Introduction
Reactive oxygen species (ROS) are a group of
molecules generated in the process of oxygen metabolism, among
which the endogenous stable oxidant, hydrogen peroxide
(H2O2), is considered as the principal ROS
member and has been the main focus of studies on ROS biology in
recent years (1). For many years
after its discovery, H2O2 was viewed as a
non-specific agent of destruction to human tissues (2); however, growing evidence over the
past few years suggests that H2O2 may act as
a ‘Jekyll and Hyde’ signaling molecule in cell proliferation,
migration, survival and death (1). At low concentrations,
H2O2 can act as a classical second messenger
with a pro-survival role by regulating kinase-driven pathways in
several physiological processes. At high concentrations,
H2O2 induces cellular injury by damaging key
cellular molecules, such as DNA and lipids, and by inducing
apoptosis, necrosis or autophagy. Several pharmacological agents
targeting H2O2 metabolism have been
demonstrated to have therapeutic potential in the treatment of
neurological disorders, ranging from acute insults, such as
ischemic and traumatic brain injury to chronic neurodegenerative
disorders, such as Alzheimer’s disease and Parkinson’s disease
(3,4).
Mitochondria are usually described as ‘cellular
power plants’ due to their ability to generate most of the chemical
energy of cells, namely adenosine triphosphate (ATP), and they can
also receive cellular signals and propagate a targeted response to
mediate several basic cellular functions (5). Due to their important role in
regulating cell metabolism and ROS generation, the damage and
ensuing dysfunction of the mitochondria in neurons has been
demonstrated to be a key factor in various types of oxidative
stress related to neurological diseases (6). Following oxidative stress, the
majority of the mitochondria develop varying degrees of swelling,
and several pro-apoptotic molecules are released or activated, such
as cytochrome c, caspase-9 and the pro-apoptotic Bcl-2
family of proteins (which includes Bax). A number of studies have
demonstrated that many pharmacological agents and
mitochondria-associated molecules exert protective effects against
neuronal injury through the preservation of mitochondrial function,
and this may be an ideal neuroprotective strategy (6,7).
The sirtuins (or Sir2-like proteins) are a conserved
family of nicotinamide adenine nucleotide
(NAD+)-dependent protein deacetylases, and have been
reported to be involved in transcriptional silencing, the genetic
control of aging and the longevity of organisms ranging from yeast
to humans (8). Among these
sirtuins, Sirt3 resides primarily in the mitochondria and serves as
a primary regulator of mitochondrial function and metabolism by
binding and deacetylating several metabolic and respiratory enzymes
(9). The increased expression of
Sirt3 protects cardiomyocytes against genotoxic and oxidative
stress-mediated cell death by hindering the translocation of Bax to
the mitochondria (10). A recent
study demonstrated that the Sirt3-mediated deacetylation of
forkhead box O3 (FOXO3) attenuates oxidative stress-induced
mitochondrial dysfunction through the coordination of mitochondrial
biogenesis, fission/fusion and mitophagy (11). However, the exact role of Sirt3 in
oxidative stress-induced neuronal cell injury has not yet been
fully elucidated. Therefore, the aim of the present study was to
investigate the effects of Sirt3 knockdown and its overexpression
in H2O2-induced neuronal injury in HT22
cells, as well as the potential mechanisms involved with focus on
mitochondrial oxidative phosphorylation, calcium metabolism and the
intrinsic apoptotic pathway.
Materials and methods
Cell culture
HT22 mouse hippocampal cells were obtained from the
Institute of Biochemistry and Cell Biology (IBCB), Shanghai
Institutes for Biological Sciences, Chinese Academy of Sciences
(CAS), Shanghai, China. The cells were grown in Dulbecco’s modified
Eagle’s medium (DMEM) plus 10% fetal bovine serum and 1%
antibiotics (penicillin/streptomycin) in a humidified incubator
with 5% CO2 and 95% air. The growth medium was removed
and replaced by medium containing H2O2 for
the induction of apoptosis.
Cell viability assay
Cell viability assay was performed using the Cell
Proliferation Reagent WST-1 following the manufacture’s
instructions (Roche, Basel, Switzerland). Briefly, the HT22 cells
were cultured at a concentration of 0.5–5×104 in
microplates in a final volume of 100 μl/well culture medium.
Following the various treatments, 10 μl of the cell proliferation
reagent, WST-1, were added to each well followed by incubation for
4 h at 37°C. Subsequently, 100 μl/well culture medium and 10 μl
WST-1 were added to one well in the absence of HT22 cells, and its
absorbance was used as a blank position for the ELISA reader. The
cells were shaken thoroughly for 1 min on a shaker and the
absorbance of the samples was measured using a microplate (ELISA)
reader (Bio-Rad Laboratories, Cambridge, MA, USA).
Lactate dehydrogenase (LDH) release
assay
Cytotoxicity was determined by the release of LDH, a
cytoplasmic enzyme released from cells, and a marker of membrane
integrity. The LDH release into the culture medium was detected
using a diagnostic kit according to the manufacturer’s instructions
(Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Briefly, 50 μl of supernatant from each well were collected to
measure the release of LDH. The samples were incubated with a
reduced form of nicotinamide adenine dinucleotide (NADH) and
pyruvate for 15 min at 37°C and the reaction was terminated by the
addition of 0.4 mol/l NaOH. The activity of LDH was calculated from
the absorbance at 440 nm and the background absorbance from the
culture medium that was not used for any cell cultures was
subtracted from all the absorbance measurements. The results were
normalized to the maximal LDH release, which was determined by
treating the control wells for 60 min with 1% Triton X-100 to lyse
all cells.
Small interfering RNA (siRNA) and
transfection
Specific siRNA targeting Sirt3 (Si-Sirt3, sc-61556)
and control siRNA (Si-Control, sc-37007), which should not knock
down any known proteins, were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). The above-mentioned
siRNA molecules were transfected into the cells using Lipofectamine
2000 (Invitrogen, Carlsbad, CA, USA) in 6-well plates for 48 h.
Following transfection, the HT22 cells were treated with
H2O2 (500 μM) for 24 h and subjected to
various measurements.
Lentivirus construction and
transfection
The coding sequence of Sirt3 was amplified by
RT-PCR. The primer sequences were as follows: forward,
5′-TACTTCCTTCGGCTGCTTCA-3′ and reverse, 5′-AAGGCGAAATCAGCCACA-3′.
The PCR fragments and the pGC-FU plasmid (Shanghai Genechem Co.,
Ltd., Shanghai, China) were digested with AgeI and then
ligated with T4 DNA ligase to produce pGC-FU-Sirt3. To generate the
recombinant lentivirus, LV-Sirt3, 293T cells were co-transfected
with the pGC-FU plasmid (20 μg) with a cDNA encoding Sirt3,
pHelper1.0 plasmid (15 μg) and pHelper 2.0 plasmid (10 μg) using
Lipofectamine 2000 (100 μl). The supernatant was harvested and the
viral titer was calculated by transducing 293T cells. As a control,
we also generated a control lentiviral vector that expresses GFP
alone (LV-Control). The HT22 cells were transfected with the
lentiviral vectors for 72 h and subjected to various
treatments.
Measurement of the NAD+/NADH
ratio
To investigate whether Sirt3 is enzymatically active
under our experimental conditions, measurement of the
NAD+/NADH ratio was performed using the
NAD+/NADH Quantification kit (BioVision, Milpitas, CA,
USA) according to the manufacturer’s instructions. Briefly,
5×105 HT22 cells seeded in 6-well plates were washed
with cold PBS, collected and centrifuged at 1,500 rpm for 5 min.
The cells were then lysed by 2 freeze/thaw cycles in NADH/NAD
extraction buffer and subsequently vortexed for 10 sec. The
cellular extracts were transferred into a 96-well plate in
duplicate, and incubated with NAD Cycling Mix for 5 min.
NAD+total was quantified by the addition of
NADH developer to each sample and by reading the plate at OD450 nm
for 30 min. To detect NADH, the extracted samples were incubated at
60°C for 30 min in order to decompose NAD+.
Subsequently, the samples were incubated with NAD Cycling Mix for 5
min and, after the addition of NADH developer, were read as
previously described (12). The
NAD+/NADH ratio was calculated as follows:
(NAD+total − NADH)/NADH.
Measurement of ROS generation
Briefly, the HT22 cells were incubated with
2,7-dichlorodihydrofluorescein diacetate (DCF-DA) (Sigma, St.
