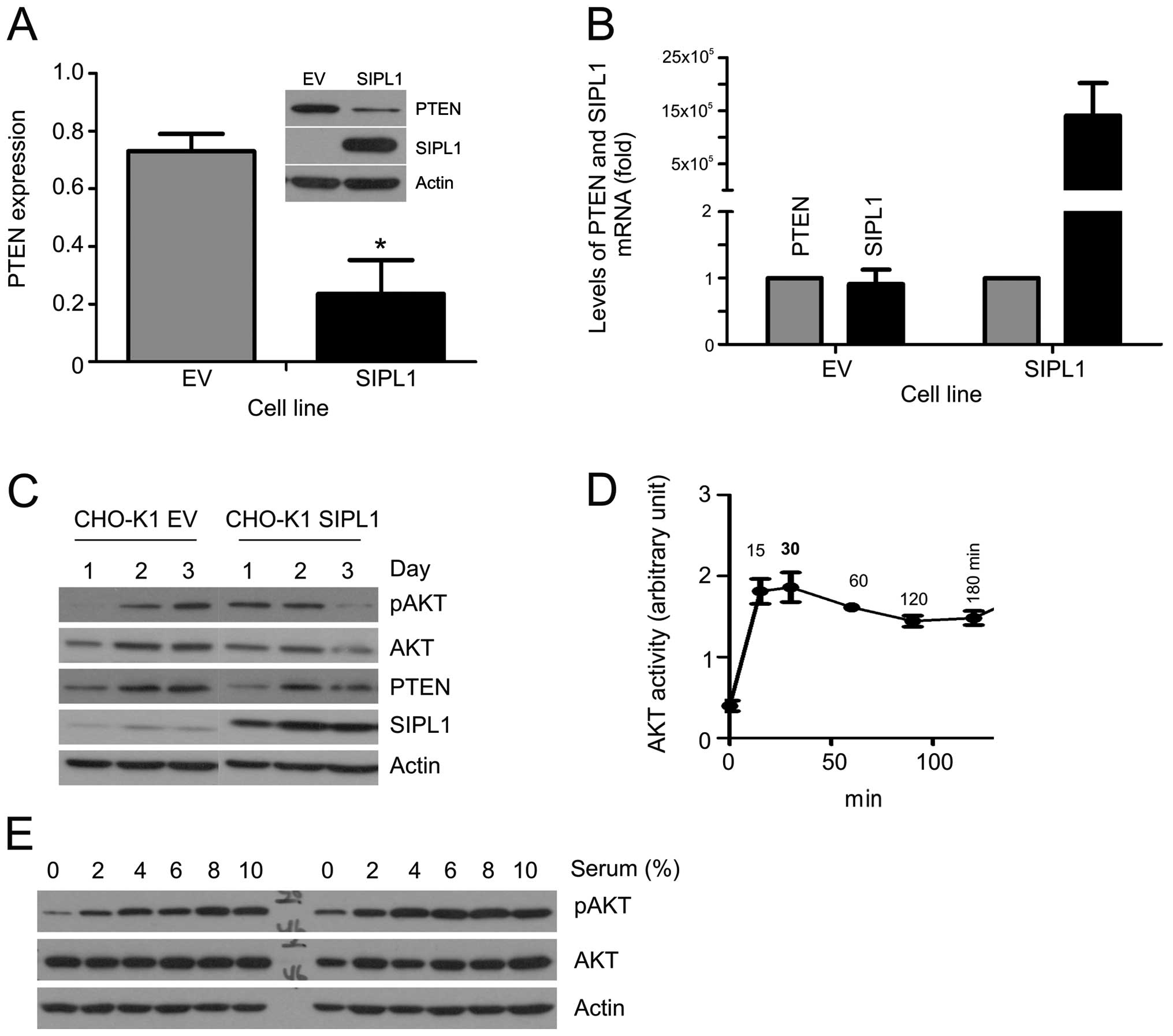
1A). SIPL1 overexpression resulted in a marked decrease of the
PTEN protein (Fig. 1A). To
determine whether the decreased PTEN protein was associated with
reduction in PTEN mRNA abundance, we quantified the PTEN mRNA in
CHO-K1 empty vector (EV) and SIPL1 cells. As expected, the
overexpression of SIPL1 in CHO-K1 SIPL1 cells was readily
demonstrated (Fig. 1B). However,
in comparison to CHO-K1 EV cells, comparable levels of PTEN mRNA
were detected in CHO-K1 SIPL1 cells (Fig. 1B). It, thus, seems unlikely that
SIPL1 reduces the PTEN protein by reducing PTEN mRNA.
