Introduction
Atherosclerosis is a complex pathological process
that possesses many features of chronic inflammation (1) and oxidative stress in the vascular
wall. It also plays a major role in atherogenesis. Reactive oxygen
species (ROS), generated as byproducts of cellular metabolism,
elicit numerous effects on cell functions in several cell types
such as endothelial cells, macrophages and vascular smooth muscle
cells (VSMCs) (2).
In endothelial cells, ROS regulate numerous
signaling pathways including those regulating cell growth,
proliferation, vaso-relaxation, inflammatory responses, barrier
function and vascular remodeling (3). The vascular ROS production by NADPH
oxidase or mitochondria is markedly enhanced in atherosclerotic
arteries, which in turn limits the activity of nitric oxide (NO),
thereby producing endothelial dysfunction (4). A number of in vitro and in
vivo studies have demonstrated a critical role for ROS or
enzyme systems involved in ROS production, including endothelial NO
synthases, xanthine oxidase, enzymes of the respiratory chain,
cytochrome P450 monooxygenases and NAD(P)H oxidase in the
vasculature (5). Upregulation of
the NAD(P)H oxidase subunits gp91phox and Nox4 increases
intracellular oxidative stress in macrophages and non-phagocytic
vascular cells of human coronary atherosclerosis, respectively
(6). Furthermore, the endothelial
cell responds to various proinflammatory mediators such as oxLDL.
oxLDL has been previously shown to upregulate the expression of
MCP-1 via activation of ROS and nuclear factor (NF)-κB (7).
In macrophages, a recent study showed that the
CD14/TLR4 (a Toll-like receptor 4)/MD-2 complex interacts with
mmLDL, inducing cytoskeletal rearrangements and the secretion of
macrophage inflammatory protein-2, MCP-1, tumor necrosis factor-α
(TNF-α) and interleukin-6 (8,9)
via ROS generation from spleen tyrosine kinase/Nox2 signaling
(10). In addition, NF-κB, the
most well-known redox-dependent transcriptional factors, regulates
a number of genes involved in inflammatory responses in macrophages
(11).
In VSMCs, ROS mediates various functions including
growth, migration, matrix regulation, inflammation and contraction
(12) which are critical factors
in the progression and complications of atherosclerosis. In
addition, in VSMCs, ROS also mediate inflammation, e.g., MCP-1
expression via TNF-α (13).
The cytokine TNF-α, characterized as a potent
pro-inflammatory cytokine, induces oxidative stress in cells and
increases intracellular ROS generation (14,15). It also leads to the activation of
NF-κB. However, antioxidants have been shown to scavenge
intracellular ROS production and block the NF-κB activation
(16). These results suggest that
the suppression of ROS-dependent intracellular signaling may be an
effective strategy for inflammatory vascular diseases.
Epoxyeicosatrienoic acids (EETs) are synthesized
predominantly by the epoxygenases of the CYP2 family, including the
2C and 2J classes. CYP2C and CYP2J are mainly expressed in
epithelial, endothelial, and smooth muscle cells, as well as
cardiomyocytes, autonomic ganglion cells, and islet cells in the
heart, vessel, kidney, lung and pancreas (17–20). Specifically, CYP2C8 is expressed
mainly in the endothelium. EETs possess a number of biological
effects in the cardiovascular and renal systems, including
anti-inflammatory (17) and
angiogenic (21) effects on
endothelial cells, and inhibition of vascular smooth muscle cell
migration (22). EETs have
recently been reported to attenuate ROS (23). However, how CYP2C8-derived EETs
affect ROS signaling pathways that lead to inflammation and
atherosclerosis remains to be determined. The focus of the present
study was CYP2C8 and its capacity to elucidate how the arachidonic
acid metabolites, EETs, attenuate TNF-α induced inflammation
through ROS in vascular endothelial cells and macrophages and
improve endothelial function and provide new insight into how
CYP2C8-derived EETs ameliorate vascular inflammatory diseases such
as atherosclerosis.
Materials and methods
Materials
Chemicals and reagents were obtained as follows:
Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum
(FBS) were purchased from HyClone Laboratories, Inc. (Logan, UT,
USA); HUVECs, VSMCs, macrophages cell lines and
2′,7′-dichlorodihydrofluorescein diacetate (HB2BDCF-DA)
were purchased from Wuhan Boster Biological Technology, Ltd.
(Wuhan, China); exogenous EETs and PPARγ-specific inhibitor GW9662
were from Cayman Chemical (Ann Arbor, MI, USA); RPMI-1640 medium
and recombinant human TNF-α were from Sigma Chemical, Co. (St.
Louis, MO, USA); pCMV-CYP2C8 plasmids from OriGene Technologies,
Inc. (Rockville, MD, USA) were introduced into cells using
Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA); antibodies
against PPARγ, lamin B1 and nuclear factor κB (NF-κB) were from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA); antibodies
against gp-91, CYP2C8 and eNOS were from Cell Signaling Technology
(Beverly, MA, USA); antibody against β-actin was from Neomarkers
(Fremont, CA, USA). All other reagents were purchased from standard
commercial suppliers.
Cell culture
Human umbilical vein endothelial cells (HUVECs) were
maintained in RPMI-1640 medium supplemented with 10% FBS, 100 U/ml
penicillin and 100 μg/ml streptomycin at 37°C under 5%
CO2. Macrophages and VSMCs were maintained in DMEM
supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml
streptomycin at 37°C under 5% CO2. HUVECs and
macrophages were seeded in a 6-well plate at a density of
3×105 cells/well. After the cells reached 60% confluence
and the medium was removed, the cells were transferred to
serum-free medium. The HUVECs and macrophages were pretreated with
exogenous EETs (1 μM) or transfected with CYP2C8 in advance for 30
min in the presence or absence of the specific PPARγ antagonist
GW9662 (1 μM), and subsequently stimulated with TNF-α (10 ng/ml)
for the indicated times.
Cell transfection and expression
The plasmids pCMV-CYP2C8 and pCMV-GFP were obtained
from OriGene Technologies Inc. (Rockville). As the density of the
cultured cells reached ~50–70%, each group was pretreated with
Lipofectamine 2000 and then interfered by adding the pCMV-CYP2C8
and pCMV-GFP plasmids, respectively. After 6 h of transfection, the
experimental medium was added to the cells followed by exposure to
TNF-α (10 ng/ml) in the absence or presence of GW9662 (1 μM). The
efficacy of transfection was obtained by examining the green
fluorescence by microscopy (Nikon, Tokyo, Japan).