Louis, MO, USA) (10 μM) for 1 h at 37°C in the dark and then
resuspended in phosphate-buffered saline (PBS). Intracellular ROS
production was detected using the fluorescence intensity of the
oxidant-sensitive probe, H2DCF-DA, in a microscope and
fluorescence was read using an excitation wavelength of 480 nm and
an emission wavelength of 530 nm.
Measurement of lipid peroxidation
Malonyldialdehyde (MDA) and 4-hydroxynonenal
(4-HNE), 2 indexes of lipid peroxidation, were determined using
assay kits from Cell Biolabs (San Diego, CA, USA) strictly
following the manufacturer’s instructions. The absorbance of the
samples was measured using a microplate (ELISA) reader.
Detection of antioxidant enzyme
activity
The enzyme activity of superoxide dismutase (SOD),
catalase (CAT), glutathione peroxidase (GPx) and glutathione
S-transferase (GST) was determined using commercially available
assay kits following the manufacturer’s instructions (Cayman
Chemical Co., Mountain View, CA, USA). The protein concentration
was determined using the BCA protein kit (Nanjing Jiancheng
Bioengineering Institute). The enzyme activities were then
normalized to the corresponding protein concentration for each
sample.
Determination of mitochondrial
respiratory chain complex activity
The mitochondria were purified by Percoll density
gradient centrifugation in extraction buffer (50 mM Tris HCl, pH
7.5, 500 mM NaCl, 0.03% reduced Triton X-100, 1 mM EDTA, 1 mM PMSF,
0.5 mM benzamidine, and 1 mg/ml each of pepstatin-A, leupeptin and
aprotinin). All the samples were subjected to 3 freeze-thaw cycles
to disrupt the membranes and expose the enzymes before analysis.
The enzymatic activity was measured at 37°C spectrophotometrically
using the following methods: complex I (NADH dehydrogenase),
complex II (succinate dehydrogenase), complex III (ubiquinol
cytochrome c reductase) and complex IV (cytochrome c
oxidase), as previously described (13–15). The data were expressed as a
percentage of the control.
Measurement of ATP synthesis
Isolated mitochondria were utilized to measure ATP
synthesis with a luciferase/luciferin-based system as previously
described (16).
Mitochondria-enriched pellets (30 μg) were resuspended in 100 μl of
buffer A (150 mM KCl, 25 mM Tris-HCl, 2 mM potassium phosphate, 0.1
mM MgCl2, pH 7.4) with 0.1% BSA, 1 mM malate, 1 mM
glutamate and buffer B (containing 0.8 mM luciferin and 20 mg/ml
luciferase in 0.5 M Tris-acetate pH 7.75). The reaction was
initiated by the addition of 0.1 mM adenosine diphosphate (ADP) and
monitored for 5 min using a microplate reader (Bio-Rad
Laboratories).
Measurement of mitochondrial
swelling
Mitochondrial swelling was measured following a
previously published protocol (17). Briefly, the isolated mitochondria
were suspended in fresh swelling buffer (0.2 M sucrose, 10 mM
Tris-MOPS, pH 7.4, 5 mM succinate, 1 mM phosphate, 2 μM rotenone
and 1 μM EGTA-Tris, pH 7.4) at 0.5 mg/ml, and the swelling of the
mitochondria was monitored by a decrease in absorbance at 540 nm in
the presence of calcium chloride (CaCl2; 200 μM).
Measurement of mitochondrial calcium
buffering capacity
Mitochondrial calcium buffering capacity was
estimated using the Ca2+ sensitive Calcium Green 5N
fluorescent dye. The incubation medium was composed of 125 mM KCl,
20 mM HEPES (pH 7.2), 2 mM KH2PO4, 2 mM
MgCl2, 5 mM succinate, 1 μM rotenone and 0.2 mM ADP,
with 1 μg/ml oligomycin and 1 μM Calcium Green 5N. Bolus additions
of CaCl2 were made to the 60 μg of mitochondria in
suspension in 30 nM increments and changes in Calcium Green 5N
fluorescence were recorded at an emission of 532 nm.
Flow cytometry
The HT22 cells were harvested 24 h following
exposure to H2O2, washed with ice-cold
Ca2+ free PBS, and re-suspended in binding buffer. Cell
suspension was transferred into a tube and double-stained for 15
min with Alexa Fluor 488-conjugated Annexin V (AV) and propidium
iodide (PI) at room temperature in the dark. After the addition of
400 μl binding buffer, the stained cells were analyzed using an
FC500 flow cytometer (Beckman-Coulter, Brea, CA USA) with the
fluorescence emission at 530 nm and >575 nm. CXP cell quest
software (Beckman-Coulter) was used to count the number of
apoptotic cells (AV+/PI+, late phase
apoptotic cells and AV+/PI−, early phase
apoptotic cells) and analyzed the results.
Quantification of cytochrome c
release
Cytochrome c release into the cytoplasm was
assessed following subcellular fraction preparation. The HT22 cells
were washed with ice-cold PBS 3 times and lysed with lysis buffer
containing protease inhibitors. The cell lysate was centrifuged for
10 min at 750 × g at 4°C, and the pellets containing the nuclei and
unbroken cells were discarded. The supernatant was then centrifuged
at 15,000 × g for 15 min. The resulting supernatant was removed and
used as the cytosolic fraction. The pellet fraction containing the
mitochondria was further incubated with PBS containing 0.5% Trition
X-100 for 10 min at 4°C. Following centrifugation at 16,000 × g for
10 min, the supernatant was collected as the mitochondrial
fraction. The levels of cytochrome c in the cytosolic and
mitochondrial fractions were measured using the Quantikine M
Cytochrome C Immunoassay kit obtained from R&D Systems
(Minneapolis, MN, USA). Data were expressed as ng/mg protein.
Measurement of caspase-3 activity
The activity of caspase-3 was measured using the
colorimetric assay kit according to the manufacturer’s instructions
(Cell Signaling Technology, Danvers, MA, USA). Briefly, after being
harvested and lysed 106 cells were mixed with 32 μl of
assay buffer and 2 μl of 10 mM Ac-DEVD-pNA substrate. Absorbance at
405 nm was measured following incubation at 37°C for 4 h. The
absorbance of each sample was determined by subtraction of the mean
absorbance of the blank and corrected by the protein concentration
of the cell lysate. The results were described as the relative
activity to that of the control group.
Western blot analysis
Equivalent amounts of protein (40 μg per lane) were
loaded and separated by 10% SDS-PAGE gels, and transferred onto
polyvinylidene difluoride (PVDF) membranes. The membranes were
blocked with 5% non-fat milk solution in Tris-buffered saline with
0.1% Triton X-100 (TBST) for 1 h, and then incubated overnight at
4°C with primary Sirt3 antibody (1:1,000), Bax (1:800), Bcl-2
(1:800), cleaved caspase-9 (1:500) or caspase-9 (1:600) antibody
dilutions in TBST. Subsequently, the membranes were washed and
incubated with secondary antibody (anti-rabbit and anti-goat IgG;
Santa Cruz Biotechnology) for 1 h at room temperature.
Immunoreactivity was detected with Super Signal West Pico
chemiluminescent substrate (Thermo Fisher Scientific, Rockford, IL,
USA). The band densities were corrected for the β-actin signals.
ImageJ analysis software (Scion Corp.) was used to quantify the
optical density of each band.
Statistical analysis
Statistical analysis was performed using the SPSS
16.0, a statistical software package. Statistical evaluation of the
data was performed by one-way analysis of variance (ANOVA) followed
by Bonferroni’s multiple comparisons. A value of P<0.05 was
considered to indicate a statistically significant difference.