PTEN downregulation results in the elevation of AKT
activation. To examine the activation of AKT, we seeded CHO-K1 EV
and SIPL1 cells at comparable densities followed by the examination
of AKT phosphorylation at serine 473 (pAKT Ser473), a
well-established surrogate marker of AKT activation (12). During the course of three days, an
increase in pAKT Ser473 was detected in CHO-K1 SIPL1 cells in
comparison to CHO-K1 EV cells at day 1, at the time when cells were
largely sub-confluent (Fig. 1C),
while the increase was not clear on days 2 and 3, when the cell
density was significantly higher (Fig. 1C). The density-dependent AKT
activation observed in CHO-K1 SIPL1 cells matched with the levels
of PTEN protein in the respective cell densities, where on day 1
PTEN CHO-K1 EV cells were higher than those of PTEN CHO-K1 SIPL1
cells. Of note, the difference was eradicated in the respective day
2 and 3 cells (Fig. 1C).
To examine SIPL1-facilitated AKT activation, CHO-K1
EV and SIPL1 cells were examined for serum-induced AKT activation.
Prior to this task, the kinetics of serum-induced AKT activation
were determined. The addition of a medium containing 10% serum to
serum-starved CHO-K1 cells resulted in peak AKT activation at 30
min (Fig. 1D). With the kinetics
determined, we treated serum-starved CHO-K1 EV and SIPL1 cells with
different doses of serum, and examined the peak-AKT activation. In
comparison to CHO-K1 EV cells, CHO-K1 SIPL1 cells maintained higher
levels of pAKT Ser473 even under serum-free conditions (Fig. 1E). Thus, the above observations
support that SIPL1-mediated downregulation of PTEN in CHO-K1 cells
facilitates AKT activation.
SIPL1 enhances CHO-K1 cell
proliferation
