Introduction
The pathogenesis of Alzheimer’s disease (AD) is
multi-aetiological, and it is likely due to these various origins
that AD induces diverse neuropathological changes. The most
prominent lesions in brains with AD are atrophy, a large number of
senile plaques (SPs) formed by amyloid β (Aβ) between neurons and
neurofibrillary tangles (NFTs) made of abnormally
hyperphosphorylated tau protein in neurons (1). Several other mechanisms have also
been proposed to explain the pathogenesis of AD. Although each of
these mechanisms may contribute to the pathogenesis of the disease,
the extent to which they drive the neurodegenerative process is
uncertain (2). This is further
complicated by the fact that two different pathways are detected in
neurons, i.e., neurodegeneration and neurofibrillary degeneration.
Neurodegeneration incorporates different abnormal signalling
pathways that lead to neuronal loss, including apoptosis and other
modes of cell death. Neurofibrillary degeneration is a specific
type of neuronal reaction marked by the accumulation of
hyperphosphorylated tau protein as paired helical filaments in
NFTs, which can also induce abnormal neuronal metabolism and death.
Of note, these two pathways share mTOR-dependent signalling as a
common key regulator (1).
The mammalian or mechanistic target of rapamycin
(mTOR) is a Ser/Thr protein kinase that functions as an adenosine
triphosphate (ATP) and amino acid sensor to balance nutrient
availability and cell growth (3,4).
mTOR is capable of forming two complexes named mTORC1 and mTORC2
(5). The rapamycin-sensitive
mTORC1 contains the following proteins: raptor, GβL (also known as
mLST8) and the proline-rich Akt substrate of 40 kDa (PRAS40). The
mTORC2 complex contains rictor, mamalian stress-activated protein
kinase (SAPK)-interacting protein 1 (mSIN1), Protor-1 and GβL5
(5). A number of recent studies
have found a strong link between mTOR and AD. For example, mTOR is
critical for long-lasting forms of synaptic plasticity and
long-term memory (LTM) formation, which is impaired in mouse models
of AD (6). The importance of mTOR
in synaptic plasticity is in agreement with the central role of
mTOR in controlling transcriptional events, since de novo
protein synthesis is involved in these long-lasting forms of
synaptic plasticity and LTM (7).
The inhibition of the mTOR pathway appears to modulate the process
of aging, a well-established risk factor for AD (8,9).
Moreover, autophagy, a pathway for organelle and protein turnover,
has been implicated in neurodegeneration. Autophagy is
constitutively active and highly efficient in healthy neurons and
rapamycin (an mTOR inhibitor) is able to induce autophagy (10). Finally, mTOR signalling has been
shown to be altered in models of AD (11,12). mTOR signalling has been shown to
be inhibited both in cultured neurons and hippocampal slices from
AD transgenic mice and in wild-type (WT) hippocampal slices exposed
to exogenous Aβ1–42, and this mTOR dysregulation
correlates with impairment in synaptic plasticity (13).
Recently, a novel endogenous inhibitor of the mTOR
pathway, termed DEP domain-containing mTOR-interacting protein
(DEPTOR), has been shown to bind to both the mTORC1 and mTORC2
complexes (14). Its precise
function has not yet been fully elucidated; however, knocking down
DEPTOR leads to the activation of signalling through mTORC1 and
mTORC2. This is demonstrated both by the observation that there is
a change in the phosphorylation status of S6 kinase 1 (S6K1) and
protein kinase B (PKB) when DEPTOR levels are decreased (by
RNA-based interference) and by the increased in vitro
activity against these substrates of mTOR complexes from cells with
decreased levels of DEPTOR (14,15). Moreover, DEPTOR has been shown to
be downregulated in malignancies of the prostate, bladder, head and
neck, cervix and thyroid (14),
whereas we have previously demonstrated the significant
upregulation of DEPTOR in two paclitaxel-resistant ovarian cancer
cell lines when compared to the parental ones (16). Evidence of the potential
involvement of DEPTOR in AD arises from studies in which
resveratrol (RSV), a naturally occurring polyphenol, has been used.
RSV inhibits mTOR signalling by promoting the interaction between
mTOR and DEPTOR in vitro (17). Recent studies have indicated that
RSV has neuroprotective properties (18,19). In an animal model, RSV has been
shown to protect rats from Aβ-induced neurotoxicity (20).
Emerging data suggest that the augmentation of mTOR
signalling is involved in the aetiopathogenesis of AD. We
hypothesised that DEPTOR is an integral modulator of both the
mTORC1 and mTORC2 complexes, and the presence or degree of binding
to the complexes may determine at which point mTOR will be
inhibited/activated, thus leading to a neuroprotective or
neurotoxic effect.
Materials and methods
Cell culture
SH-SY5Y cells (ATCC; Manassas, VA, USA) were
cultured in 1:1 EMEM and Hams F12 (Sigma-Aldrich, Gillingham, UK)
supplemeted with 10% FBS (50 ml; Gibco, Paisley, UK), 1%
non-essential amino acids, 1% 200 mM L-glutamine, 1%
penicillin/streptomycin (Gibco) at 37°C with 5% CO2. The
SH-SY5Y cells were neuronally differentiated for 6 days by
treatment with 10 μM retinoic acid (RA) (Sigma-Aldrich).