Results
Expression of Sirt3 following
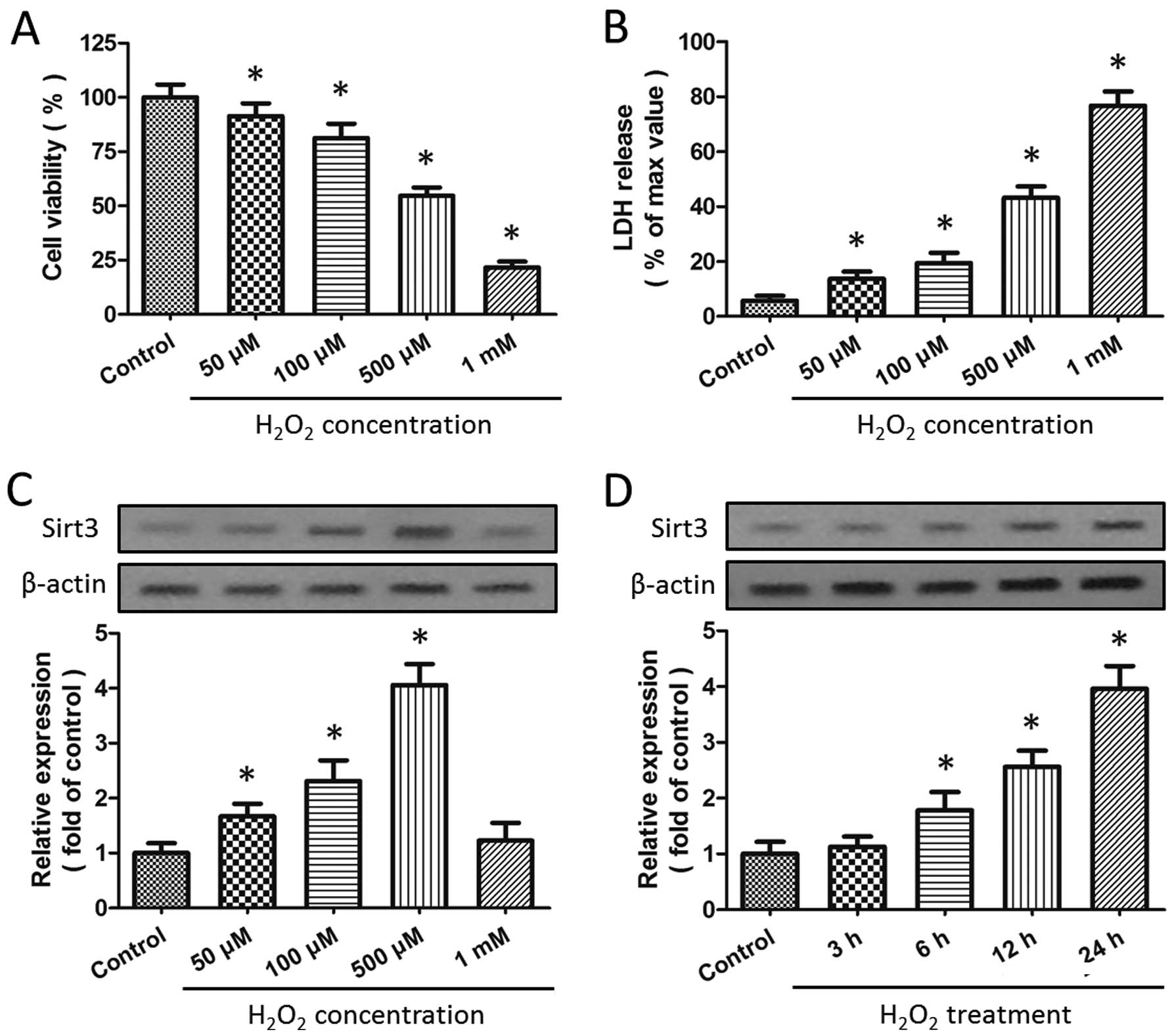
H2O2-induced injury in HT22 cells
The HT22 cells were incubated in the presence of
H2O2 at various concentrations (50, 100, 500
μM or 1 mM) for 24 h, and the cytotoxicity was determined by WST-1
assay and LDH release assay. The results revealed that incubation
with H2O2 significantly decreased cell
viability (Fig. 1A) and increased
LDH release (Fig. 1B) (both
P<0.05) in a dose-dependent manner. As the exposure of the cells
to 500 μM H2O2 caused almost half of the
cells to die, it was used in the subsequent experiments. Western
blot analysis was used to investigate the effects of
H2O2 insults on Sirt3 expression, and the
results revealed that incubation with H2O2
significantly increased the expression of Sirt3 in a dose-dependent
manner (P<0.05; Fig. 1C). As
shown in Fig. 1D, a
time-dependent increase in Sirt3 expression was also observed
following exposure to 500 μM H2O2.
H2O2-induced Sirt3
expression promotes HT22 cell survival
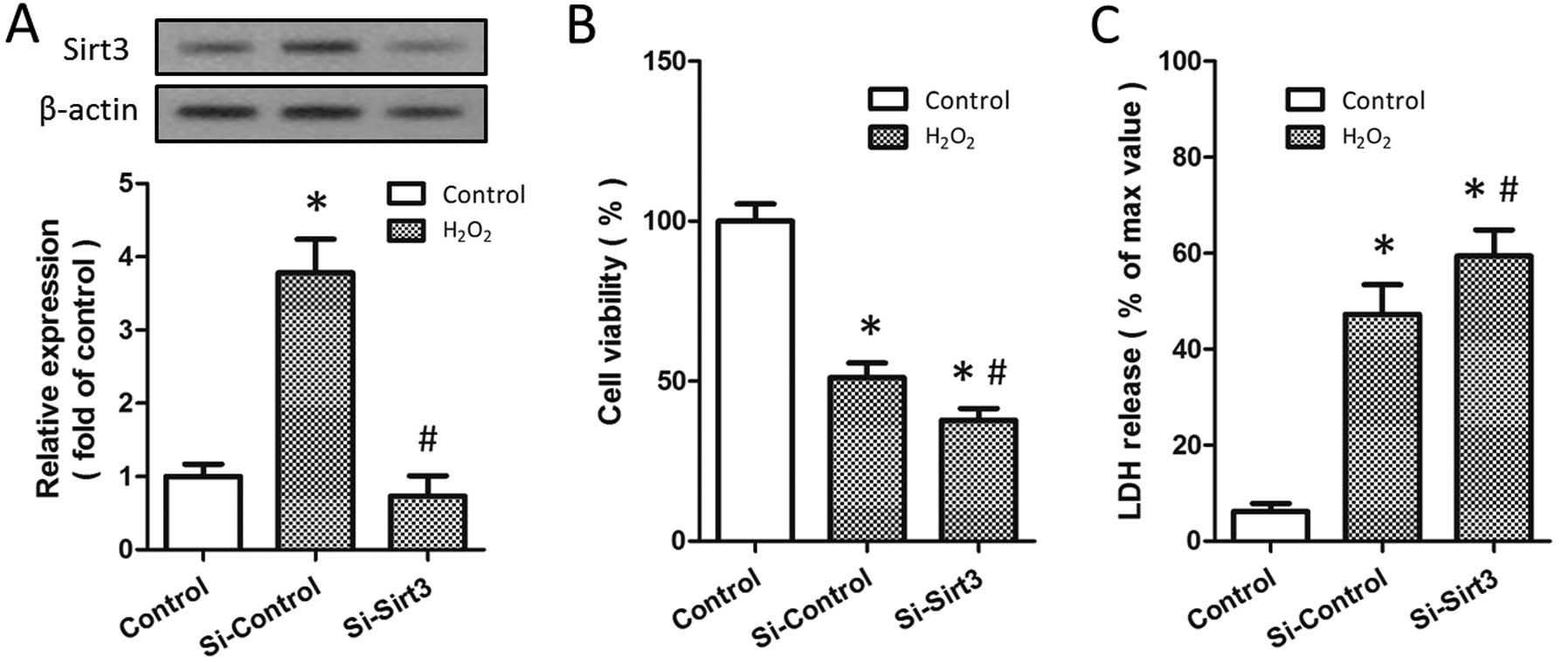
To investigate the biological functions of Sirt3 in
H2O2-induced neurotoxicity, the HT22 cells
were transfected with Sirt3-specific siRNA (Si-Sirt3) or control
siRNA (Si-Control). Western blot analysis indicated that Sirt3
expression was significantly reduced in the cells following
transfection with Si-Sirt3 (P<0.05; Fig. 2A). Following treatment with 500 μM
H2O2 for 24 h, the viability of the cells
transfected with Si-Sirt3 was lower than that of the cells
transfected with Si-Control (Fig.
2B). By contrast, the knockdown of Sirt3 further increased the
release of LDH induced by H2O2 treatment in
the HT22 cells (Fig. 2C). These
data suggest that the knockdown of Sirt3 aggravates
H2O2-induced neuronal injury and that
H2O2-induced Sirt3 expression may be an
endogenous protective mechanism.
