Introduction
Aplastic anemia (AA) is a bone marrow failure
syndrome characterized by peripheral pancytopenia and bone marrow
hypoplasia. The damage to the bone marrow may be triggered by
environmental exposures, such as chemicals and drugs, or viral
infections, and possibly endogenous antigens generated by
genetically altered bone marrow cells. However, in approximately
half of the cases, the cause is unknown. Based on our previous
study (1), in the present study,
we aimed to analyze the entire mitochondrial DNA (mtDNA) nucleotide
sequences and telomere length of patients with AA.
While most of the genetic material of a cell is
contained within the nucleus, the mitochondria possess their own
circular DNA. Additionally, mitochondria have their own molecular
machinery for protein synthesis and replicate by the process of
fission, in the same way as bacteria. Mitochondria are considered
to be the ‘powerhouses of the cell’, as they produce adenosine
triphosphate (ATP) by systematically extracting energy from
nutrient molecules (substrates) (2). Moreover, mtDNA is replicated with a
high mutation rate, since it lacks protective histones and an
effective DNA repair system.
mtDNA is located near the inner mitochondrial
membrane, where it is far more likely to be exposed to oxygen-free
radicals generated by the respiratory chain than is nuclear DNA
(3,4). A number of studies have reported
unexpectedly large numbers of somatic mutations in patients with
leukemia and myelodysplastic syndromes (MDS) (5–7).
Acquired deletions of mtDNA in the hematopoietic compartment have
also been found to occur in association with severe pancytopenia
and reticulocytopenia (8).
Among the etiologies which are thought to promote
AA, exposure to radiation, and some drugs and chemicals apparently
give rise to mtDNA abnormalities (8,9).
In the present study, we hypothesized that AA may be associated
with mtDNA aberrations.
Telomeres were first measured more than a decade
ago, and found to be short in approximately one third of patients
with acquired AA. Those individuals with the shortest telomeres
appeared to have a longer disease duration and more likely to
develop late malignant clonal complications. Granulocytes were
subsequently found to be mainly affected by telomere erosion in
acquired marrow failure; patients with short telomeres were
predominantly those who did not respond well to immunosuppression
(10–12). Telomere shortening was thought to
be the result of a ‘stressed’ hematopoietic stem cell, which
over-proliferates in response to marrow failure.
Subjects and methods
Patients
Between September 2010 and February 2013, 40
patients, 23 males and 17 females (median age, 39.95 years; range,
11–64 years) were compared with 40 healthy controls, 17 males and
23 females (median age, 38.50 years; range, 15–68 years) (Table I). These patients were diagnosed
at a single institution (Department of Hematology, Affiliated
Hospital of Shandong University of Traditional Chinese Medicine,
Jinan, China). The diagnosis of AA was based on the bone marrow and
blood-count criteria of the International Agranulocytosis and
Aplastic Anemia Study (13).
These patients had no family history of hematologic disease, and
the majority of the patients presented with fatigue and petechiae.
Bone marrow and oral epithelial samples were collected from the
patients with AA for the examination of mtDNA mutations and
telomere length. Bone marrow samples were also collected from 40
healthy volunteers as the controls for the examination of telomere
length. This study was approved by the Institutional Review Board
of the Affiliated Hospital of Shandong University of Traditional
Chinese Medicine, and written informed consent was obtained from
all participants in accordance with the Declaration of
Helsinki.
 | Table IComparison of patient characteristics
between groups. |
Table I
Comparison of patient characteristics
between groups.
| Characteristic | Normal group
(n=40) | AA group (n=40) |
|---|
| Age (mean ± SD,
years) | 38.50±14.47 | 39.95±11.46 |
| Gender
(male/female) | 17/23 | 23/17 |
| WBC count
(×109/l) | - | 2.18±0.46 |
| Hemoglobin count
(g/l) | - | 67.25±11.56 |
| Platelet count
(×109/l) | - | 48.98±11.72 |
| mtDNA mutations
(without silent mutation) | -a | 2.68±2.31 |
| Relative T/S
value | 1.26±0.31b | 1.06±0.38 |
Extraction of genomic DNA
Bone marrow cells were collected from the patients
and oral epithelial cells were collected for normal tissue
comparison. Bone marrow cells were collected from the healthy
volunteers. Total DNA from the bone marrow of patients and healthy
volunteers was extracted using an EasyPure Genomic DNA Extraction
kit (Beijing TransGen Biotech Co., Ltd., China), and oral
epithelial samples from patients were extracted using an TIANamp
Swab DNA kit (Tiangen Biotech Co., Beijing, China). The extracted
DNA was resuspended in Tris-EDTA buffer (10 mM Tris-HCl, 1 mM EDTA,
pH 7.5) and stored at −20°C prior to use.