Quantitative RT-PCR
Anterior hippocampus with entorhinal cortex samples
from adults with AD [n=10; 5 with early-onset familial AD (EOFAD);
median age, 61 years; and 5 with late-onset AD (LOAD); median age,
84 years] were provided in collaboration with Brains for Dementia
Research (BDR), University of Newcastle, Newcastle upon Tyne, UK.
The relative expression of the genes of interest was assessed by
quantitative PCR (qPCR) on an ABI PRISM® 7900HT Sequence
detection system (Applied Biosystems, Foster City, CA, USA) using
SYBR®-Green PCR reaction mixture (Sigma-Aldrich) and the
primers for mTOR and DEPTOR as previously described (21). As a negative control for all the
reactions, distilled water was used in place of the cDNA. RNAs were
assayed from 3 independent biological replicates. The RNA levels
were expressed as a relative quantification using the housekeeping
gene, 18S rRNA (RQ) value. The ΔCt method was employed for
comparing relative expression results between treatments in qPCR,
as previously described (22).
Western blot analysis
Protein lysates in 1X Laemmli buffer (Sigma-Aldrich)
were separated on an SDS-10% polyacrylamide gel (Sigma-Aldrich) and
the proteins were transferred onto nitrocellulose membranes (GE
Healthcare, Buckinghamshire, UK). The membranes were blocked in TBS
(Fisher Scientific, Loughborough, UK) containing 5% dried milk
powder (w/v) and 0.1% Tween-20 (Fisher Scientific), for 1 h at room
temperature. Following 3 washes with TBS-0.1% Tween-20, the
nitrocellulose membranes were incubated with primary antibodies
against Aβ42 and GAPDH (both from Cell Signalling
Technology, Danvers, MA, USA). The primary antisera were used at a
1:1,000 dilution overnight at 4°C. The membranes were washed
thoroughly for 30 min with TBS-0.1% Tween-20 prior to incubation
with the secondary antibody, HRP-conjugated immunoglobulin
(1:2,000; Sigma-Aldrich), for 1 h at room temperature and further
washing for 30 min with TBS-0.1% Tween-20. Antibody complexes were
visualised as previously described (21).
Immunofluorescence staining of AD
sections
Three brain regions (precentral gyrus, postcentral
gyrus and occipital lobe) from a single brain with AD and a normal
brain were obtained as a tissue microarray from LifeSpan
Biosciences Inc. (LSBio; Seattle, WA, USA). The ages of the
patients were 75 and 54 years, respectively, and they were both
male. Following a series of deparaffinisations and dehydrations,
the slides were incubated with 10% bovine serum albumin (BSA;
Sigma-Aldrich) for 1 h. This was followed by incubation for 1 h
with an antibody against DEPTOR (Millipore, Abingdon, UK), at a
1,200 dilution in 1% BSA/PBS. The cells were then washed with PBS
prior to an incubation with a fluorescein isothiocyanate
(FITC)-conjugated secondary antibody (Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA) for 1 h. The slides were washed with PBS
and mounted in VECTASHIELD® Mounting Medium (Vector
Laboratories, Inc., Burlingame, CA, USA) containing the dye,
4,6-diamido-2-phenylindole (DAPI) to counterstain the nuclei.
Images were captured using a Plan Apo Neofluar 63X NA 1.25 oil
objective (Zeiss, Thornwood, NY, USA) on a Zeiss Axiovert 200 M
microscope and viewed using AxioVision software. The images were
then analyzed using ImageJ 1.34 image analysis software (National
Institutes of Health, Bethesda, MD, USA).
Immunofluorescence staining of SH-SY5Y
cells
The neuronally differentiated SH-SY5Y cells were
fixed in 4% paraformaldehyde (Sigma-Aldrich) for 10 min prior to
washes in PBS and incubation with 10% BSA for 1 h. The cells were
incubated for 1 h with DEPTOR (Millipore), mTOR (Cell Signalling
Technology) and pan-neuronal marker (Millipore) antibodies at a
1:100 dilution in 1% BSA/PBS. The cells were then washed with PBS
prior to a further incubation with secondary antibodies as
previously described (21).
Images were captured using a Plan Apo Neofluar 63X NA 1.25 oil
objective (Zeiss) on a Zeiss Axiovert 200 M microscope and viewed
using AxioVision software.
Statistical analysis
qPCR data are reported as the means ± standard error
of the mean (SEM). Statistical analysis was performed using the
Student’s t-test. A value of p<0.05 was considered to indicate a
statistically significant difference.