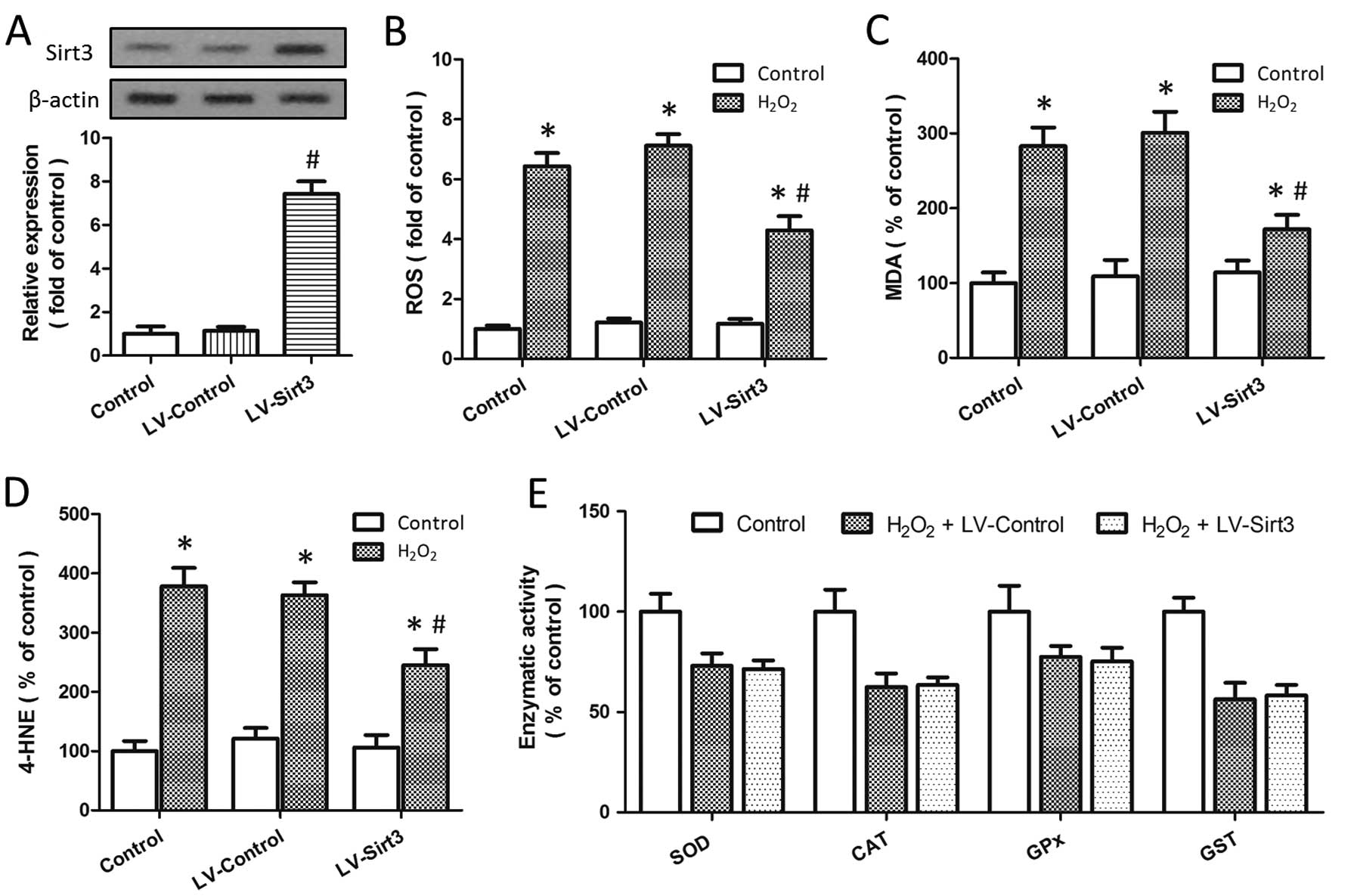
Overexpression of Sirt3 reduces ROS
generation and lipid peroxidation
To determine whether Sirt3 affects the generation of
intracellular ROS, the HT22 cells were transfected with lentivirus
expressing Sirt3 (LV-Sirt3) or a control lentivirus (LV-Control).
The results of western blot analysis indicated that the expression
of Sirt3 was significantly increased by transfection with LV-Sirt3
as compared to transfection with the LV-Control (P<0.05;
Fig. 3A). ROS generation induced
by H2O2 treatment was reduced by transfection
with LV-Sirt3, but not by transfection with LV-Control (Fig. 3B). We also measured the expression
levels of MDA and 4-HNE, 2 bioactive markers of lipid peroxidation,
following H2O2-induced injury, and the
results revealed that the knockdown of Sirt3 significantly
decreased the levels of MDA (Fig.
3C) and 4-HNE (Fig. 3D) (both
P<0.05). As shown in Fig. 3E,
treatment with H2O2 significantly inhibited
the enzymatic activity of SOD, CAT, GPx and GST; however, neither
LV-Sirt3 nor LV-Control had any effect on the enzymatic activity of
these endogenous antioxidant enzymes, indicating the presence of an
endogenous antioxidant system independent of the neuroprotective
mechanisms.
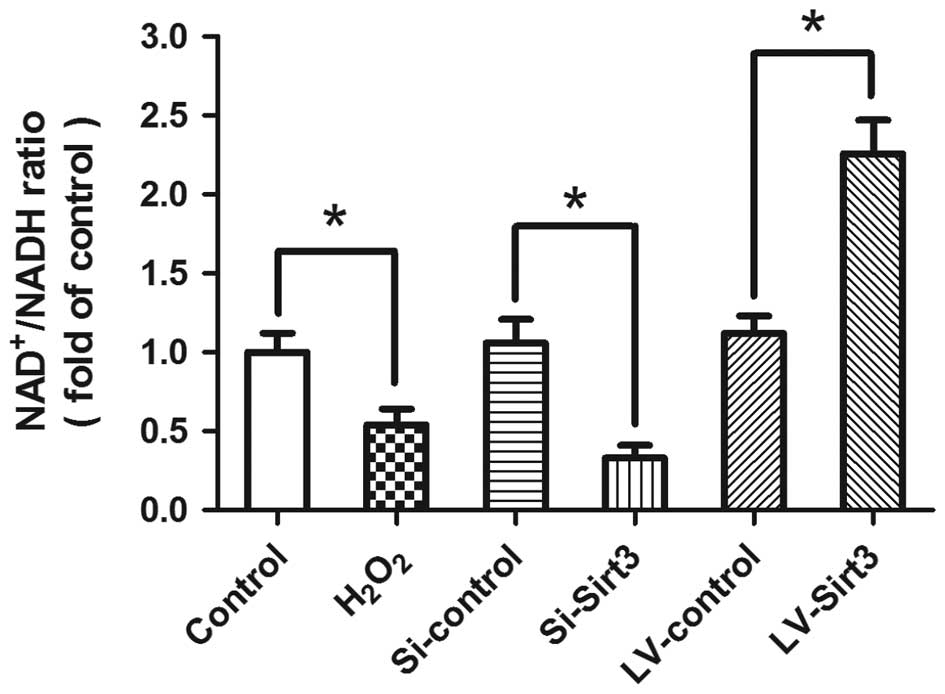
To assess whether Sirt3 is enzymatically active
under our experimental conditions, we detected the
NAD+/NADH ratio following transfection and/or
H2O2 treatment (Fig. 4). The results revealed that
treatment with H2O2 significantly decreased
the NAD+/NADH ratio compared with the control group,
indicating that the enzymatic activity of Sirt3 was inhibited by
oxidative stress in our in vitro model. The knockdown of
Sirt3 decreased the ratio of NAD+/NADH, whereas the
overexpression of Sirt3 by LV-Sirt3 transfection significantly
increased the NAD+/NADH ratio.