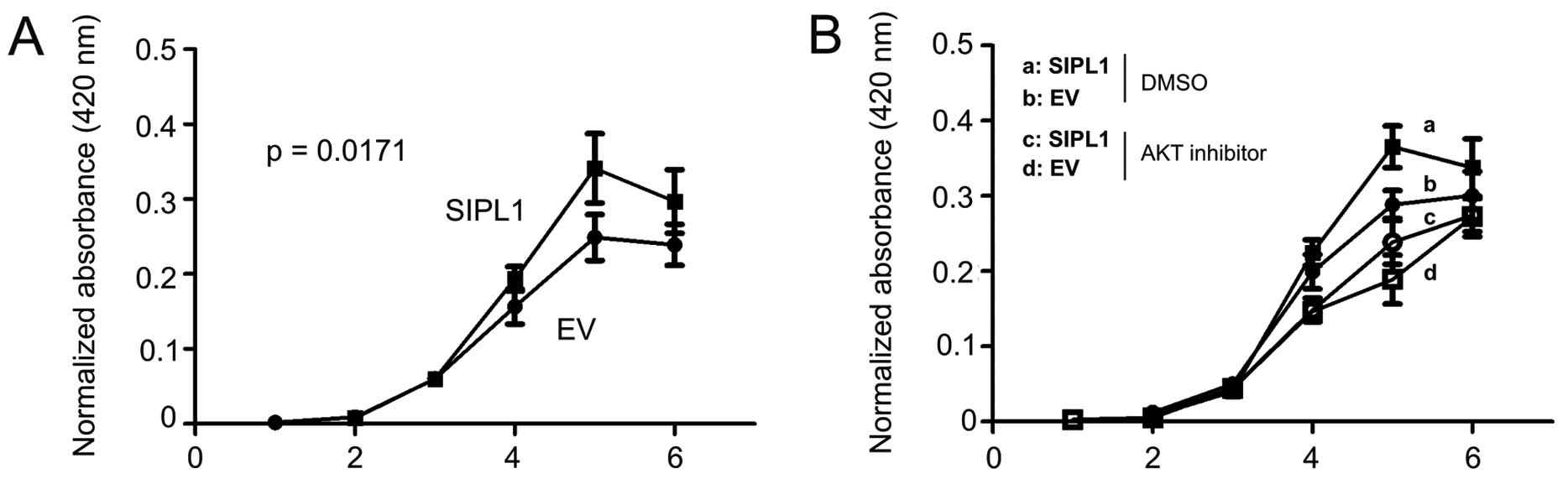
AKT activation promotes cell proliferation (21). To examine whether facilitaion of
AKT activation of SIPL1 would enhance CHO-K1 cell proliferation,
CHO-K1 EV and SIPL1 cells were seeded in 96-well plates and their
growth was monitored over a course of six days using a WST assay.
SIPL1 cells proliferated significantly more rapidly as compared to
EV cells (Fig. 2A). The increase
in CHO-K1 SIPL1 cell proliferation over CHO-K1 EV cells was only
observed within a certain time frame (Fig. 2A), an observation that is in
concordance with SIPL1-facilitating AKT activation in these cells
in a density-dependent manner (Fig.
1C). To consolidate the contributions of AKT activation to the
SIPL1-enhanced proliferation of CHO-K1 cells, CHO-K1 EV and SIPL1
cells were treated with an inhibitor of AKT, AKT inhibitor VIII. We
have previously demonstrated that the AKT inhibitor VIII potently
inhibited AKT activation (24,25). As expected, CHO-K1 SIPL1 cells
proliferated at a faster rate compared to CHO-K1 EV cells in the
vehicle (DMSO) treatment group (Fig.
2B). Inhibition of AKT activation significantly reduced the
proliferation of CHO-K1 EV and SIPL1 cells (Fig. 2B). Of note, there was no
significant difference between SIPL1-expressing cells and EV cells
treated with the AKT inhibitor (Fig.
2B). Taken together, the above results demonstrate a critical
role of AKT activation in SIPL1-accelerated CHO-K1 cell
proliferation.
SIPL1 expression increases the attachment
and spread of CHO-K1 cells
SIPL1 facilitates AKT activation, particularly at
lower densities (Fig. 1C).
Additionally, SIPL1 enhances CHO-K1 cell proliferation in an AKT
activity-dependent manner (Fig.