Results
Development of an in vitro neuronal
model
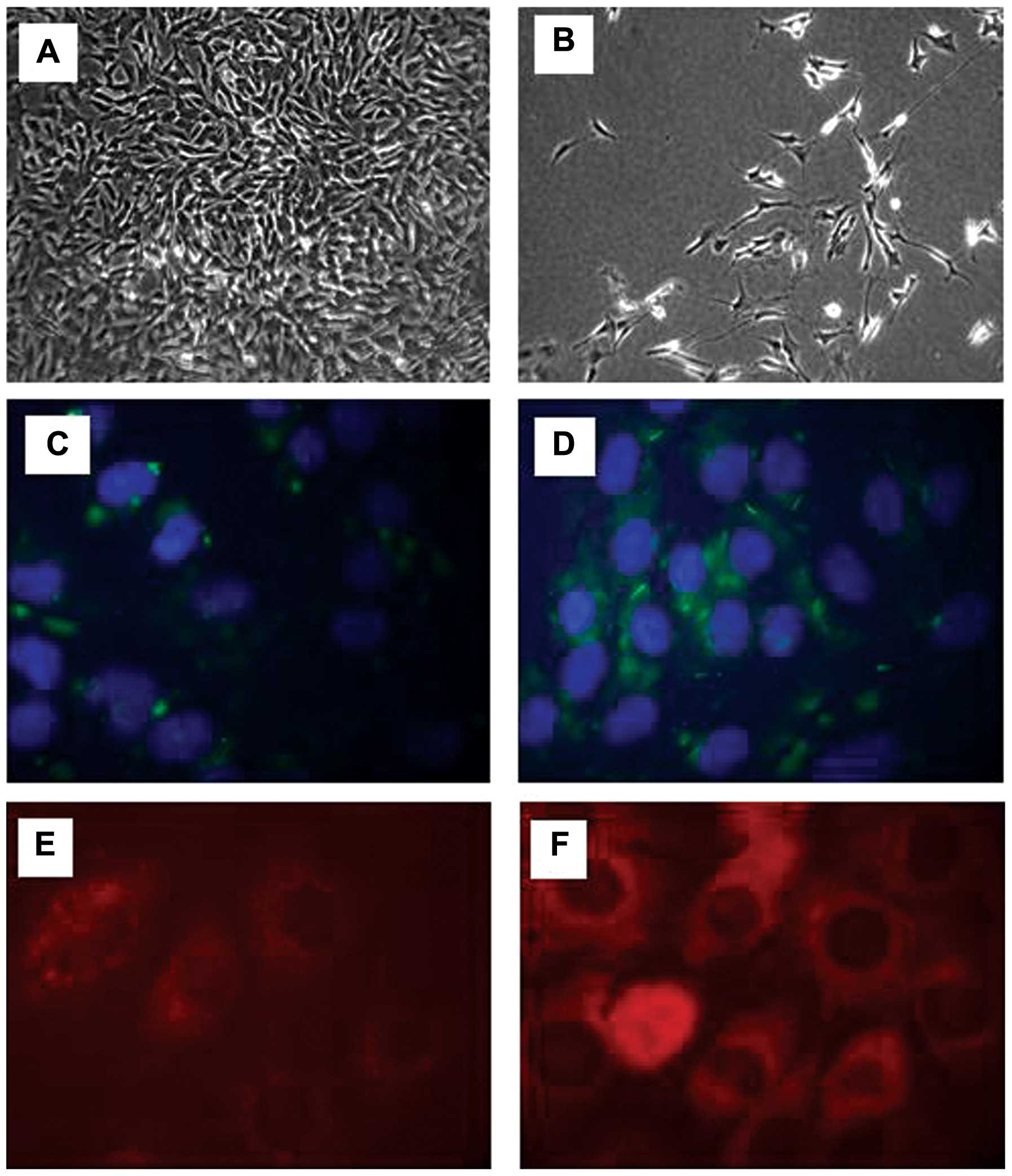
The morphological changes of the SH-SY5Y were
monitored during the differentiation process using a microscope.
The SH-SY5Y cells were seeded at 1×106 and the
undifferentiated SH-SY5Y cells were fast-growing and rounded in
shape (Fig. 1A). Following
differentiation, the cells did not reach confluence and by day 6,
neurite extensions were prominent (Fig. 1B). A pan-neuronal marker was then
used in the differentiated and undifferentiated cells in order to
observe the changes occurring in fundamental somatic, nuclear,
dendritic and axonal proteins. In the undifferentiated cells, there
was a low expression of neuronal proteins, as SH-SY5Y is a
neuroblastoma cell line (Fig.
1C). Following differentiation using RA for 6 days, the SH-SY5Y
cells demonstrated a notable increase in the pan-neuronal marker
signal, further confirming the acquisition of a neuronal phenotype
(Fig. 1D). We then assessed the
protein expression of mTOR and DEPTOR in the neuronally
differentiated cells. Using immunofluorescence, an intense
cytoplasmic staining was observed for both mTOR (Fig. 1E) and DEPTOR (Fig. 1F).