Overexpression of Sirt3 preserves
mitochondrial respiration and ATP production
The activity assays of electron transfer complexes
I–IV in the cell homogenates were performed to investigate the
effects of Sirt3 overexpression on mitochondrial respiration. The
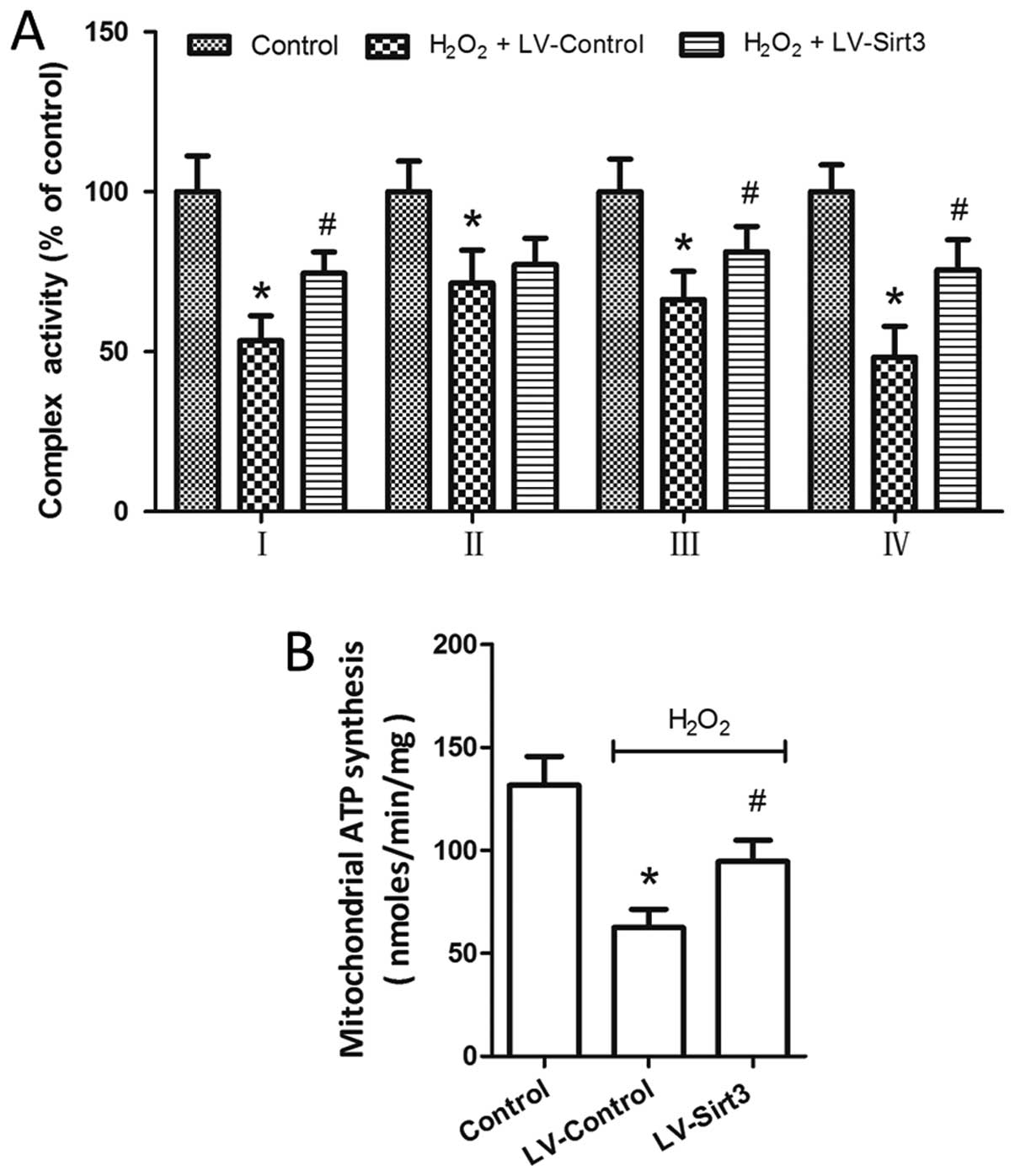
results revealed that the activity of complexes I–IV was markedly
inhibited by H2O2 treatment (Fig. 5A). The overexpression of Sirt3 by
LV-Sirt3 transfection significantly increased the activity of
complexes I, III and IV following H2O2
insult, whereas the activity of complex II was not affected by
Si-Sirt3 as compared to Si-Control (P>0.05). A dominant role for
the mitochondria is the production of ATP through the electron
transport chain. Thus, we measured ATP synthesis with a
luciferase/luciferin-based system following mitochondrial isolation
and purification. As shown in Fig.
5B, compared to the LV-Control-transfected cells, the
overexpression of Sirt3 in the HT22 cells reversed the decrease in
mitochondrial ATP production which was observed following treatment
with H2O2.
Overexpression of Sirt3 blocks
H2O2-induced mitochondrial dysfunction
To characterize the effects of Sirt3 on
mitochondrial calcium homeostasis, we examined the calcium
buffering capacity in isolated mitochondria following transfection
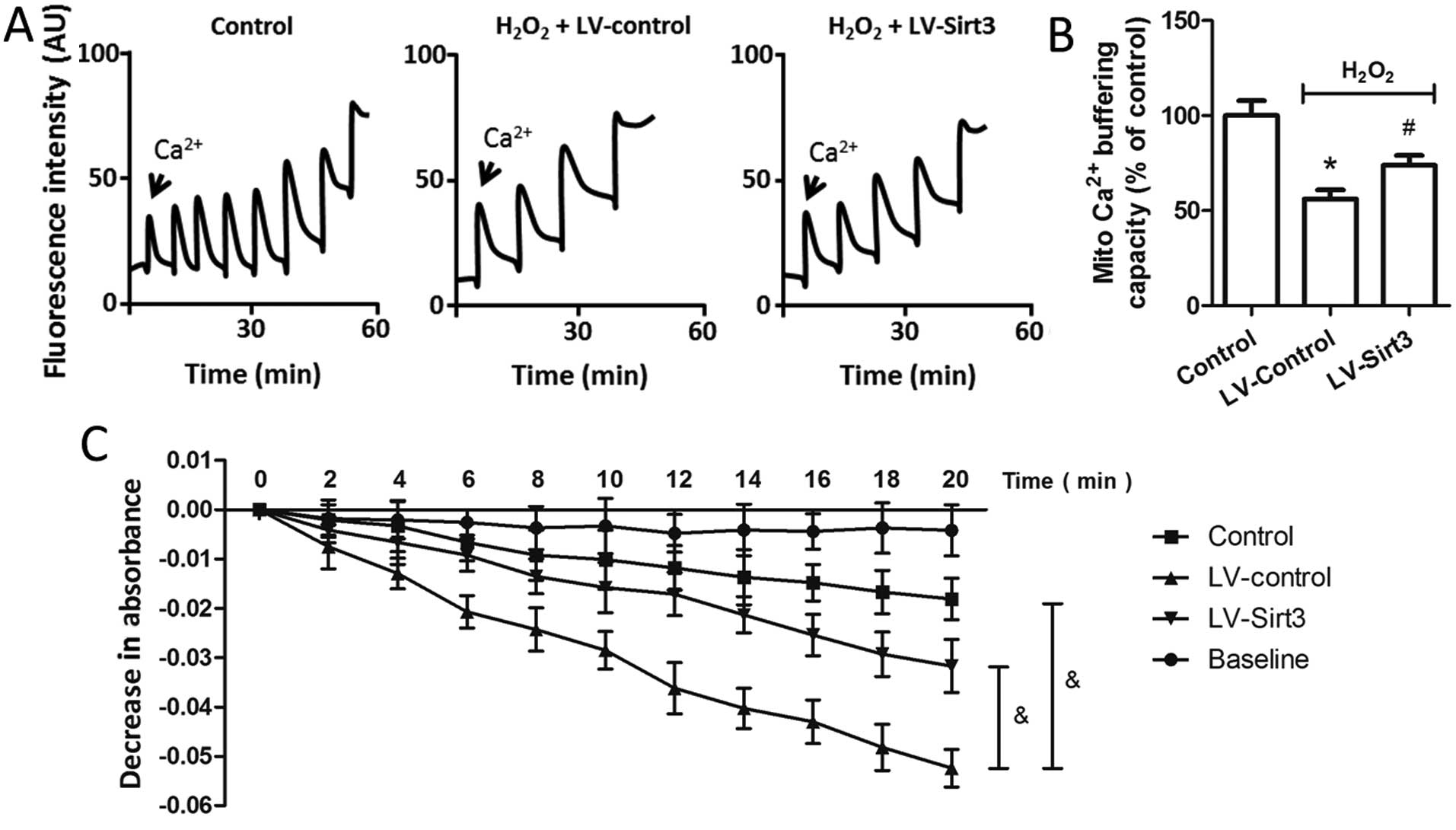
and treatment with H2O2. As shown in Fig. 6A, the peaks corresponded to
sequential bolus additions of 30 nM of Ca2+, and the
downward deflections reflected mitochondrial Ca2+
uptake. Treatment with H2O2 resulted in a
~50% reduction in Ca2+ buffering capacity in the
isolated mitochondria, whereas the overexpression of Sirt3
significantly preserved the Ca2+ buffering capacity
compared to that in the LV-Control-transfected cells (Fig. 6B). Mitochondrial swelling under
oxidative stress resulted in damage to the organelle, and is a
signature of mitochondrial dysfunction. Thus, we also examined the
effects of Sirt3 on mitochondrial swelling, which was induced by
the addition of 200 μM Ca2+ into isolated mitochondria
(Fig. 6C). The results revealed
that the decreased absorbance at 540 nm induced by
H2O2 treatment was partly prevented by
LV-Sirt3 transfection in the HT22 cells, indicating that Sirt3
overexpression attenuated mitochondrial swelling following neuronal
oxidative damage.