2B). These observations suggest that the property of cell
adhesion may be altered in SIPL1-overexpressing cells. This
possibility is in concordance with the importance of cell adhesion
to cell proliferation (26). To
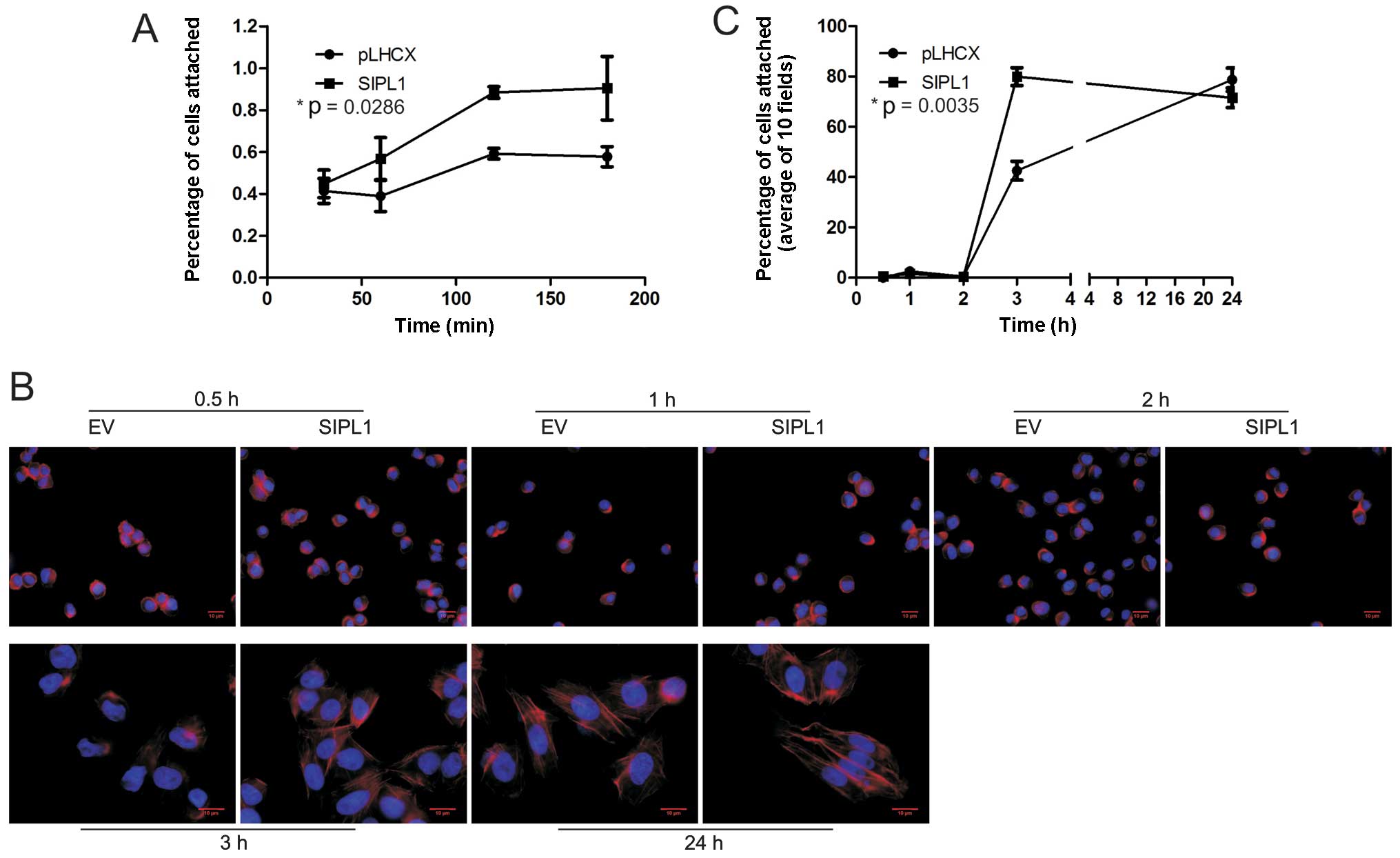
examine the effect of SIPL1 expression on cell attachment, CHO-K1
EV and SIPL1 cells were seeded in culture dishes at comparable
densities and allowed to adhere for 30–180 min. At each time-point,
any unattached cells were removed, and the remaining cells were
trypsinized and counted using a haemocytometer. CHO-K1 SIPL1 cells
were able to attach to the culture dish at a significantly higher
rate compared to CHO-K1 EV cells (Fig. 3A).