Treatment of differentiated SH-SY5Y cells
with Aβ42 to mimic an AD milieu in vitro: effects on
mTOR and DEPTOR
As already mentioned in the Introduction, one of the
hallmarks of AD is the large number of SPs formed by an
accumulation of toxic Aβ between neurons. Aβ42 in
patients with AD has been shown to reach concentrations lower than
10 nM, but can reach μM ranges (23–25). Our aim was to treat fully
differentiated SH-SY5Y cells with 1 μM Aβ42 in an
attempt to mimic an AD milieu and assess its effects on mTOR and
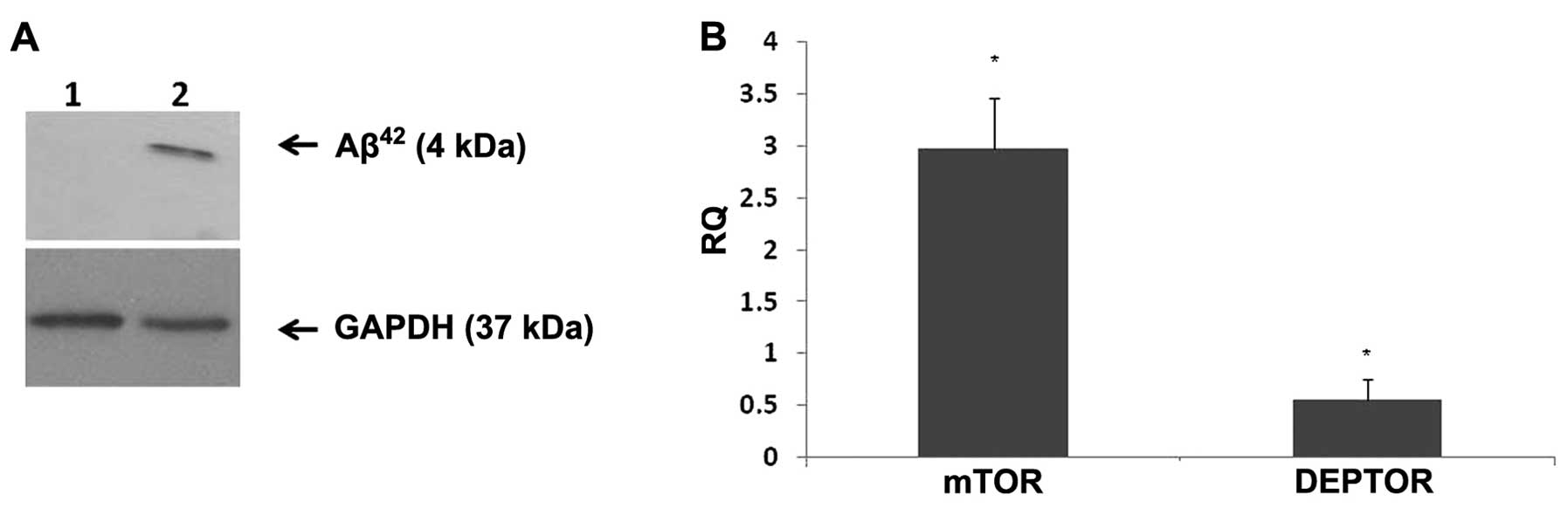
DEPTOR expression in vitro. Western blot analysis was
performed to ensure that Aβ42 was being deposited. In
the cells treated with Aβ42, there was protein
deposition which was detected at 4 kDa (Fig. 2A). Fully differentiated SH-SY5Y
cells were seeded at a density of 1×105/well,
serum-starved for 4 h and treated with Aβ42 (1 μM) for
24 h. mTOR epxression was markedly increased (2-fold) upon 24 h of
Aβ42 treatment, whereas DEPTOR expression was markedly
decreased compared to the control (Fig. 2B).
Differential expression of DEPTOR in
brains with AD
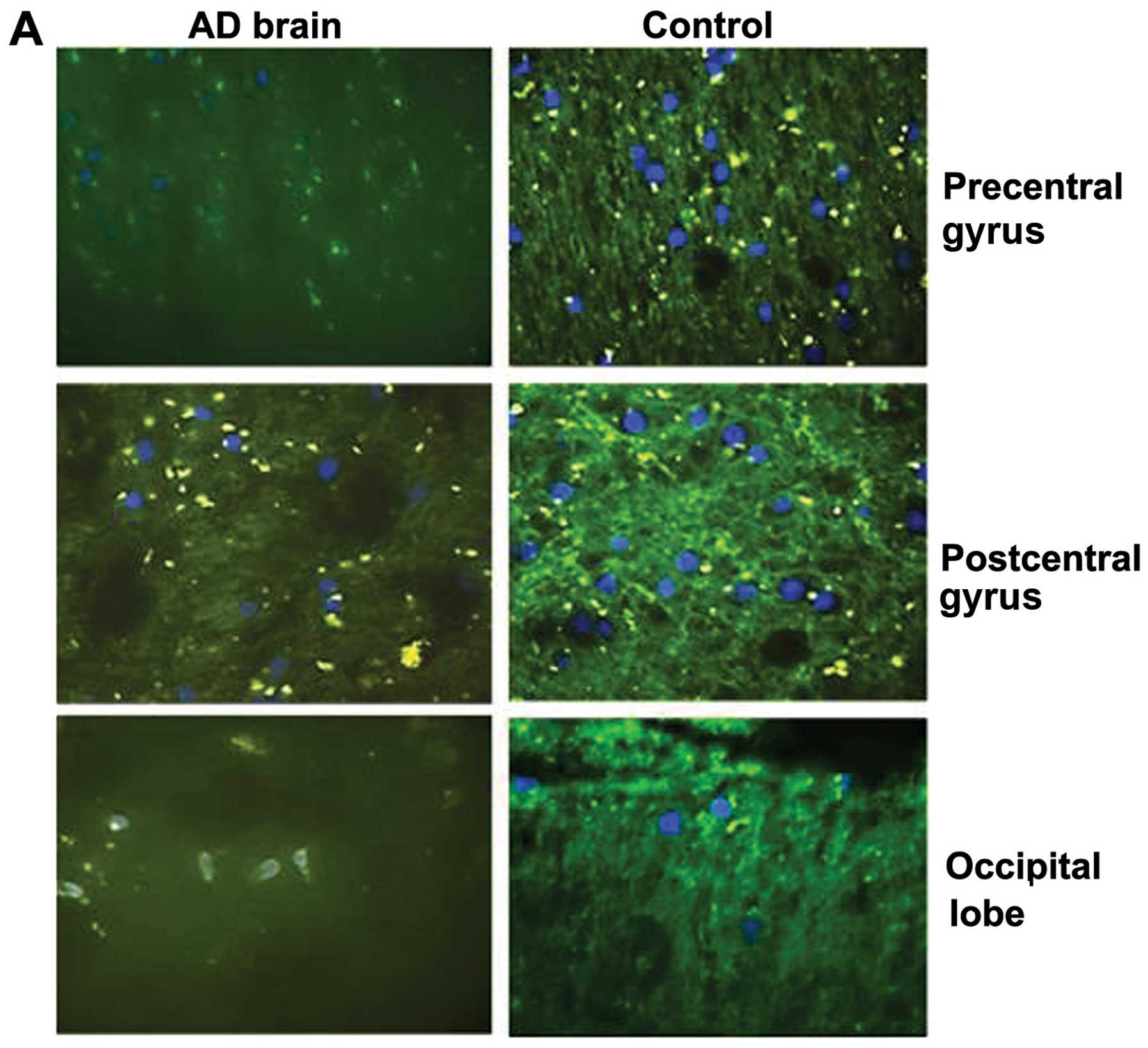
We then assessed the protein expression of DEPTOR
from 3 brain regions from a single brain with AD and a normal
(healthy) brain. The ages of the patients were of 75 and 54 years,
respectively, and they were both male. In all 3 different regions,
i.e., precentral gyrus, postcentral gyrus and occipital lobe, the
expression of DEPTOR was markedly decreased in the patient with AD
compared to the same region of the healthy control (Fig. 3A).
Using clinical samples from patients with EOFAD
(median age, 61 years) and LOAD (median age, 84 years) we examined
the expression of DEPTOR at the mRNA level using qPCR. There was a
significant (~4-fold) decrease in the expression of DEPTOR in the
patients with LOAD when compared with the patients with EOFAD
(Fig. 3B).
Discussion
To the best of our knowledge, in this study, we
demonstrate for first time the expression of DEPTOR in human
brains, as well as the mechanisms through which its expression is
affected by Aβ42 accumulation. Several disease
mechanisms involved in AD affect the structure, as well as the
signalling experienced by neurons; thus, it is essential to obtain
a model that is more similar to an in vivo situation so as