Overexpression of Sirt3 inhibits
mitochondrial-associated apoptosis
To determine the protective effects of Sirt3 against
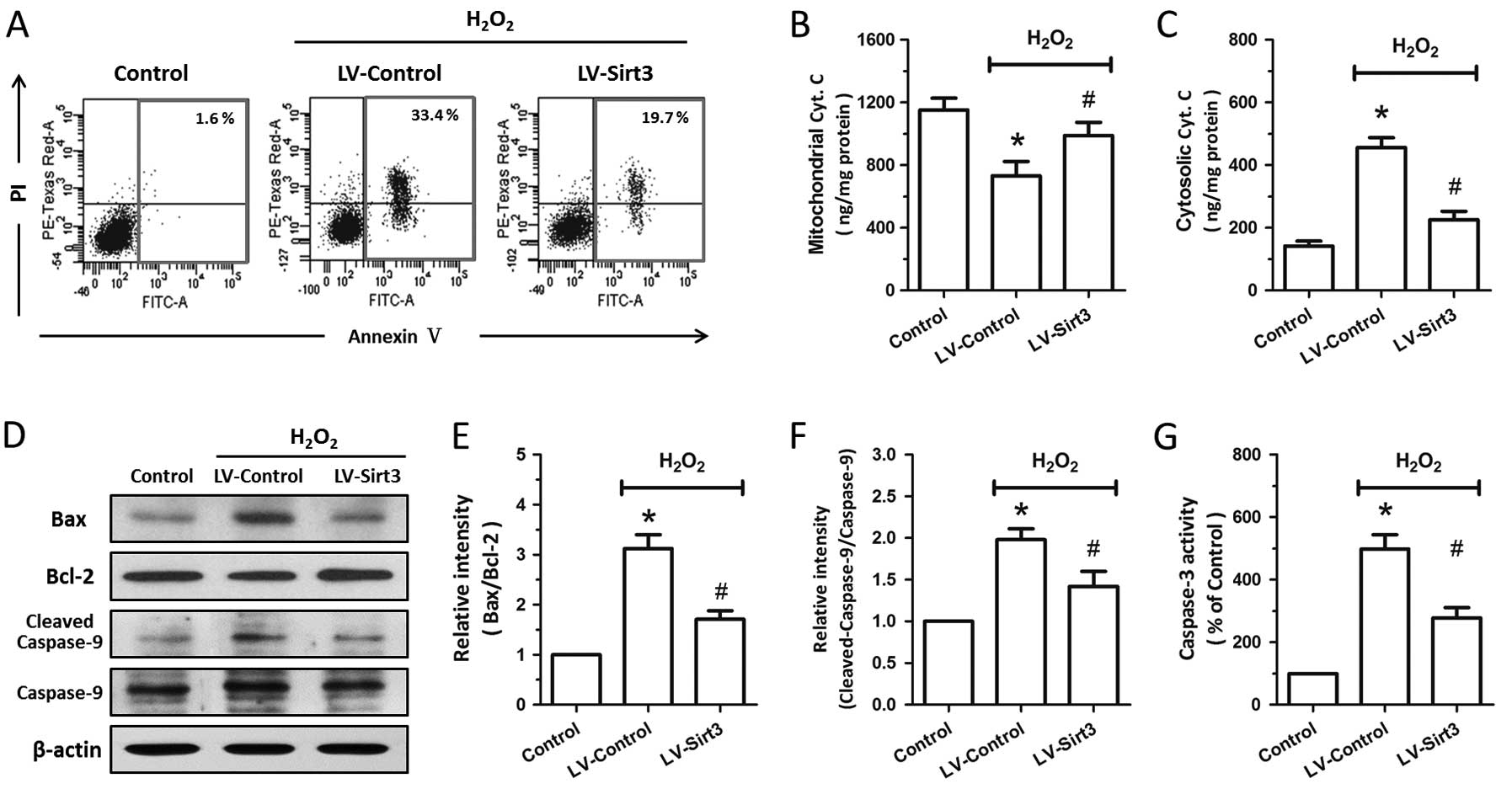
apoptotic neuronal death induced by oxidative stress, flow
cytometric analysis was performed at 24 h following treatment with
H2O2. As shown in Fig. 7A, treatment with
H2O2 resulted in apparent apoptotic cell
death (evidenced by AV+/PI+ and
AV+/PI− cells) in the HT22 cells, and SIRT3
overexpression markedly decreased the number of early apoptotic
cells and late apoptotic cells. We also measured the release of
cytochrome c into the cytoplasm by an immunoassay kit
following subcellular fraction preparation. The results revealed
that treatment with H2O2 induced a
significant decrease in mitochondrial cytochrome c and a
marked increase in cytosolic cytochrome c; these effects
were partly reversed by transfection with LV-Sirt3 (Fig. 7B and C). To further confirm the
anti-apoptotic activity of Sirt3, we examined the expression of
Bax, Bcl-2 and caspase-9 by western blot analysis (Fig. 7D). The Bax/Bcl-2 ratio and
cleaved-caspase-9/caspase-9 ratio significantly increased following
treatment with H2O2 (Fig. 7E and F), whereas Sirt3
overexpression exerted a significant inhibitory effect on the
H2O2-induced increase in the Bax/Bcl-2 ratio
and cleaved-caspase-9/caspase-9 ratio. The results of caspase-3
activity assay indicated that Sirt3 overexpression also attenuated
the activation of caspase-3 induced by treatment with
H2O2 (Fig.
7G).
Discussion
Mitochondrial proteins are acetylated at a high
frequency (approximately 20%), and acetylation is an important
mechanism for the regulation of mitochondrial function through the
modulation of protein-protein interactions, altering complex
stability or affecting enzyme activities (18). Among the 3 members of the sirtuin
family localized in the mitochondria (Sirt3, Sirt4 and Sirt5),
which are referred to as mitochondrial stress sensors (19), only Sirt3 has been demonstrated to
robustly deacetylate mitochondrial proteins (20). The increased expression of Sirt3
has been shown to be associated with the extended lifespan of
humans, and the Sirt3-mediated reprogramming of cellular metabolism
may be of particular importance under oxidative stress conditions
(21,22). As previously demonstrated, cells
in organs with high ATP demands (such as the heart, liver and
brain) present higher levels of Sirt3 expression (23), which was also found in our in
vitro neuronal model. The increased expression of Sirt3 has
also been observed in the heart under stress conditions, whereas
Sirt3 levels are reduced in hypertrophied or failing hearts
(24,25). In the present study, we found that
the expression of Sirt3 was increased following treatment with
H2O2 in a dose- and time-dependent manner,
and the knockdown of Sirt3 by transfection with specific siRNA
aggravated the H2O2-induced neuronal injury.
These data indicate that Sirt3 may be an endogenous protective
mechanism under oxidative stress conditions, and that the
overexpression of Sirt3 may provide an incremental protective
effect against H2O2 injury.
H2O2 is often considered to be
a toxic molecule since it was discovered by Thénard in 1818. It has
been implicated in several neuropathological conditions, such as
brain trauma, cerebral ischemia and neurodegenerative diseases
(3,4). In previous studies, a marked and
rapid increase in H2O2 levels was recorded in
the reperfusion phase following transient brain ischemia (26), and the concentrations of
H2O2 in rat vascular smooth muscle cells
following ischemia and reperfusion injury were higher than 1 mM
(27).