Intracellular ROS production assay
Confluent HUVECs in 6-well plates were pretreated
with EETs for 1 h. Following removal of the EETs from the wells,
the cells were incubated with 20 μM HB2BDCF-DA for 30
min and then stimulated with TNF-α (10 ng/ml) for 1 h. The
fluorescence intensity was measured at an excitation and emission
wavelength of 485 and 530 nm, respectively, using a fluorescence
spectrophotometer or a fluorescence microscopy (Nikon). Values for
each treatment group were recorded as the mean fluorescence
intensity.
VSMCs Transwell assay
Transwsell 12-well plates were obtained from Costar
Corp. (Cambridge, MA, USA). Monolayers of serum-starved adherent
cells were trypsinized and cell suspensions were placed in
serum-free medium in the upper well of a Transwell filter
apparatus. The filter was suspended in a well of a 12-well plate
and the lower reservoir was filled with the same medium (no cells)
plus added TNF-α. The cells were incubated under basal condition
for 12 h. The cells were stained with DAPI (Wuhan Boster Biological
Technology) and cells found on the underside of the filter were
counted by microscopy (Nikon).
Western blot analysis
HUVECs and macrophages were pre-treated with
exogenous EETs (1 μM) and PPARγ inhibitor GW9662 for 1 h, and then
stimulated with or without TNF-α (10 ng/ml). Cultured HUVECs and
macrophage were lysed in RIPA buffer containing a mixture of
protease inhibitors, and the total protein concentration was
determined by protein assay. Proteins (50 μg) from cell lysates
were electrophoresed by SDS-PAGE, proteins and nuclear extracts
were then transferred to a PVDF membrane (Roche Diagnostics,
Mannheim, Germany). The membrane was blocked with blocking buffer
(1× TBS, 0.1% Tween-20, 5% non-fat dry milk), and washed and
incubated overnight at 4°C with anti-NF-κB p-65 (1:1,000 dilution),
anti-IκBα (1:1,000 dilution), anti-eNOS (1:1,000 dilution),
anti-gp-91 (1:1,000 dilution), anti-β-actin (1:1,000 dilution) or
anti-lamin B-1 (1:1,000 dilution) primary antibodies. The membrane
was subsequently washed with TBS-T (10 mmol/l Tris-HCl, 150 mmol/l
NaCl, and 0.1% Tween-20) and incubated with horseradish
peroxidase-conjugated secondary antibodies at 37°C for 1 h. The
immune complex was detected with an enhanced chemiluminescence
system (Beyotime Institute of Biotechnology, Jiangsu, China),
exposed to X-ray film, and analyzed using ImageJ2x software.
β-actin and lamin B-1 were used as an internal control.
Measurement of MCP-1 and IL-6 level by
enzyme-linked immunosorbent assay (ELISA)
The level of MCP-1 and IL-6 in the supernatants was
measured using a commercially available kit from Wuhan Boster
Biological Technology according to the manufacturer’s instructions.
Optical densities were recorded on a universal microplate reader
(Bio Tek Instruments, VT, Winooski, USA) at 450 nm.
Statistical analysis
Data are presented as the mean ± SEM. In
vitro experiments were performed a total of 4–6 times, and each
experiment was carried out in triplicate for each treatment group.
Statistical comparisons among groups were performed by the Wilcoxon
test, the Student’s t-test or ANOVA as appropriate. In all cases,
statistical significance was defined as P<0.05.
Results
Transfection with CYP2C8 attenuated TNF-α
induced inflammation by decreasing the levels of inflammatory
factor MCP-1 and IL-6
The levels of inflammatory factors MCP-1 and IL-6
were examined in macrophages and HUVECs. pCMV-CYP2C8 delivery led
to an abundant CYP2C8 expression and increased EETs generation.
ELISA analysis showed that transfection with CYP2C8 markedly
suppressed the expression of inflammatory cytokines including
interleukin (IL)-6 and MCP-1 induced by TNF-α, which was reversed
by CYP2C8 inhibitor C26 (Fig. 1A and
B). As shown in Fig. 1C and
D, 8,9-EET, 11,12-EET and 14,15-EET markedly reduced the IL-6
and MCP-1 expression induced by TNF-α in HUVECs and macrophages,
with 11,12-EET exerting the most significant effect. Therefore, we
used 11,12-EET as the representative of EET in the subsequent
experiments.