As cytoskeleton reorganization is essential for cell
attachment, we examined the formation of actin fibres in both
CHO-K1 EV and SIPL1 cells. While actin stress fibres were clearly
visible 3 h after attachment for CHO-K1 SIPL1 cells, they were not
clearly present in CHO-K1 EV cells (Fig. 3B). When quantified, 79.9% of
SIPL1-expressing cells and 42.5% of EV cells were able to form
defined actin cytoskeletons within 3 h (Fig. 3C). At the 24-h time-point, there
were no differences between actin stress fibres formed between
CHO-K1 EV and SIPL1 cells (Fig. 3B
and C). This suggests an increase in the rate at which
SIPL1-expressing cells spread and form defined actin cytoskeletons
(Fig. 3B and C). Taken together,
we have demonstrated that SIPL1 enhances cell attachment by
increasing the kinetics of forming actin stress fibres.
SIPL1 promotes the motility of CHO-K1
cells
The dynamics of cytoskeleton organization affects
the migration ability of a cell. By facilitating the rapid
formation of actin stress fibres, SIPL1 may also play a role in
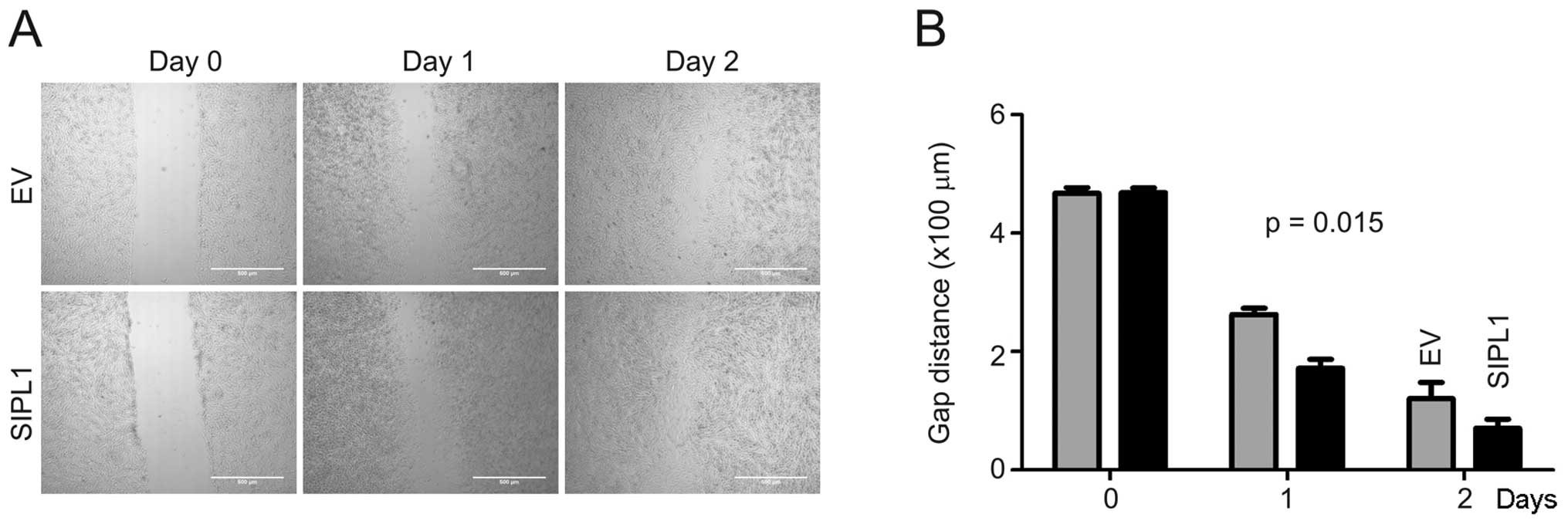
cell motility. To investigate this possibility, a wound healing
assay was performed on CHO-K1 EV and SIPL1 cells. A ‘wound’ was
created by making a scratch across the culture dish followed by
monitoring the rate of gap-closing. At day 1 and 2 post-scratching,
CHO-K1 SIPL1 cells closed the gaps with faster kinetics compared to
the CHO-K1 EV cells (Fig. 4).