to study more comparable results. Immortalised cell lines are an
appropriate solution to study this; however, they lack some of the
key characteristics of human neurons, such as morphology, cessation
of mitosis and the expression of neuronal markers. The transition
to a more neuronal phenotype is vital for mimicking a disease or a
signalling milieu in vitro. Due to extensive study
previously performed on SH-SY5Y cells, combined with the
comprehensive use of this cell line in previous research on AD, we
decided that this would be the most appropriate cell line to use
(26,27). Neuronal differentiation was
accomplished by the addition of RA and validated by observing cell
morphology, neurite extension and the expression of neuronal
proteins. In this model, both mTOR and DEPTOR appeared to be
expressed at the protein level, mainly localised in the cytoplasm.
This finding corroborates findings from previous studies performed
in our laboratory, demonstrating that the cellular distribution of
mTOR is primarily cytoplasmic (21). mTOR can also shuttle between the
nucleus and cytoplasm (28), and
in the SH-SY5Y cells, DEPTOR was also primarily localised in the
cytoplasm; however, there was some immunofluorescence staining in
the nucleus. This could be suggestive of potential trafficking to
the nucleus alone or as part of the mTORC1 complex.
As already mentioned, one of most important
hallmarks of AD is the accumulation of Aβ42, causing the
formation of Aβ plaques. When the SH-SY5Y cells were treated with
the ‘toxic’ Aβ42 peptide, this led to a decrease in the
expression of DEPTOR and a marked upregulation in mTOR gene
expression. This is the first time that Aβ42 appears to
differentially affect key mTOR signalling components in
vitro. We then expanded our observations using clinical
samples, where there was a notable decrease in DEPTOR
immunostaining in certain regions of the brain with AD when
compared to a healthy control. This is of increasing importance, as
microstructural damage to the precentral gyrus of patients with AD,
as well as a thinning of the postcentral gyrus, have been recently
demonstrated (29,30). It is also thought that damage
incurred by the occipital lobe in AD can greatly contribute to
psychotic symptoms, as a result of deficits in attention and visual
systems (31,32).