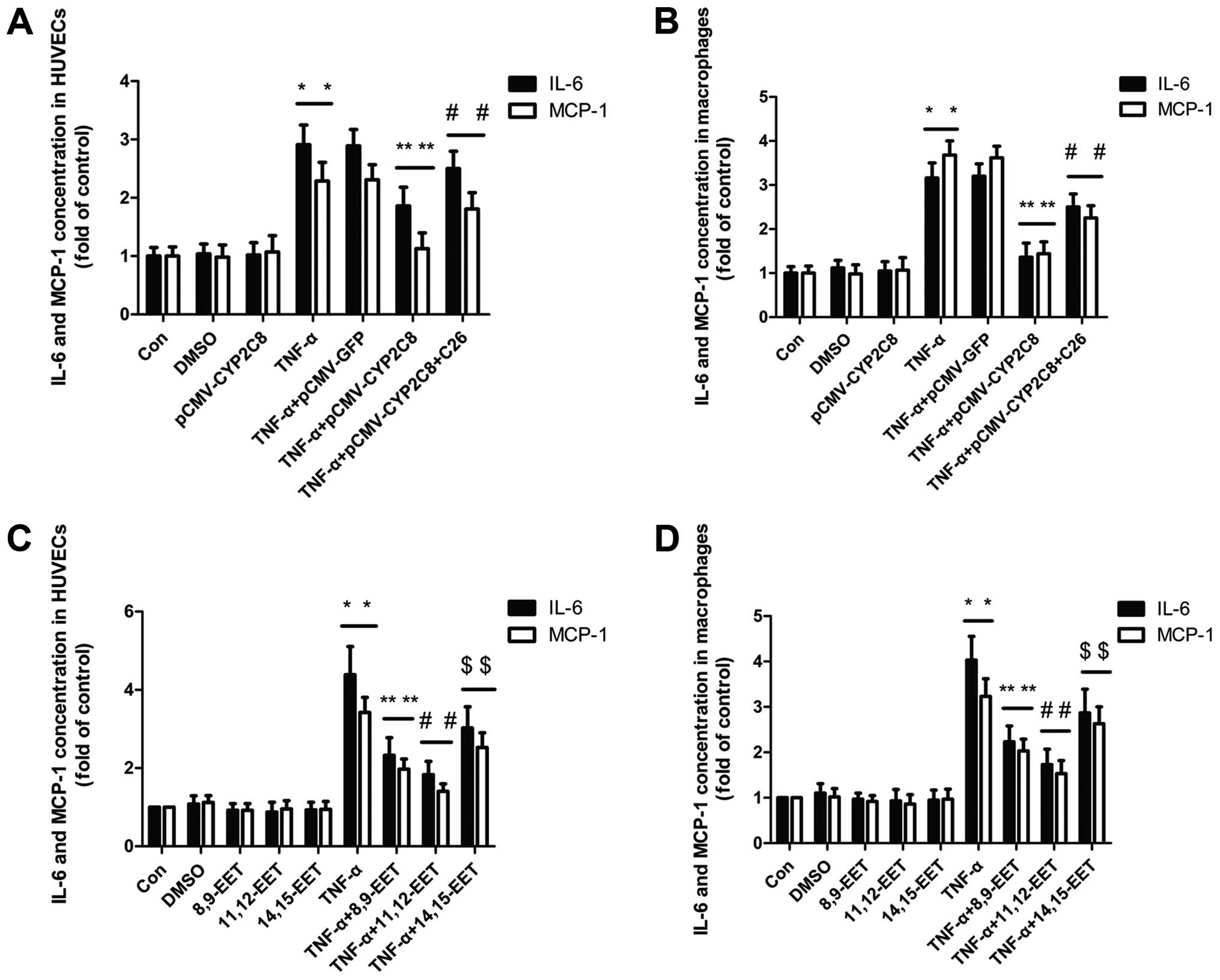 | Figure 1CYP2C8-derived EETs attenuated TNF-α
induced inflammation by decreasing the levels of inflammatory
factor MCP-1 and IL-6. (A) Primary cultures of HUVECs were
transfected with CYP2C8 and administered with CYP2C8 inhibitor C26
for 60 min and stimulated with TNF-α (10 ng/ml) for 24 h.
(*P<0.05 vs. control group; **P<0.05
vs. TNF-α treatment group; #P<0.05 vs. no inhibitor
treatment group). (B) Macrophages were transfected as described in
(A). (*P<0.05 vs. control group;
**P<0.05 vs. TNF-α treatment group;
#P<0.05 vs. no inhibitor treatment group). (C)
Primary cultures of HUVECs were pre-treated with exogenous 8,9-EET
(1 μM), 11,12-EET (1 μM) and 14,15-EET (1 μM), respectively, for 60
min and stimulated with TNF-α (10 ng/ml) for 24 h.
(*P<0.05 vs. control group; **P<0.05,
#P<0.05 and $P<0.05 vs. TNF-α treatment
group). (D) Macrophages were pre-treated with exogenous 8,9-EET (1
μM), 11,12-EET (1 μM) and 14,15-EET (1 μM), respectively, for 60
min and stimulated with TNF-α (10 ng/ml) for 24 h.
(*P<0.05 vs. control group; **P<0.05,
#P<0.05 and $P<0.05 vs. TNF-α treatment
group). Data are the mean ± SEM of results from at least five
independent experiments, each performed in triplicate. |
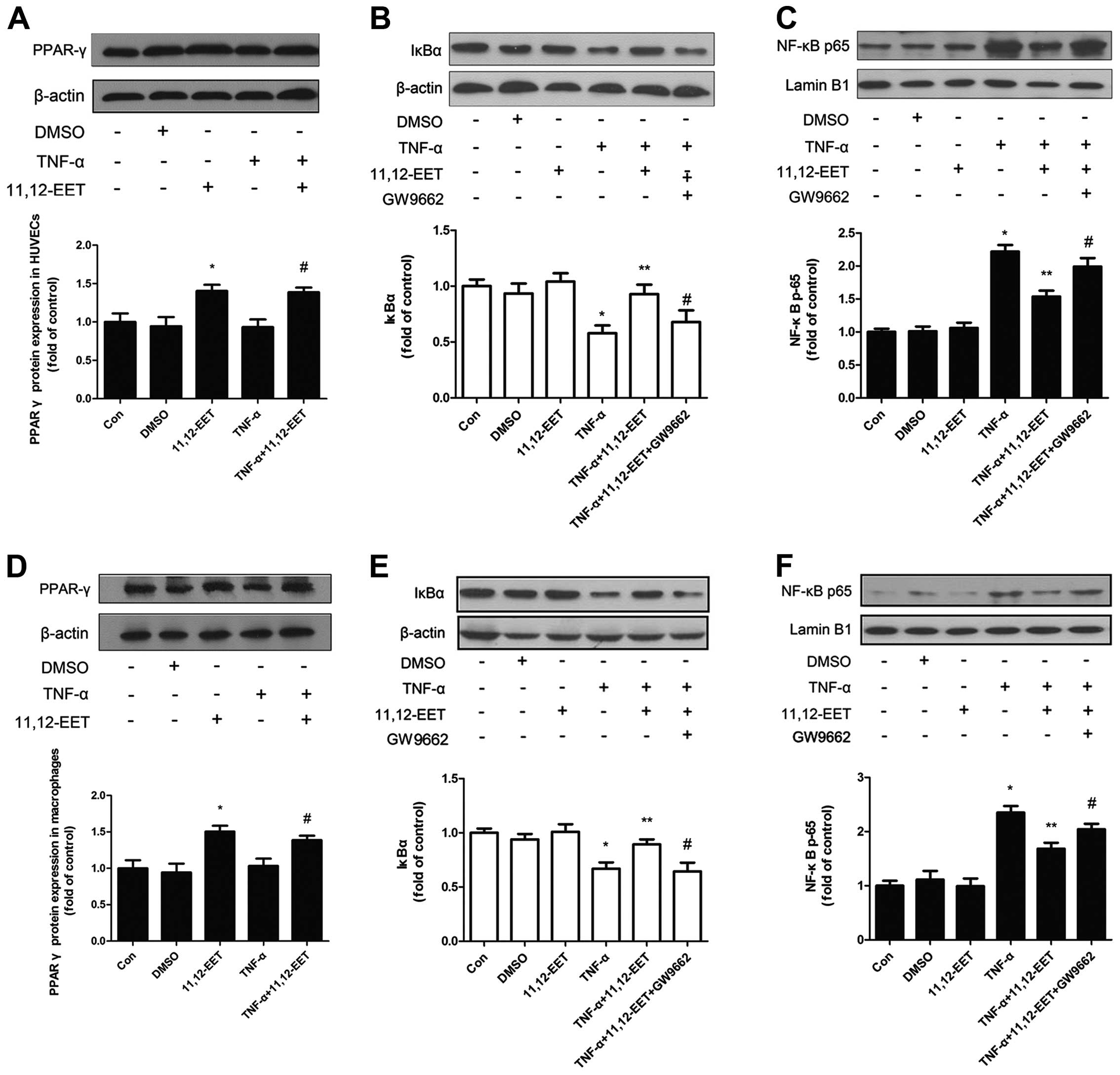
CYP2C8-derived EETs inhibited TNF-α
induced expression of NF-κB through PPARγ in HUVECs and
macrophages
To elucidate how CYP2C8 plays a role on
anti-inflammation, it is necessary to examine the possible
mechanism of CYP2C8-derived EET on anti-inflammation. Results of
the western blot analysis revealed that 11,12-EET increased the
protein expression of PPARγ, which was the possible acceptor of
EETs and associated with inflammation (24) (Fig.