Discussion
To examine the impact of SIPL1 on PTEN activity,
SIPL1 was overexpressed in CHO-K1 cells. CHO-K1 cells were selected
for a number of reasons: SIPL1 expression in CHO-K1 cells had
previously been shown to increase their tumorigenic potential
(27); SIPL1 is highly expressed
in ovary tissues (27); and
CHO-K1 cells are commonly used to express ectopic proteins
(28). Overexpression of SIPL1
resulted in a marked decrease in the amount of PTEN protein
(Fig. 1A). NEDD4-1 has been shown
to poly-ubiquitinate PTEN and reduces the amount of PTEN present in
the cell lysate. This reduction is reversed following treatment
with MG132 (29), a potent
inhibitor of the proteasome (30). It was postulated that the SIPL1
protein may also promote the degradation of PTEN via the
proteasome. Recently, SIPL1 was shown to form part of the LUBAC, a
complex of proteins that form linear ubiquitin chains on NF-κB and
promotes its activity (16–18). The SIPL1 protein also possesses a
ubiquitin-like (UBL) domain and a NFZ domain required for its
binding to ubiquitin (15,18),
lending credence to this possibility. However, we were unable to
detect any effect of MG132 on the SIPL1-induced reduction of the
PTEN protein in CHO-K1 cells (data not shown), suggesting that the
ubiquitin proteasome system may not be significant in SIPL1-induced
downregulation of the PTEN protein. As SIPL1-overexpressing CHO-K1
cells did not exhibit reduction in PTEN mRNA (Fig. 1B), it is possible that a mechanism
independent of the protease system and mRNA abundance may be
involved in SIPL1-induced reduction of the PTEN protein.
Previous studies have concluded that SIPL1 promotes
the activation of AKT through the direct binding to and
inactivation of PTEN (15).
Results of the present study show that by reducing the cellular
PTEN protein concentration, SIPL1 was able to elevate AKT
activation and induce CHO cell proliferation in an AKT-dependent
manner. AKT activity is critical in a number of cell processes
including cell proliferation, cell adhesion, migration and
cytoskeleton reorganization (31). While we have shown that AKT
activity is critical for SIPL1-enhanced CHO-K1 cell proliferation,
whether AKT is also critical in CHO-K1 cell adhesion and migration
remains unclear, owing to the essential role of FAK in cell
adhesion and migration. Future studies should therefore address the
impact of SIPL1 on FAK function.
SIPL1 promotes the activation of NF-κB (16–18) through its association with the
LUBAC. NF-κB can have a direct impact on PTEN expression by binding
the promoter region of PTEN, thereby repressing its transcription
(32). Results of this study show
that reduction in the PTEN protein was not due to changes in the
proteasomal degradation or mRNA transcription of PTEN, indicating
the SIPL1 regulation of PTEN may be independent of its NF-κB
activating activity as part of the LUBAC.
SIPL1 expression appears to regulate PTEN at a
greater extent at lower cell densities. This is in agreement with
our observations that SIPL1 facilitated cell migration and adhesion
concomitantly with enhancing the formation of the actin stress
fibres. This is in agreement with previous studies indicating that
the overexpression of PTEN impaired cell migration, spreading and
attachment (33,34). High cell densities have been shown
to promote a mild hypoxic condition in cell cultures (35), a process in which the PI3K/AKT
pathway induces changes in gene expression through its effects on
HIF1α and Redd1 and promotes cell survival (35). While SIPL1 may be able to inhibit
PTEN to a greater extent at lower cell densities, its role in
inactivating PTEN may become reduced at high densities, where the
PI3K/AKT pathway may overpower PTEN function. In addition to
regulating PTEN, and participating in the LUBAC to regulate NF-κB
signaling, SIPL1 has also been shown to inhibit β1-integrin
activation (19). This is
contrary to our understanding of activities of SIPL1. The
differences may be attributable to different assay conditions.