H2O2-induced oxidative stress and ROS
generation contributes to cell death by the oxidation of several
important lipids, proteins and nucleic acids, which further damage
mitochondrial membrane integrity and inhibit energy production. In
the present study, we found that the overexpression of Sirt3
attenuated the H2O2-induced ROS generation
and lipid peroxidation, suggesting that Sirt3 activates the
endogenous antioxidant system through the recruitment of
antioxidants, such as SOD, CAT, GPx and GST, thereby attenuating
H2O2-induced neuronal injury. To confirm this
assumption, we also measured the enzymatic activity of SOD, CAT,
GPx and GST following H2O2-induced injury in
cells with or without Sirt3 overexpression. SOD, in its 3 isoforms
(cytosolic Cu, Zn-SOD, extracellular Cu, Zn-SOD and mitochondrial
Mg-SOD), is responsible for H2O2 production
from •O2− (28), and H2O2 is
subsequently converted to H2O by GPx or decomposed in
peroxisomes to H2O and O2 by CAT (29,30). Accumulating evidence has suggested
that the elevation of these endogenous antioxidants plays a
protective role against H2O2-induced
oxidative stress. Intriguingly, our results demonstrated that the
overexpression of Sirt3 had no effect on the decreased enzymatic
activity of SOD, CAT, GPx and GST induced by
H2O2 insults, indicating the presence of an
endogenous antioxidant system which is independent of the
protective mechanisms of Sirt3 overexpression; this requires
further investigation.
The electron transport chain (ETC) plays important
roles in oxidative stress, not only due to its function in creating
a transmembrane proton gradient, which is required for the
generation of ATP through protons, but also as the dysregulated ECT
function results in electron leak and increased ROS production
through a series of protein complexes (I–IV) (9). Sirt3 physically interacts with the
39-kDa protein, NADH dehydrogenase (ubiquinone) 1 alpha subcomplex
9 (NDUFA9), one of the known subunits of complex I, and regulates
the complex I acetylation level and activity (23). Both the increased expression of
Sirt3 and complex II activity have been observed in K562 cell lines
following treatment with kaempferol (31). Recently, a 56-kDa core I subunit
of complex III was identified as a potential target of Sirt3 by
immunoprecipitation with anti-acetyl-lysine antibody from
mitochondrial lysates (32). Our
results revealed that the overexpression of Sirt3 preserved the
decreased activity of mitochondrial complex I, II and IV, and
increased mitochondrial ATP synthesis following
H2O2-induced injury. It is conceivable that
the upregulation of Sirt3 preserves ATP generation in injured
mitochondria to meet the cellular energy requirements, and this
action of Sirt3 on mitochondrial energy metabolism may contribute
to its neuroprotective effects against
H2O2-induced oxidative injury.
Even though Ca2+ is well known as a
crucial second messenger regulating cellular physiological
functions, there are two opposite sides to the effects of
Ca2+ on mitochondrial functions: it may either be
beneficial for mitochondrial function in the processes of oxidative
phosphorylation and ATP synthesis or it may be detrimental in
instigating subsequent pathological cascades (33). Although the endoplasmic reticulum
(ER) is considered to be the main site of intracellular
Ca2+ storage, the mitochondria also serve as an
important intracellular calcium buffer shaping Ca2+
signaling (34). Recent evidence
has validated that mitochondrial Ca2+ loading plays
crucial roles in apoptotic cell death under oxidative stress,
possibly through nitric oxide production, cytochrome c
dissociation, mitochondrial permeability transition pore (mPTP)
opening with the release of cytochrome c and
Ca2+-calmodulin dependent protein kinase activation
(35). A previous study
demonstrated that when exposed to Ca2+ under normoxic
conditions, mitochondria isolated from rat cerebral cortex generate
reactive hydroxyl (•OH), even in the absence of respiratory chain
inhibitors (36). In the present
study, we found that treatment with H2O2
resulted in a ~50% reduction in Ca2+ buffering capacity
in the isolated mitochondria, whereas the overexpression of Sirt3
significantly preserved the Ca2+ buffering capacity
compared to that in the LV-Control-transfected cells. Considering
the present data that the Ca2+ buffering capacity in the
mitochondria decreases with the process of mitochondrial
Ca2+ loading and the subcellular localization of Sirt3
in the mitochondria (37,38), a hypothetical molecular mechanism
explaining the relation between Sirt3 and the mitochondrial
Ca2+ flux is that Sirt3 is involved in the
Ca2+ transfer from the cytoplasm to the mitochondria
through the regulation of mitochondrial calcium uniporters (mCU),
such as uncoupling proteins (UCPs) and voltage-dependent anion
channels (VDAC) (39). These data
suggest that Sirt3 acts as a mitochondrial Ca2+
regulator through its interactions with other mitochondrial
proteins; this requires confirmation in in vitro neuronal
models.
The mitochondrion plays a prominent role in the
induction of apoptotic cell death following oxidative stress. The
mitochondria-associated intrinsic apoptotic pathway is mediated by
the interplay between anti-apoptotic and pro-apoptotic Bcl-2 family
proteins, which is initiated by the translocation of the
pro-apoptotic protein, Bax, from the cytoplasm to the mitochondria
(40). The disruption of
mitochondrial membrane integrity and the opening of mPTP result in
the release of cytochrome c, which further activates
caspase-9 and in turn causes the cleavage of caspase-3 (40,41). Previous studies have indicated
that under caloric restriction (CR) conditions, the upregulated
expression of Sirt3 directly deacetylates cyclophilin D (CypD),
preventing its association with the adenine nucleotide translocator
(ANT) and therefore blocking mPTP formation (42,43). SIRT3 is required for the
regulation of cytochrome c superoxide-scavenging capacity
through deacetylation and the activation of complex IV (44). However, the exact role of Sirt3 in
apoptosis is contradictory, largely depending upon cell types.
Previous studies have demonstrated the pro-apoptotic effects of
Sirt3 in leucocythemia and colorectal cancer cells (45,46), whereas the anti-apoptotic activity
of Sirt3 and the Sirt3-mediated anti-apoptotic mechanisms have been
reported by several other studies (10,47,48). Recent observations also hint at
additional neuroprotective effects of SIRT3, involving the
regulation of mitochondrial dynamics (9,49,50). In the present study, we found that
the overexpression of Sirt3 significantly inhibited cytochrome
c release, the increase in the Bax/Bcl-2 ratio, as well as
caspase-3 and caspase-9 activation, and attenuated neuronal
apoptosis following treatment with H2O2.
These data indicate that the protective effects of Sirt3 in
oxidative stress may partly be mediated by mitochondrial-associated
apoptosis, and the anti-apoptotic activity of Sirt3 following
treatment with H2O2 was confirmed in our
in vitro neuronal injury model.
In conclusion, our results demonstrate that Sirt3
not only reduces H2O2-induced ROS
overgeneration and lipid peroxidation, but also attenuates the
mitochondrial dysfunction and subsequent activation of apoptosis.
The increased expression of Sirt3 induced by oxidative stress may
be an endogenous protective mechanism, which is partly dependent on
the preservation of mitochondrial calcium homeostasis. Thus,
metabolic rescue observed upon the overexpression of Sirt3 may
represent an appropriate strategy to avoid neuronal death in a
broad range of neuronal disorders, where
H2O2-related oxidative stress may play a
major role, such as in cerebral ischemia-reperfusion injury and
neurodegenerative diseases.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (nos. 81071034,
81371447, 81301037, 81200949 and 30930093), research funds from the
government (nos. AWS11J008 and 2012BAI11B02) and the Program for
Changjiang Scholars and Innovative Research Team in University (no.
IRT1053).
References
|
1
|
Gough DR and Cotter TG: Hydrogen peroxide:
a Jekyll and Hyde signalling molecule. Cell Death Dis. 2:e2132011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Plaine HL: The effect of oxygen and of
hydrogen peroxide on the action of a specific gene and on tumor
induction in drosophila melanogaster. Genetics. 40:268–280.
1955.
|
|
3
|
Halliwell B, Clement MV and Long LH:
Hydrogen peroxide in the human body. FEBS Lett. 486:10–13. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Armogida M, Nistico R and Mercuri NB:
Therapeutic potential of targeting hydrogen peroxide metabolism in
the treatment of brain ischaemia. Br J Pharmacol. 166:1211–1224.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McBride HM, Neuspiel M and Wasiak S:
Mitochondria: more than just a powerhouse. Curr Biol. 16:R551–R560.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chan PH: Mitochondria and neuronal
death/survival signaling pathways in cerebral ischemia. Neurochem
Res. 29:1943–1949. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ye R, Yang Q, Kong X, et al: Ginsenoside
Rd attenuates early oxidative damage and sequential inflammatory
response after transient focal ischemia in rats. Neurochem Int.
58:391–398. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Michan S and Sinclair D: Sirtuins in
mammals: insights into their biological function. Biochem J.