2A and D), both in the basal and inflammatory condition.
Therefore, we hypothesized that the overexpression of CYP2C8 in
vitro to increase EETs may prevent the development of
inflammation potentially through PPARγ activation. Moreover, we
assessed the vital transcription factor NF-κB and the conclusion
was consistent in HUVECs and macrophages. IκBα expression was
decreased under the stimulation of TNF-α, which could be reversed
by 11,12-EET (Fig. 2B and E). The
NF-κB subunit p-65 expression was opposite to the effect of IκBα
(Fig. 2C and F). Notably, after
adding PPARγ-specific inhibitor GW9662, the beneficial effects
caused by exogenous 11,12-EET were attenuated, indicating that
11,12-EET may be involved through the PPARγ pathway in blocking the
activation of NF-κB.
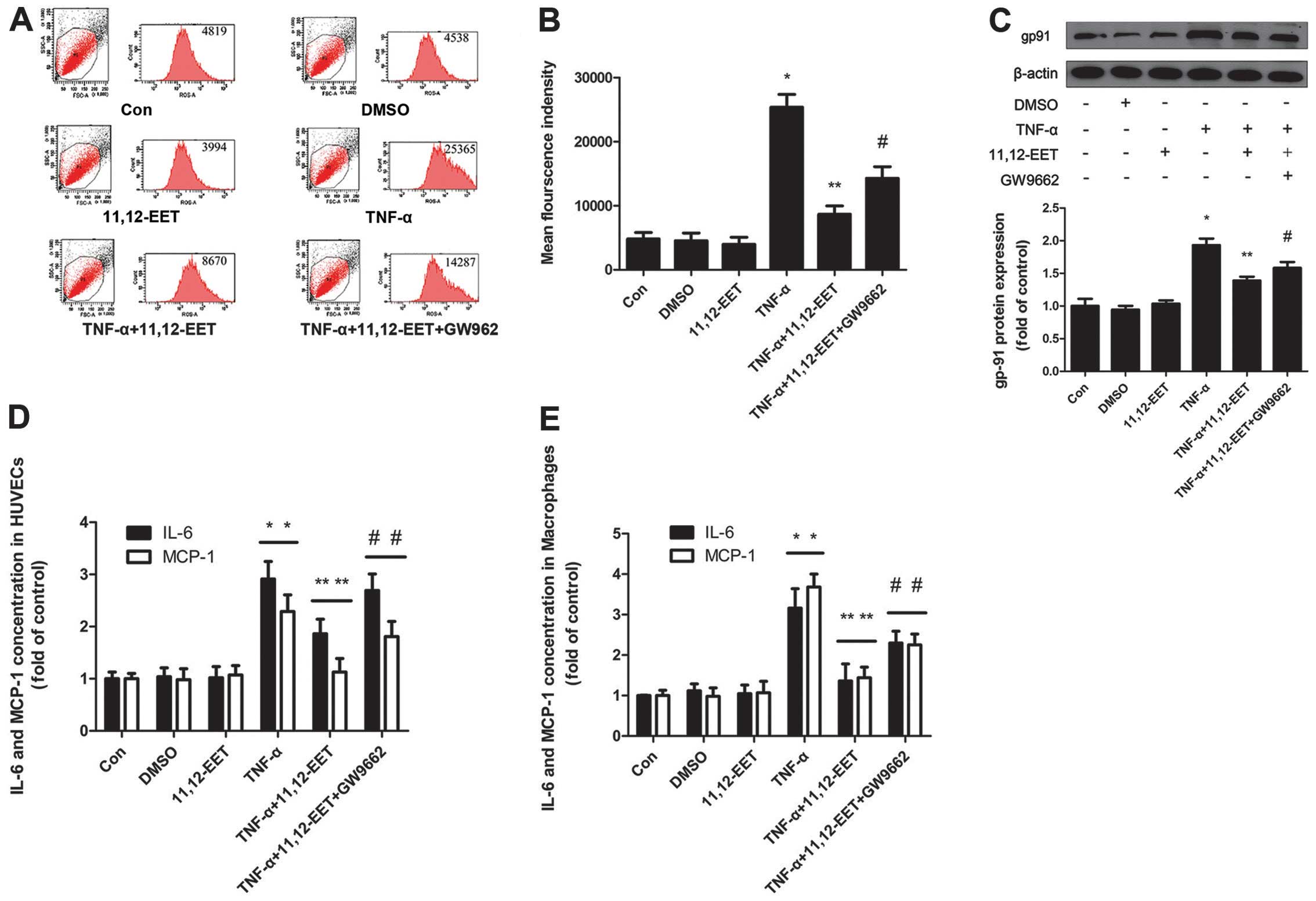
11,12-EET decreased TNF-α induced
intracellular ROS production
Mounting evidence has indicated that the induction
of ROS was necessary for the development of atherosclerosis and
EETs possessed the ability of anti-atherosclerosis. To confirm the
beneficial effects of 11,12-EET on atherosclerosis, intracellular
ROS production induced by TNF-α in HUVECs were measured. The
results showed that 11,12-EET decreased the ROS production induced
by TNF-α and suppressed the TNF-α-induced mean fluorescence
intensity of the dye to a level that was comparable with that of
the control. However, following the addition of the PPARγ-specific
inhibitor GW9662, the anti-oxidative effect caused by 11,12-EET was
also attenuated (Fig. 3A and B).