We also provide novel data regarding the
differential expression of DEPTOR in early- and late-onset AD. It
is evident that, in LOAD, there is a marked downregulation in
DEPTOR expression, in agreement with the diminished expression of
this protein in brain regions with AD. EOFAD presents before 65
years of age and there are multiple cases within a family usually
attributable to mutations in the amyloid precursor protein
(APP) gene, the presenilin (PSEN)1 gene or the
PSEN2 gene (33). By
contrast, LOAD occurs after 65 years of age and is sporadic with no
known single targetable cause (34). We would like to acknowledge that,
due to ethical restrictions, we were unable to obtain more brain
samples in order to investigate the expression and cellular
distribution of other components of the mTOR signalling
pathway.
Our study provides new insight into the higher order
of complexity regarding the involvement of DEPTOR and mTOR in the
aetiopathogenesis of AD. Caccamo et al (35) demonstrated that Aβ accumulation
causes mTOR hyperactivity by regulating PRAS40 phosphorylation,
whereas RSV promotes the non-amyloidogenic cleavage of the amyloid
precursor protein, the enhanced clearance of Aβ-peptides and
reduced neuronal damage (19). Of
note, RSV exerts its effects by tightly complexing DEPTOR with
mTOR. DEPTOR depletion enhances the in vitro kinase activity
of the mTORC1 and mTORC2 complexes. More specifically, mTORC1
immunopurified from cells depleted of DEPTOR has increased in
vitro kinase activity towards two known substrates, S6K1 and
4EBP1. Of note, these molecules control the synthesis of β- or
α-secretase and, therefore, Aβ generation (36).
Treatment of differentiated mouse N2a neuroblastoma
cells with Aβ42 has been shown to induce the
phosphorylation of mTOR at S2448 and of p70S6K at T38927.
Similarly, in differentiated human SH-SY5Y neuroblastoma cells, a
significant upregulation of p-p70S6K at T389 and T421/S424 sites
was induced following treatment with Aβ42 (11).
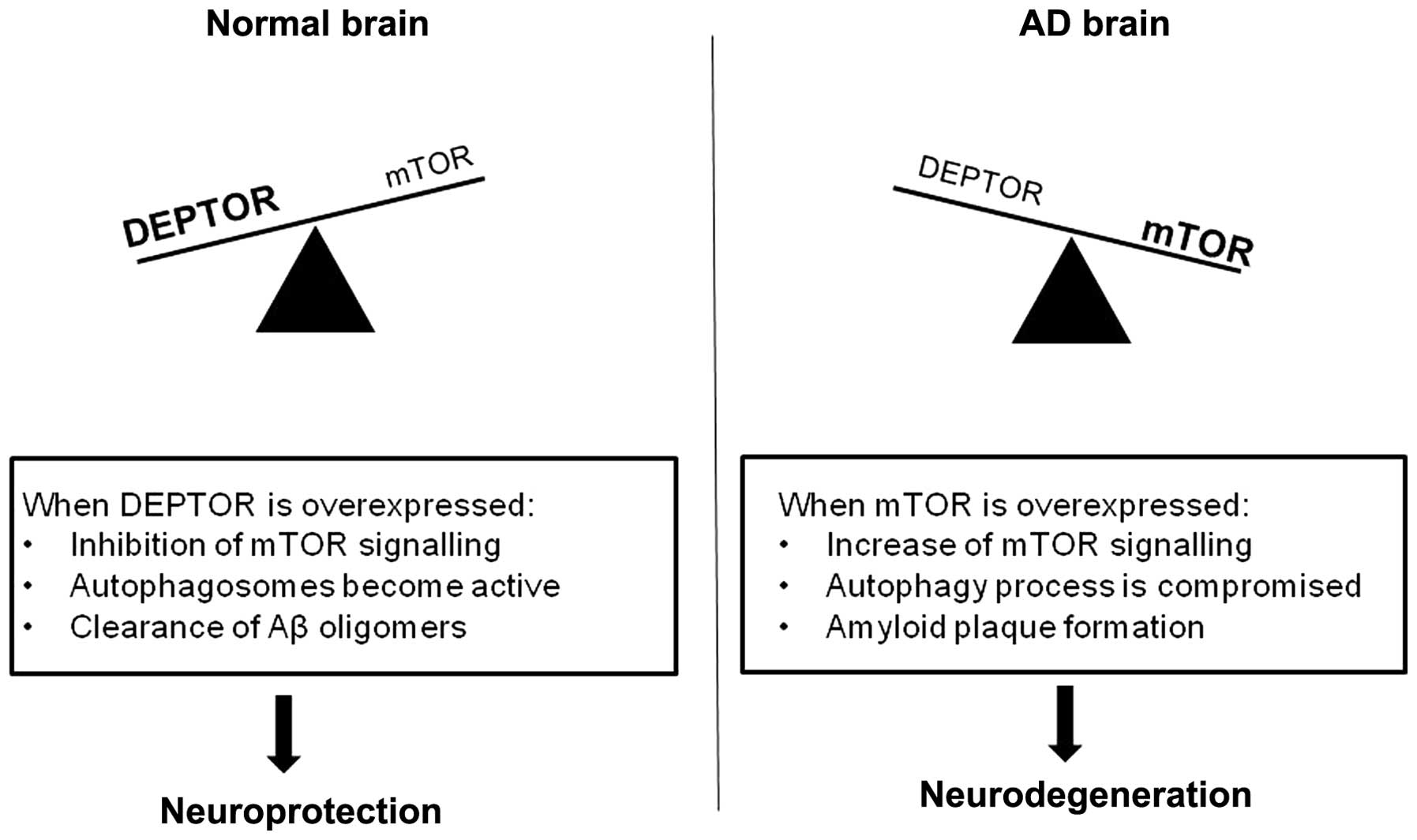
Taken together, we would like to propose the
following (Fig. 4) that builds
upon the model proposal shown in the study by Cai et al
(36): under normal conditions,
mTOR activity is modulated by DEPTOR. This will allow
autophagosomes to clear any toxic Aβ oligomers, thus favouring a
neuroprotective environment. In patients with AD, the mTOR pathway
can be overactive, leading to a dysfunction of autophagy and,
consequently, a lack of Aβ clearance. In turn, the excess of Aβ
will cause a further decrease in DEPTOR and an increase in mTOR
expression, augmenting the neurodegenerative process.
References
|
1
|
Pei JJ and Hugon J: mTOR-dependent
signalling in Alzheimer’s disease. J Cell Mol Med. 12:2525–2532.
2008.PubMed/NCBI
|
|
2
|
Blennow K, de Leon MJ and Zetterberg H:
Alzheimer’s disease. Lancet. 368:387–403. 2006.