Taken together, the present study has shown that
SIPL1 plays a role in the reduction of PTEN, leading to AKT
activation. AKT activity enhances CHO-K1 cell proliferation, and
may also contribute to increased cell attachments and motility.
Although it is suspected that SIPL1 induces PTEN reduction
independently of its role in NF-κB activation, this possibility
cannot be excluded. Furthermore, the ability of SIPL1 to promote
FAK activation via PTEN inactivation should be investigated in the
future.
Acknowledgements
The present study was supported by the Canadian
Institute of Health Research (CIHR) grant (COP-107971) to D.T.
References
|
1
|
Steck PA, Pershouse MA, Jasser SA, et al:
Identification of a candidate tumour suppressor gene, MMAC1,
at chromosome 10q23.3 that is mutated in multiple advanced cancers.
Nat Genet. 15:356–362. 1997.PubMed/NCBI
|
|
2
|
Li J, Yen C, Liaw D, et al: PTEN, a
putative protein tyrosine phosphatase gene mutated in human brain,
breast, and prostate cancer. Science. 275:1943–1947. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Myers MP, Stolarov JP, Eng C, et al:
P-TEN, the tumor suppressor from human chromosome 10q23, is a
dual-specificity phosphatase. Proc Natl Acad Sci USA. 94:9052–9057.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maehama T and Dixon JE: The tumor
suppressor, PTEN/MMAC1, dephosphorylates the lipid second
messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem.
273:13375–13378. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li DM and Sun H: PTEN/MMAC1/TEP1
suppresses the tumorigenicity and induces G1 cell cycle
arrest in human glioblastoma cells. Proc Natl Acad Sci USA.
95:15406–15411. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Robertson GP, Furnari FB, Miele ME, et al:
In vitro loss of heterozygosity targets the PTEN/MMAC1 gene
in melanoma. Proc Natl Acad Sci USA. 95:9418–9423. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Duerr EM, Rollbrocker B, Hayashi Y, et al:
PTEN mutations in gliomas and glioneuronal tumors. Oncogene.
16:2259–2264. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mallory SB: Cowden syndrome (multiple
hamartoma syndrome). Dermatol Clin. 13:27–31. 1995.PubMed/NCBI
|
|
9
|
Liaw D, Marsh DJ, Li J, et al: Germline
mutations of the PTEN gene in Cowden disease, an inherited
breast and thyroid cancer syndrome. Nat Genet. 16:64–67. 1997.
|
|
10
|
Di Cristofano A, Pesce B, Cordon-Cardo C,
Pandolfi PP, Cristofano AD and PP: Pten is essential for embryonic
development and tumour suppression. Nat Genet. 19:348–355.
1998.PubMed/NCBI
|
|
11
|
Yuan TL and Cantley LC: PI3K pathway
alterations in cancer: variations on a theme. Oncogene.
27:5497–5510. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stambolic V, Suzuki A, Mirtsos C, et al:
Negative regulation of PKB/Akt-dependent cell survival by the tumor
suppressor PTEN. Cell. 95:29–39. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tamura M, Gu J, Matsumoto K, Aota S,
Parsons R and Yamada KM: Inhibition of cell migration, spreading,
and focal adhesions by tumor suppressor PTEN. Science.
280:1614–1617. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhao X and Guan JL: Focal adhesion kinase
and its signaling pathways in cell migration and angiogenesis. Adv
Drug Deliv Rev. 63:610–615. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
He L, Ingram A, Rybak AP and Tang D:
Shank-interacting protein-like 1 promotes tumorigenesis via PTEN
inhibition in human tumor cells. J Clin Invest. 120:2094–2108.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gerlach B, Cordier SM, Schmukle AC, et al:
Linear ubiquitination prevents inflammation and regulates immune
signalling. Nature. 471:591–596. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tokunaga F, Nakagawa T, Nakahara M, et al:
SHARPIN is a component of the NF-κB-activating linear ubiquitin
chain assembly complex. Nature. 471:633–636. 2011.