Results of the western blot analysis revealed that the
ROS-associated NAD(P)H subunit gp-91 was increased almost 2-fold
compared with the control when stimulated with TNF-α. However, this
effect disappeared following the addition of 11,12-EET (Fig. 3C). Therefore, 11,12-EET performed
the coincident anti-inflammatory effect through the exogenous
administration and CYP2C8 gene transfection. 11,12-EET
pre-incubation reduced the expression of IL-6 and MCP-1 induced by
TNF-α in HUVECs and macrophages (Fig.
3D and E). Moreover, the PPARγ-specific inhibitor GW9662
partially eliminated the beneficial effects of CYP2C8 transfection
or the exogenous supply with 11,12-EET. NF-κB was one of the major
transcription factors regulating the TNF-α-induced expression of
inflammatory biomarkers in HUVECs and macrophages, while ROS was
crucial in inflammation. Thus, the CYP2C8/EET/PPARγ/ROS/NF-κB
pathway may regulate the TNF-α induced inflammatory cytokine
expression in HUVECs and macrophages.
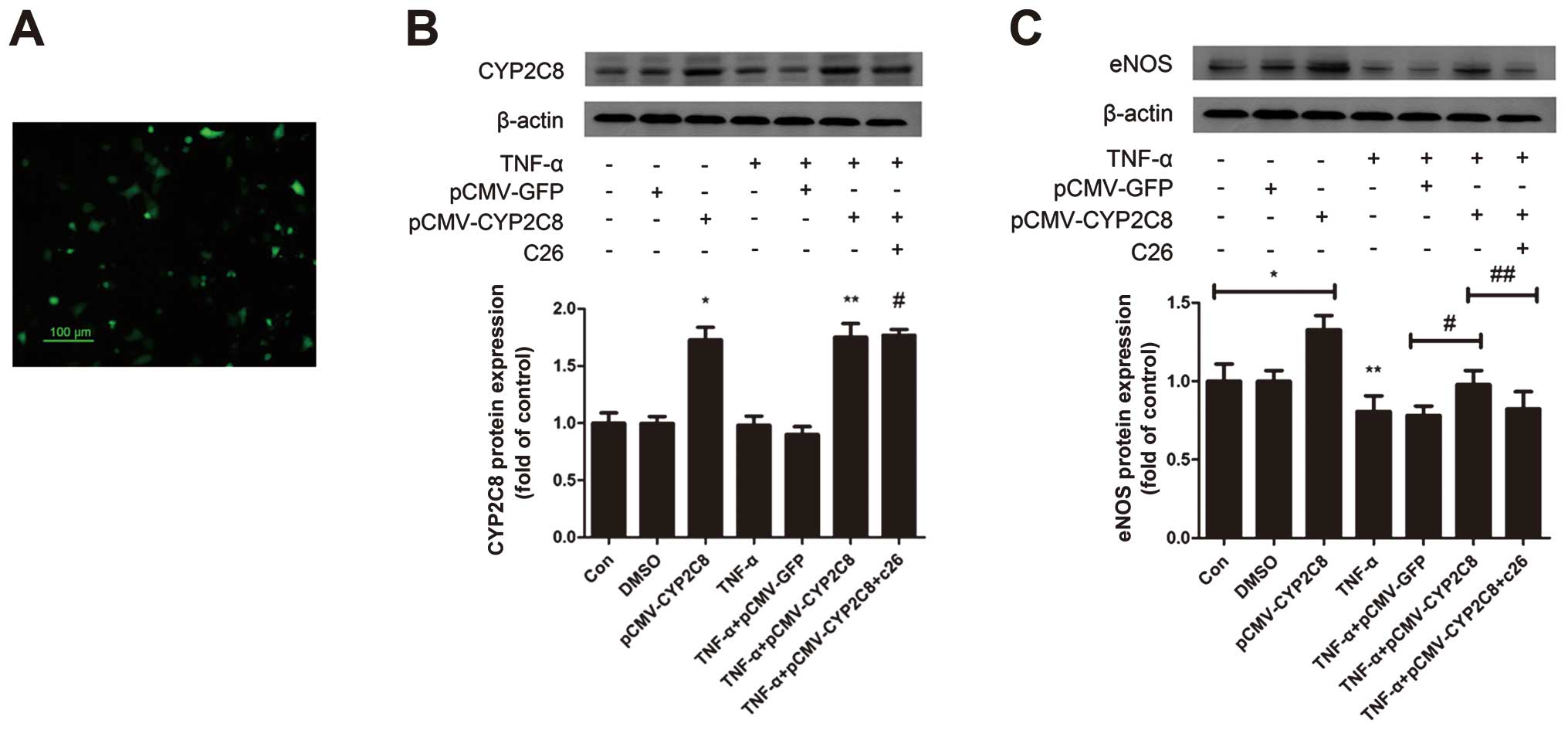
CYP2C8-derived EETs improve endothelial
function through the upregulation of eNOS
In addition, eNOS catalyzed the formation of NO
characterized as an anti-atherosclerosis effect. To examine how
CYP2C8-derived EETs affect the eNOS expression, CYP2C8 transfection
was performed. pCMV-CYP2C8 transfection led to a substantial
expression of CYP2C8 in HUVECs. Moreover, western blot analysis
revealed a CYP2C8 protein overexpression in HUVECs (Fig. 4A and B) and eNOS was upregulated
by CYP2C8 overexpression, which was decreased significantly under
the stimulation of TNF-α (Fig.
4C). This effect suggested that CYP2C8 overexpression could
increase the eNOS protein expression, which contributed to
improving endothelial function and anti-atherosclerosis.