|
|
3
|
Brown EJ, Albers MW, Shin TB, Ichikawa K,
Keith CT, Lane WS, et al: A mammalian protein targeted by
G1-arresting rapamycin-receptor complex. Nature. 369:756–758. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dennis PB, Jaeschke A, Saitoh M, Fowler B,
Kozma SC and Thomas G: Mammalian TOR: a homeostatic ATP sensor.
Science. 294:1102–1105. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wullschleger S, Loewith R and Hall MN: TOR
signaling in growth and metabolism. Cell. 124:471–484. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hoeffer CA and Klann E: mTOR signaling: at
the crossroads of plasticity, memory and disease. Trends Neurosci.
2:67–75. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sutton MA and Schuman EM: Dendritic
protein synthesis, synaptic plasticity, and memory. Cell.
127:49–58. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Harrison DE, Strong R, Sharp ZD, Nelson
JF, Astle CM, Flurkey K, et al: Rapamycin fed late in life extends
lifespan in genetically heterogeneous mice. Nature. 460:392–395.
2009.PubMed/NCBI
|
|
9
|
Selman C, Tullet JM, Wieser D, Irvine E,
Lingard SJ, Choudhury AI, et al: Ribosomal protein S6 kinase 1
signaling regulates mammalian life span. Science. 326:140–144.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Boland B, Kumar A, Lee S, Platt FM, Wegiel
J, Yu WH, et al: Autophagy induction and autophagosome clearance in
neurons: relationship to autophagic pathology in Alzheimer’s
disease. J Neurosci. 28:6926–6937. 2008.PubMed/NCBI
|
|
11
|
Lafay-Chebassier C, Paccalin M, Page G,
Barc-Pain S, Perault-Pochat MC, Gil R, et al: mTOR/p70S6k
signalling alteration by Abeta exposure as well as in APP-PS1
transgenic models and in patients with Alzheimer’s disease. J
Neurochem. 94:215–225. 2005.PubMed/NCBI
|
|
12
|
Spilman P, Podlutskaya N, Hart MJ, Debnath
J, Gorostiza O, Bredesen D, et al: Inhibition of mTOR by rapamycin
abolishes cognitive deficits and reduces amyloid-beta levels in a
mouse model of Alzheimer’s disease. PLoS One.
5:e99792010.PubMed/NCBI
|
|
13
|
Ma T, Hoeffer CA, Capetillo-Zarate E, Yu
F, Wong H, Lin MT, et al: Dysregulation of the mTOR pathway
mediates impairment of synaptic plasticity in a mouse model of
Alzheimer’s disease. PLoS One. 5:e128452010.PubMed/NCBI
|
|
14
|
Peterson TR, Laplante M, Thoreen CC,
Sancak Y, Kang SA, Kuehl WM, et al: DEPTOR is an mTOR inhibitor
frequently overexpressed in multiple myeloma cells and required for
their survival. Cell. 137:873–886. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Proud CG: Dynamic balancing: DEPTOR tips
the scales. J Mol Cell Biol. 1:61–63. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Foster H, Coley HM, Goumenou A, Pados G,
Harvey A and Karteris E: Differential expression of mTOR signalling
components in drug resistance in ovarian cancer. Anticancer Res.