404:1–13. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bause AS and Haigis MC: SIRT3 regulation
of mitochondrial oxidative stress. Exp Gerontol. 48:634–639. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sundaresan NR, Samant SA, Pillai VB,
Rajamohan SB and Gupta MP: SIRT3 is a stress-responsive deacetylase
in cardiomyocytes that protects cells from stress-mediated cell
death by deacetylation of Ku70. Mol Cell Biol. 28:6384–6401. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tseng AH, Shieh SS and Wang DL: SIRT3
deacetylates FOXO3 to protect mitochondria against oxidative
damage. Free Radic Biol Med. 63:222–234. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
D’Aquila P, Rose G, Panno ML, Passarino G
and Bellizzi D: SIRT3 gene expression: a link between
inherited mitochondrial DNA variants and oxidative stress. Gene.
497:323–329. 2012.
|
|
13
|
Estornell E, Fato R, Pallotti F and Lenaz
G: Assay conditions for the mitochondrial NADH:coenzyme Q
oxidoreductase. FEBS Lett. 332:127–131. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dave KR, DeFazio RA, Raval AP, et al:
Ischemic preconditioning targets the respiration of synaptic
mitochondria via protein kinase C epsilon. J Neurosci.
28:4172–4182. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Krahenbuhl S, Chang M, Brass EP and Hoppel
CL: Decreased activities of ubiquinol:ferricytochrome c
oxidoreductase (complex III) and ferrocytochrome c:oxygen
oxidoreductase (complex IV) in liver mitochondria from rats with
hydroxycobalamin[c-lactam]-induced methylmalonic aciduria. J Biol
Chem. 266:20998–21003. 1991.PubMed/NCBI
|
|
16
|
Parone PA, Da Cruz S, Han JS, et al:
Enhancing mitochondrial calcium buffering capacity reduces
aggregation of misfolded SOD1 and motor neuron cell death without
extending survival in mouse models of inherited amyotrophic lateral
sclerosis. J Neurosci. 33:4657–4671. 2013. View Article : Google Scholar
|
|
17
|
Yu Z, Liu N, Li Y, Xu J and Wang X:
Neuroglobin overexpression inhibits oxygen-glucose
deprivation-induced mitochondrial permeability transition pore
opening in primary cultured mouse cortical neurons. Neurobiol Dis.
56:95–103. 2013. View Article : Google Scholar
|
|
18
|
Kim SC, Sprung R, Chen Y, et al: Substrate
and functional diversity of lysine acetylation revealed by a
proteomics survey. Mol Cell. 23:607–618. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Huang JY, Hirschey MD, Shimazu T, Ho L and
Verdin E: Mitochondrial sirtuins. Biochim Biophys Acta.
1804:1645–1651. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Verdin E, Hirschey MD, Finley LW and
Haigis MC: Sirtuin regulation of mitochondria: energy production,
apoptosis, and signaling. Trends Biochem Sci. 35:669–675. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bellizzi D, Rose G, Cavalcante P, et al: A
novel VNTR enhancer within the SIRT3 gene, a human homologue
of SIR2, is associated with survival at oldest ages.
Genomics. 85:258–263. 2005.PubMed/NCBI
|
|
22
|
Rose G, Dato S, Altomare K, et al:
Variability of the SIRT3 gene, human silent information
regulator Sir2 homologue, and survivorship in the elderly.
Exp Gerontol. 38:1065–1070. 2003.
|
|
23
|
Ahn BH, Kim HS, Song S, et al: A role for
the mitochondrial deacetylase Sirt3 in regulating energy
homeostasis. Proc Natl Acad Sci USA. 105:14447–14452. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pillai VB, Sundaresan NR, Kim G, et al:
Exogenous NAD blocks cardiac hypertrophic response via activation
of the SIRT3-LKB1-AMP-activated kinase pathway. J Biol Chem.
285:3133–3144. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sundaresan NR, Gupta M, Kim G, Rajamohan
SB, Isbatan A and Gupta MP: Sirt3 blocks the cardiac hypertrophic
response by augmenting Foxo3a-dependent antioxidant defense
mechanisms in mice. J Clin Invest. 119:2758–2771. 2009.PubMed/NCBI
|
|
26
|
Lei B, Adachi N and Arai T: Measurement of
the extracellular H2O2 in the brain by
microdialysis. Brain Res Brain Res Protoc. 3:33–36. 1998.
View Article : Google Scholar
|
|
27
|
Sundaresan M, Yu ZX, Ferrans VJ, Irani K
and Finkel T: Requirement for generation of
H2O2 for platelet-derived growth factor
signal transduction. Science. 270:296–299. 1995.
|
|
28
|
Fridovich I: Superoxide radical and
superoxide dismutases. Annu Rev Biochem. 64:97–112. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dringen R: Oxidative and antioxidative
potential of brain microglial cells. Antioxid Redox Signal.
7:1223–1233. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dringen R, Pawlowski PG and Hirrlinger J:
Peroxide detoxification by brain cells. J Neurosci Res. 79:157–165.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cimen H, Han MJ, Yang Y, Tong Q, Koc H and
Koc EC: Regulation of succinate dehydrogenase activity by SIRT3 in
mammalian mitochondria. Biochemistry. 49:304–311. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jing E, Emanuelli B, Hirschey MD, et al:
Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin
signaling via altered mitochondrial oxidation and reactive oxygen
species production. Proc Natl Acad Sci USA. 108:14608–14613. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brookes PS, Yoon Y, Robotham JL, Anders MW
and Sheu SS: Calcium, ATP, and ROS: a mitochondrial love-hate
triangle. Am J Physiol Cell Physiol. 287:C817–C833. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rizzuto R, De Stefani D, Raffaello A and
Mammucari C: Mitochondria as sensors and regulators of calcium
signalling. Nat Rev Mol Cell Biol. 13:566–578. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Peng TI and Jou MJ: Oxidative stress
caused by mitochondrial calcium overload. Ann NY Acad Sci.
1201:183–188. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dykens JA: Isolated cerebral and
cerebellar mitochondria produce free radicals when exposed to
elevated CA2+ and Na+: implications for
neurodegeneration. J Neurochem. 63:584–591. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Contreras L, Drago I, Zampese E and Pozzan
T: Mitochondria: the calcium connection. Biochim Biophys Acta.
1797:607–618. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Giralt A and Villarroya F: SIRT3, a
pivotal actor in mitochondrial functions: metabolism, cell death
and aging. Biochem J. 444:1–10. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shi T, Wang F, Stieren E and Tong Q:
SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial
function and thermogenesis in brown adipocytes. J Biol Chem.
280:13560–13567. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Del Gaizo Moore V and Letai A: BH3
profiling - measuring integrated function of the mitochondrial
apoptotic pathway to predict cell fate decisions. Cancer Lett.
332:202–205. 2013.PubMed/NCBI
|
|
41
|
Hacker G and Paschen SA: Therapeutic
targets in the mitochondrial apoptotic pathway. Expert Opin Ther
Targets. 11:515–526. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hafner AV, Dai J, Gomes AP, et al:
Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at
lysine 166 suppresses age-related cardiac hypertrophy. Aging.
2:914–923. 2010.PubMed/NCBI
|
|
43
|
Shulga N, Wilson-Smith R and Pastorino JG:
Sirtuin-3 deacetylation of cyclophilin D induces dissociation of
hexokinase II from the mitochondria. J Cell Sci. 123:894–902. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kong X, Wang R, Xue Y, et al: Sirtuin 3, a
new target of PGC-1alpha, plays an important role in the
suppression of ROS and mitochondrial biogenesis. PloS One.
5:e117072010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Marfe G, Tafani M, Indelicato M, et al:
Kaempferol induces apoptosis in two different cell lines via Akt
inactivation, Bax and SIRT3 activation, and mitochondrial
dysfunction. J Cell Biochem. 106:643–650. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Allison SJ and Milner J: SIRT3 is
pro-apoptotic and participates in distinct basal apoptotic
pathways. Cell Cycle. 6:2669–2677. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yang H, Yang T, Baur JA, et al:
Nutrient-sensitive mitochondrial NAD+ levels dictate
cell survival. Cell. 130:1095–1107. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kim SH, Lu HF and Alano CC: Neuronal Sirt3
protects against excitotoxic injury in mouse cortical neuron
culture. PloS One. 6:e147312011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kincaid B and Bossy-Wetzel E: Forever
young: SIRT3 a shield against mitochondrial meltdown, aging, and
neurodegeneration. Front Aging Neurosci. 5:482013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Rato L, Duarte AI, Tomas GD, et al:
Pre-diabetes alters testicular PGC1-alpha/SIRT3 axis modulating
mitochondrial bioenergetics and oxidative stress. Biochim Biophys
Acta. 1837:335–344. 2014. View Article : Google Scholar : PubMed/NCBI
|