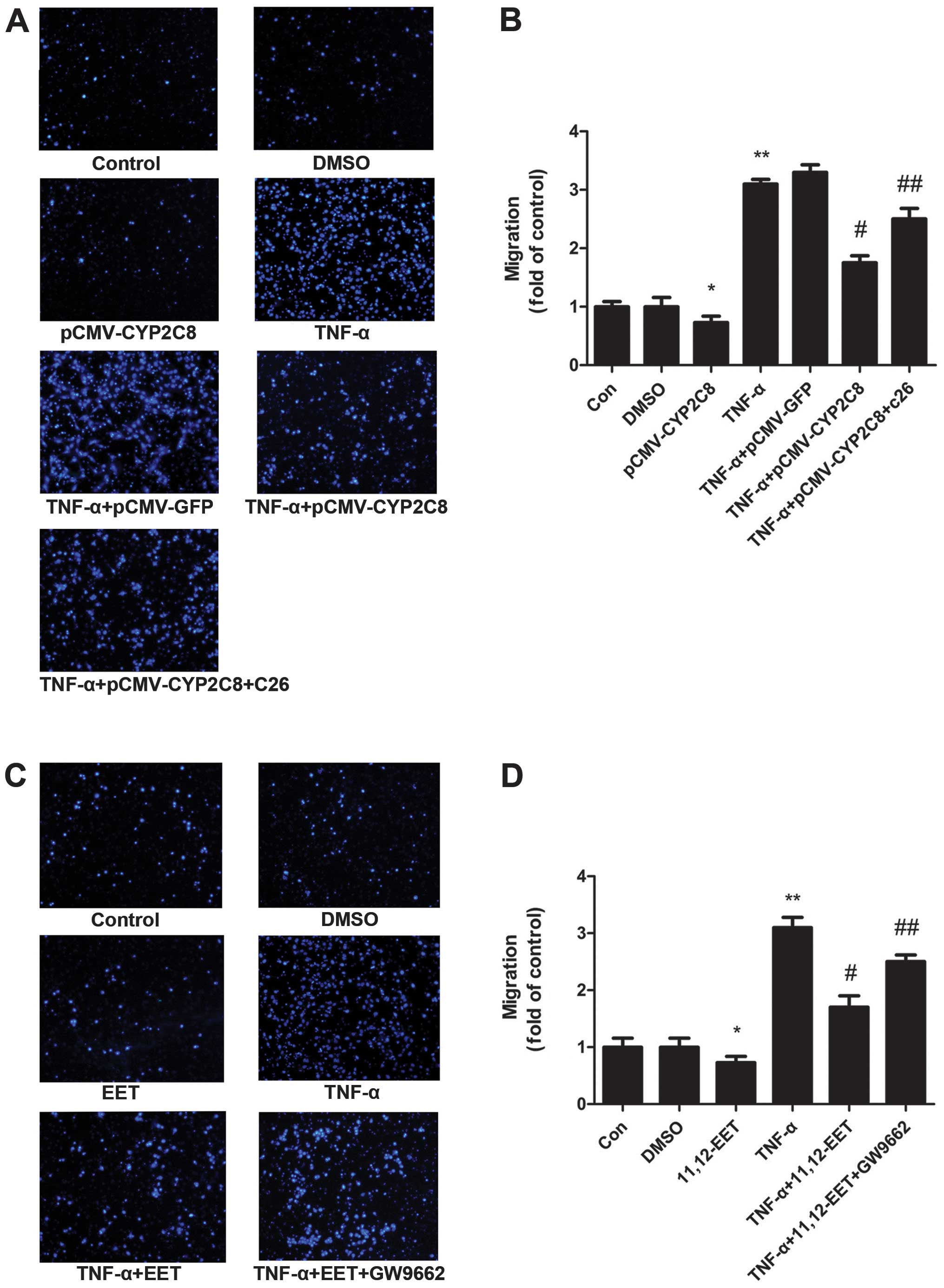
CYP2C8-derived EETs increased the
migration of VSMCs
To assess the effect of CYP2C8-derived EETs on VSMC
migration, we applied the Transwell migration model. pCMV-CYP2C8
transfection led to an abundant expression of CYP2C8 in VSMCs
(Fig. 5A and B). The Transwell
results showed that TNF-α increased the ability of the migration of
VSMCs significantly. Additionally, EETs reversed the increased
migration by TNF-α. TNF-α+EET+GW9662 also exerted a similar effect
as TNF-α on migration (Fig. 5C and
D). These results indicated that CYP2C8-derived EETs possess an
anti-TNF-α effect on cell migration and may play a role through the
PPARγ pathways.
Discussion
The present study has demonstrated that
CYP2C8-derived EETs significantly suppressed TNF-α-induced
intracellular ROS formation and the redox-sensitive NF-κB
activation via the suppression of IκB degradation and
phosphorylation. We investigated CYP2C8 gene-alleviated
vascular inflammation through the CYP2C8/EET/PPARγ/ROS/NF-κB
signaling pathway and improved endothelial function through the
upregulation of eNOS and it may ameliorate the migration of VSMCs.
These results have demonstrated that CYP2C8-derived EETs exerted
antivascular inflammatory and anti-oxidative effects and it may be
useful in the prevention and treatment of vascular inflammatory
diseases.
Our previous study indicated that CYP2C8 exerted a
protective effect on atherosclerosis induced by a western-type diet
in APOEKO+/−CYP2C8Tg+/− and
CYP2C8Tg+/− mice (25). The formation and area of
atherosclerosis plaque was decreased significantly in teh aortic
artery in the CYP2C8 gene overexpression group. To the best
of our knowledge, the present study provided the first evidence
that CYP2C8-derived EETs suppressed TNF-α-induced inflammation
through the suppression of NF-κB activation and ROS in HUVECs.
Thus, we revealed that CYP2C8-derived EETs prevented the early
pathogenesis of atherosclerosis by modulating vascular inflammation
and ROS.
The production of intercellular ROS induced by
cytokines such as TNF-α has been suggested in the activation of
NF-κB and expression of inflammatory biomarkers (26–28). In the present study, TNF-α
increased intercellular ROS production and we confirmed that
CYP2C8-derived EETs decreased ROS generation in TNF-α-stimulated
HUVECs. Thus, the inhibition of ROS generation by CYP2C8-derived
EETs may contribute to the inhibition NF-κB activation and
expression of inflammatory biomarkers.