|
|
18
|
Ikeda F, Deribe YL, Skånland SS, et al:
SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB
activity and apoptosis. Nature. 471:637–641. 2011.PubMed/NCBI
|
|
19
|
Rantala JK, Pouwels J, Pellinen T, et al:
SHARPIN is an endogenous inhibitor of β1-integrin activation. Nat
Cell Biol. 13:1315–1324. 2011.PubMed/NCBI
|
|
20
|
Liang Y: Chronic proliferative dermatitis
in mice: NFκB activation autoinflammatory disease. Patholog Res
Int. 2011:9367942011.
|
|
21
|
Gijbels MJ, Hogenesch H, Blauw B, Roholl P
and Zurcher C: Ultrastructure of epidermis of mice with chronic
proliferative dermatitis. Ultrastruct Pathol. 19:107–111. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Seymour RE, Hasham MG, Cox GA, Shultz LD,
Hogenesch H, Roopenian DC and Sundberg JP: Spontaneous mutations in
the mouse Sharpin gene result in multiorgan inflammation, immune
system dysregulation and dermatitis. Genes Immun. 416–421. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Manning BD and Cantley LC: AKT/PKB
signaling: navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xie Y, Yan J, Cutz JC, et al: IQGAP2, A
candidate tumour suppressor of prostate tumorigenesis. Biochim
Biophys Acta. 1822:875–884. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yan J, Wong N, Hung C, Chen WX and Tang D:
Contactin-1 reduces E-cadherin expression via activating AKT in
lung cancer. PLoS One. 8:e654632013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gilmore AP and Romer LH: Inhibition of
focal adhesion kinase (FAK) signaling in focal adhesions decreases
cell motility and proliferation. Mol Biol Cell. 7:1209–1224. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jung J, Kim JM, Park B, et al: Newly
identified tumor-associated role of human Sharpin. Mol Cell
Biochem. 340:161–167. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Greene G, Gilna P, Waterfield M, Baker A,
Hort Y and Shine J: Sequence and expression of human estrogen
receptor complementary DNA. Science. 231:1150–1154. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang X, Trotman LC, Koppie T, et al:
NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell.
128:129–139. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zanotto-Filho A, Braganhol E, Battastini
AM and Moreira JC: Proteasome inhibitor MG132 induces selective
apoptosis in glioblastoma cells through inhibition of PI3K/Akt and
NFkappaB pathways, mitochondrial dysfunction, and activation of
p38-JNK1/2 signaling. Invest New Drugs. 30:2252–2262. 2012.
View Article : Google Scholar
|
|
31
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ghosh-Choudhury N, Mandal CC,
Ghosh-Choudhury N and Ghosh Choudhury G: Simvastatin induces
derepression of PTEN expression via NFκB to inhibit breast cancer
cell growth. Cell Signal. 22:749–758. 2010.PubMed/NCBI
|
|
33
|
Attwell S, Mills J, Troussard A, Wu C and
Dedhar S: Integration of cell attachment, cytoskeletal
localization, and signaling by integrin-linked kinase (ILK),
CH-ILKBP, and the tumor suppressor PTEN. Mol Biol Cell.
14:4813–4825. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mondal S, Subramanian KK, Sakai J, Bajrami
B and Luo HR: Phosphoinositide lipid phosphatase SHIP1 and PTEN
coordinate to regulate cell migration and adhesion. Mol Biol Cell.
23:1219–1230. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jin HO, An S, Lee HC, et al: Hypoxic
condition- and high cell density-induced expression of Redd1 is
regulated by activation of hypoxia-inducible factor-1α and Sp1
through the phosphatidylinositol 3-kinase/Akt signaling pathway.
Cell Signal. 19:1393–1403. 2007.PubMed/NCBI
|