30:3529–3534. 2010.PubMed/NCBI
|
|
17
|
Liu M, Wilk SA, Wang A, Zhou L, Wang RH,
Ogawa W, et al: Resveratrol inhibits mTOR signaling by promoting
the interaction between mTOR and DEPTOR. J Biol Chem.
285:36387–36394. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lopez-Miranda V, Soto-Montenegro ML, Vera
G, Herradon E, Desco M and Abalo R: Resveratrol: a neuroprotective
polyphenol in the Mediterranean diet. Rev Neurol. 54:349–356.
2012.PubMed/NCBI
|
|
19
|
Li Z, Pang L, Fang F, Zhang G, Zhang J,
Xie M, et al: Resveratrol attenuates brain damage in a rat model of
focal cerebral ischemia via up-regulation of hippocampal Bcl-2.
Brain Res. 1450:116–124. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang TC, Lu KT, Wo YY, Wu YJ and Yang YL:
Resveratrol protects rats from Abeta-induced neurotoxicity by the
reduction of iNOS expression and lipid peroxidation. PLoS One.
6:e291022011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mparmpakas D, Zachariades E, Goumenou A,
Gidron Y and Karteris E: Placental DEPTOR as a stress sensor during
pregnancy. Clin Sci (Lond). 122:349–359. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li X, An WL, Alafuzoff I, Soininen H,
Winblad B and Pei JJ: Phosphorylated eukaryotic translation factor
4E is elevated in Alzheimer brain. Neuroreport. 15:2237–2240. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Steinerman JR, Irizarry M, Scarmeas N,
Raju S, Brandt J, Albert M, et al: Distinct pools of beta-amyloid
in Alzheimer disease-affected brain: a clinicopathologic study.
Arch Neurol. 65:906–912. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kuo YM, Emmerling MR, Vigo-Pelfrey C,
Kasunic TC, Kirkpatrick JB, Murdoch GH, et al: Water-soluble Abeta
(N-40, N-42) oligomers in normal and Alzheimer disease brains. J
Biol Chem. 271:4077–4081. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang J, Dickson DW, Trojanowski JQ and Lee
VM: The levels of soluble versus insoluble brain Abeta distinguish
Alzheimer’s disease from normal and pathologic aging. Exp Neurol.
158:328–337. 1999.
|
|
26
|
Dwane S, Durack E and Kiely PA: Optimising
parameters for the differentiation of SH-SY5Y cells to study cell
adhesion and cell migration. BMC Res Notes. 6:3662013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Petratos S, Li QX, George AJ, Hou X, Kerr
ML, Unabia SE, et al: The beta-amyloid protein of Alzheimer’s
disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP
mechanism. Brain. 131:90–108. 2008.
|
|
28
|
Bachmann RA, Kim JH, Wu AL, Park IH and
Chen J: A nuclear transport signal in mammalian target of rapamycin
is critical for its cytoplasmic signaling to S6 kinase 1. J Biol
Chem. 281:7357–7363. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sanchez-Espinosa MP, Atienza M and Cantero
JL: Sleep deficits in mild cognitive impairment are associated with
increased plasma amyloid-beta levels and cortical thinning.
Neuroimage. 98:395–404. 2014. View Article : Google Scholar
|
|
30
|
Canu E, McLaren DG, Fitzgerald ME, Bendlin
BB, Zoccatelli G, Alessandrini F, et al: Mapping the structural
brain changes in Alzheimer’s disease: The independent contribution
of two imaging modalities. J Alzheimers Dis. 26(Suppl 3):
S263–S274. 2011.
|
|
31
|
Banno K, Nakaaki S, Sato J, Torii K,
Narumoto J, Miyata J, et al: Neural basis of three dimensions of
agitated behaviors in patients with Alzheimer disease.
Neuropsychiatr Dis Treat. 10:339–348. 2014.PubMed/NCBI
|
|
32
|
Nagahama Y, Okina T, Suzuki N and Matsuda
M: Neural correlates of psychotic symptoms in dementia with Lewy
bodies. Brain. 133:557–567. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bagyinszky E, Youn YC, An SS and Kim S:
The genetics of Alzheimer’s disease. Clin Interv Aging. 9:535–551.
2014.
|
|
34
|
Borovecki F, Klepac N, Muck-Seler D,
Hajnsek S, Mubrin Z and Pivac N: Unraveling the biological
mechanisms in Alzheimer’s disease - lessons from genomics. Prog
Neuropsychopharmacol Biol Psychiatry. 35:340–37. 2010.
|
|
35
|
Caccamo A, Maldonado MA, Majumder S,
Medina DX, Holbein W, Magri A, et al: Naturally secreted
amyloid-beta increases mammalian target of rapamycin (mTOR)
activity via a PRAS40-mediated mechanism. J Biol Chem.
286:8924–8932. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cai Z, Zhao B, Li K, Zhang L, Li C, Quazi
SH, et al: Mammalian target of rapamycin: a valid therapeutic
target through the autophagy pathway for Alzheimer’s disease? J
Neurosci Res. 90:1105–1118. 2012.PubMed/NCBI
|