To identify the molecular mechanism by which
CYP2C8-derived EETs exerted its anti-inflammatory effect on
TNF-α-stimulated endothelial cells and macrophage, we examined the
activation of NF-κB, one of the major transcription factors
regulating the TNF-α-induced expression of inflammatory biomarkers
in endothelial cells and macrophage (29,30). NF-κB is present in the cytosol as
a heterodimer composed of p50 and p65 subunits bound to the
inhibitor protein IκB family in unstimulated cells. Following
stimulation with cytokine, the IκB proteins are phosphorylated and
degraded, which allows NF-κB to translocate into the nucleus and
initiates gene transcription (31). Our present results show that
CYP2C8-derived EETs significantly suppressed TNF-α-induced IκBα
degradation. We also observed that CYP2C8-derived EETs inhibited
the TNF-α-induced phosphorylation of NF-κB p65 at serine 536 and
nuclear translocation of NF-κB p65. Furthermore, the activation of
NF-κB has been associated with the transcription factor PPARγ
(peroxisome proliferator-activated receptor γ) (32–34). PPARγ is a ligand-activated nuclear
receptor, binding with the PPAR response element of the target gene
promoter, that is involved in the transcription of the related gene
(35,36). We also confirmed that
CYP2C8-derived EETs decreased the levels of inflammatory factors
such as MCP-1 and IL-6 in HUVECs and macrophage. Thus, our results
suggest that CYP2C8-derived EETs attenuated the TNF-α-induced NF-κB
activation that would be critical for the timely inhibition of
inflammatory mediator expression.
Additionally, results of this study have
demonstrated that CYP2C8-derived EETs improved endothelial function
through the upregulation of eNOS. These results were consistent
with those of a previous study (37). eNOS catalyzes the formation of NO,
thus it may be characterized as exerting an anti-atherosclerotic
effect.
Moreover, we confirm that CYP2C8-derived EETs are
involved in the migration of VSMCs. In our experiments TNF-α
increased the migration of VSMCs significantly and CYP2C8-derived
EETs decreased their migration. As previous studies have
demonstrated that the migration of VSMCs was associated with the
formation of atherosclerosis (38–41), the decrease of the migration of
VSMCs by CYP2C8-derived EETs may contribute to the improvement of
atherosclerosis.
In conclusion, vascular inflammation induced by
cytokine occurs early in the development of atherosclerosis and
leads to endothelial dysfunction. Thus, these data have provided
insight on the actions of CYP2C8-derived EETs and its potential
benefits on preventing inflammatory diseases such as
atherosclerosis.
Acknowledgements
The present study was supported by the National
Nature Science Foundation of China (no. 81170259) and the Key
Project of Health Ministration.
Abbreviations:
|
CYP2C8
|
cytochrome P450 enzymes 2C8
|
|
EET
|
epoxyeicosatrienoic acid
|
|
ROS
|
reactive oxygen species
|
|
HUVEC
|
human umbilical vein endothelial
cell
|
|
VSMC
|
vascular smooth muscle cell
|
|
NF-κB
|
nuclear factor κB
|
|
IκBα
|
inhibitor of NF-κBα
|
|
IL
|
interleukin
|
|
MCP-1
|
monocyte chemotactic protein-1
|
|
PPARγ
|
peroxisome proliferator-activated
receptor γ
|
|
C26
|
compound 26
|
References
|
1
|
Ross R: Atherosclerosis - an inflammatory
disease. N Engl J Med. 340:115–126. 1999. View Article : Google Scholar
|
|
2
|
Guzik TJ, Korbut R and Adamek-Guzik T:
Nitric oxide and superoxide in inflammation and immune regulation.
J Physiol Pharmacol. 54:469–487. 2003.PubMed/NCBI
|
|
3
|
Cai H: Hydrogen peroxide regulation of
endothelial function: origins, mechanisms, and consequences.
Cardiovasc Res. 68:26–36. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Madamanchi NR, Hakim ZS and Runge MS:
Oxidative stress in atherogenesis and arterial thrombosisthe
disconnect between cellular studies and clinical outcomes. J Thromb
Haemost. 3:254–267. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Forstermann U: Oxidative stress in
vascular disease: causes, defense mechanisms and potential
therapies. Nat Clin Pract Cardiovasc Med. 5:338–349. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Park JG and Oh GT: The role of peroxidases
in the pathogenesis of atherosclerosis. BMB Rep. 44:497–505. 2011.
View Article : Google Scholar
|
|
7
|
Cominacini L, Pasini AF, Garbin U, et al:
Oxidized low density lipoprotein (ox-LDL) binding to ox-LDL
receptor-1 in endothelial cells induces the activation of NF-κB
through an increased production of intracellular reactive oxygen
species. J Biol Chem. 275:12633–12638. 2000.
|
|
8
|
Miller YI, Viriyakosol S, Binder CJ,
Feramisco JR, Kirkland TN and Witztum JL: Minimally modified LDL
binds to CD14, induces macrophage spreading via TLR4/MD-2, and
inhibits phagocytosis of apoptotic cells. J Biol Chem.
278:1561–1568. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miller YI, Viriyakosol S, Worrall DS,
Boullier A, Butler S and Witztum JL: Toll-like receptor 4-dependent
and -independent cytokine secretion induced by minimally oxidized
low-density lipoprotein in macrophages. Arterioscler Thromb Vasc
Biol. 25:1213–1219. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bae YS, Lee JH, Choi SH, et al:
Macrophages generate reactive oxygen species in response to
minimally oxidized low-density lipoprotein: toll-like receptor 4-
and spleen tyrosine kinase-dependent activation of NADPH oxidase 2.
Circ Res. 104:210–218. 2009. View Article : Google Scholar
|
|
11
|
Forman HJ and Torres M: Reactive oxygen
species and cell signaling: respiratory burst in macrophage
signaling. Am J Respir Crit Care Med. 166:S4–S8. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Taniyama Y and Griendling KK: Reactive
oxygen species in the vasculature: molecular and cellular
mechanisms. Hypertension. 42:1075–1081. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
De Keulenaer GW, Ushio-Fukai M, Yin Q, et
al: Convergence of redox-sensitive and mitogen-activated protein
kinase signaling pathways in tumor necrosis factor-α-mediated
monocyte chemoattractant protein-1 induction in vascular smooth
muscle cells. Arterioscler Thromb Vasc Biol. 20:385–391.
2000.PubMed/NCBI
|
|
14
|
Hulsmans M and Holvoet P: The vicious
circle between oxidative stress and inflammation in
atherosclerosis. J Cell Mol Med. 14:70–78. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sury MD, Frese-Schaper M, Muhlemann MK, et
al: Evidence that N-acetylcysteine inhibits TNF-α-induced
cerebrovascular endothelin-1 upregulation via inhibition of
mitogen- and stress-activated protein kinase. Free Radic Biol Med.
41:1372–1383. 2006.
|
|
16
|
Yang WS, Lee JM, Han NJ, Kim YJ, Chang JW
and Park SK: Mycophenolic acid attenuates tumor necrosis
factor-α-induced endothelin-1 production in human aortic
endothelial cells. Atherosclerosis. 211:48–54. 2010.
|
|
17
|
Node K, Huo Y, Ruan X, et al:
Anti-inflammatory properties of cytochrome P450 epoxygenase-derived
eicosanoids. Science. 285:1276–1279. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fisslthaler B, Popp R, Kiss L, et al:
Cytochrome P450 2C is an EDHF synthase in coronary arteries.
Nature. 401:493–497. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu S, Chen W, Murphy E, et al: Molecular
cloning, expression, and functional significance of a cytochrome
P450 highly expressed in rat heart myocytes. J Biol Chem.
272:12551–12559. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zeldin DC, Foley J, Boyle JE, et al:
Predominant expression of an arachidonate epoxygenase in islets of
Langerhans cells in human and rat pancreas. Endocrinology.
138:1338–1346. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Michaelis UR, Fisslthaler B, Medhora M,
Harder D, Fleming I and Busse R: Cytochrome P450 2C9-derived
epoxyeicosatrienoic acids induce angiogenesis via cross-talk with
the epidermal growth factor receptor (EGFR). FASEB J. 17:770–772.
2003.PubMed/NCBI
|
|
22
|
Sun J, Sui X, Bradbury JA, Zeldin DC,
Conte MS and Liao JK: Inhibition of vascular smooth muscle cell
migration by cytochrome p450 epoxygenase-derived eicosanoids. Circ
Res. 90:1020–1027. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu L, Chen C, Gong W, et al:
Epoxyeicosatrienoic acids attenuate reactive oxygen species level,
mitochondrial dysfunction, caspase activation, and apoptosis in
carcinoma cells treated with arsenic trioxide. J Pharmacol Exp
Ther. 339:451–463. 2011. View Article : Google Scholar
|
|
24
|
Wray J and Bishop-Bailey D: Epoxygenases
and peroxisome proliferator-activated receptors in mammalian
vascular biology. Exp Physiol. 93:148–154. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu J, Chen C, Zeng HS and Wang DW: The
protective effects of cytochrome P450 gene on atherosclerosis
induced by high fat diet in different week old mice. Mol Cardiol
Chin. 11:37–41. 2011.
|
|
26
|
Sun J, Huang SH, Zhu YC, et al:
Anti-oxidative stress effects of Herba leonuri on ischemic rat
hearts. Life Sci. 76:3043–3056. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Simoncini T, Maffei S, Basta G, et al:
Estrogens and glucocorticoids inhibit endothelial vascular cell
adhesion molecule-1 expression by different transcriptional
mechanisms. Circ Res. 87:19–25. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu X, Pan L, Gong Q and Zhu Y: Leonurine
(SCM-198) improves cardiac recovery in rat during chronic
infarction. Eur J Pharmacol. 649:236–241. 2010. View Article : Google Scholar
|
|
29
|
Wilson SH, Best PJ, Edwards WD, et al:
Nuclear factor-κB immunoreactivity is present in human coronary
plaque and enhanced in patients with unstable angina pectoris.
Atherosclerosis. 160:147–153. 2002.
|
|
30
|
Collins T and Cybulsky MI: NF-κB: pivotal
mediator or innocent bystander in atherogenesis? J Clin Invest.
107:255–264. 2001.
|
|
31
|
Brasier AR: The nuclear
factor-κB-interleukin-6 signalling pathway mediating vascular
inflammation. Cardiovasc Res. 86:211–218. 2010.
|
|
32
|
Marx N, Duez H, Fruchart JC and Staels B:
Peroxisome proliferator-activated receptors and atherogenesis:
regulators of gene expression in vascular cells. Circ Res.
94:1168–1178. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ricote M, Li AC, Willson TM, Kelly CJ and
Glass CK: The peroxisome proliferator-activated receptor-γ is a
negative regulator of macrophage activation. Nature. 391:79–82.
1998.
|
|
34
|
Cipollone F, Fazia M, Iezzi A, et al:
Balance between PGD synthase and PGE synthase is a major
determinant of atherosclerotic plaque instability in humans.
Arterioscler Thromb Vasc Biol. 24:1259–1265. 2004. View Article : Google Scholar
|
|
35
|
Dehmer T, Heneka MT, Sastre M, Dichgans J
and Schulz JB: Protection by pioglitazone in the MPTP model of
Parkinson’s disease correlates with IκBα induction and block of NF
κB and iNOS activation. J Neurochem. 88:494–501. 2004.PubMed/NCBI
|
|
36
|
Castrillo A and Tontonoz P: PPARs in
atherosclerosis: the clot thickens. J Clin Invest. 114:1538–1540.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang H, Lin L, Jiang J, et al:
Up-regulation of endothelial nitric-oxide synthase by
endothelium-derived hyperpolarizing factor involves
mitogen-activated protein kinase and protein kinase C signaling
pathways. J Pharmacol Exp Ther. 307:753–764. 2003. View Article : Google Scholar
|
|
38
|
Lee PC, Ho IC and Lee TC: Oxidative stress
mediates sodium arsenite-induced expression of heme oxygenase-1,
monocyte chemoattractant protein-1, and interleukin-6 in vascular
smooth muscle cells. Toxicol Sci. 85:541–550. 2005. View Article : Google Scholar
|
|
39
|
Zhang T, Zhang X, Yu W, et al: Effects of
chemokine-like factor 1 on vascular smooth muscle cell migration
and proliferation in vascular inflammation. Atherosclerosis.
226:49–57. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Caglayan E, Romeo GR, Kappert K, et al:
Profilin-1 is expressed in human atherosclerotic plaques and
induces atherogenic effects on vascular smooth muscle cells. PLoS
One. 5:e136082010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nakagawa M, Ohno T, Maruyama R, et al:
Sesquiterpene lactone suppresses vascular smooth muscle cell
proliferation and migration via inhibition of cell cycle
progression. Biol Pharm Bull. 30:1754–1757. 2007. View Article : Google Scholar : PubMed/NCBI
|