Sequencing of mtDNA and data
analysis
For the direct sequencing of the entire mtDNA
genome, we used 8 primer pairs based on a modification of a
published protocol to obtain 8 partially overlapping segments. The
amplified mtDNA polymerase chain reaction (PCR) products were
directly sequenced using the BigDye Terminator v3.1 ready reaction
kit and the ABI Prism 3100 Genetic Analyzer (both from Applied
Biosystems, Foster City, CA, USA). The sequencing primers used and
the electropherogram of 8 fragments were described in our previous
study (1). Experimentally
obtained mtDNA sequences were compared with the revised Cambridge
Reference Sequence (rCRS) (found at http://www.mitomap.org/mitomap/mitoseq.html) (14) and using the Blast2 program
(www.ncbi.nlm.nih.gov/blast/bl2seq/wblast2.cgi) and the
database search tool MitoAnalyzer (www.cstl.nist.gov/biotech/strbase/mitoanalyzer.html)
(15) to determine which
polymorphisms and mutations differed from the rCRS, and to
determine whether the differences caused amino acid changes in the
resultant polypeptides. Nucleotide changes that were present in
both the bone marrow and the oral epithelial cells of the same
patient were counted as polymorphisms or homoplasmic mutations.
Those that had not already been included in the databases (MITOMAP,
mtDB or GenBank) were considered as novel polymorphisms. Changes
that were only present in the bone marrow were counted as mutations
or heteroplasmic mutations.
Measurement of telomeres
The method used for determining relative telomere
length by real-time PCR has been previously described (16), and is hereafter referred to as
Tel-PCR. For the PCR assay, 2 μl of each DNA dilution were prepared
in a total reaction volume of 20 μl vol using the Ultra SYBR
Two-Step qRT-PCR kit (Beijing CoWin Biotech Co., Ltd., Beijing,
China). The final telomere primer concentrations were as follows:
tel 1, 270 nM; and tel 2, 900 nM. In the control gene, the master
mix primers, gamma globin gene (HBG)1 and HBG2, were used at a
concentration of 400 nM. The primer sequences were as follows: tel
1, 5′-GGTTTTTGAGGGTGAGGGTGA GGGTGAGGGTGAGGGT-3′; tel 2,
5′-TCCCGACTATCCC TATCCCTATCCCTATCCCTATCCCTA-3′; HBG1, 5′-GCTT
CTGACACAACTGTGTTCACTAGC-3′; and HBG2, 5′-CACC
AACTTCATCCACGTTCACC-3′.
All sequences were obtained from Sangon Biotech Co.,
Ltd. (Shanghai, China). Telomere sequences were amplified in an ABI
7300 real-time PCR system (Applied Biosystems) under the following
conditions: 95°C, 10 min, to activate Taq polymerase; 35 cycles of
denaturation at 95°C, 15 sec, and annealing/extension, which was
carried out at 54°C, 2 min. The conditions for the amplification of
the HBG gene were as follows: 95°C, 10 min; 40 cycles at 95°C, 15
sec, and 58°C, 1 min. ABI Prism 7300 SDS software was used for the
data analysis. The telomere length (x) for each sample was based on
the telomere to single copy gene ratio (T/S ratio), which was based
on the calculation of the ΔCt [Ct(telomeres)/Ct(HBG)].
Telomere length was expressed as the relative T/S
value, which was normalized to the average T/S value of the
reference sample defined as [2−(ΔCtx−ΔCtr) =
2−ΔΔCt], which was used for the construction of the
standard curve, as the reference sample, and as the validation
sample. To make the results obtained from different plate runs
comparable, the results of each plate were approved only if the
relative T/S ratios of the validation reference sample fell within
a 3% variation. Laboratory personnel conducting the telomere length
assay were blinded to the clinical outcomes of the patients prior
to statistical analysis.
Statistical analysis
Pearson correlation coefficient was used for the
analysis of an association between the variables. We analyzed the
association between the data such as mtDNA mutations (without
silent mutation), relative T/S value and the white blood cell
(WBC)/hemoglobin/platelet counts. Paired t-test was used to assess
the differences between the variables. The tests were performed at
the 0.05 level of significance. SPSS version 19.0 (SPSS Inc.,
Chicago, IL, USA) was used for the statistical analysis.
Results
Heteroplasmic mutations of mtDNA
We detected mutations in all 40 entire mtDNA genomes
obtained from both bone marrow and oral epithelial cells of the
same patient. Overall, we detected 146 mutations in 18 genes,
including 39 silent mutations (39/26.7%) and 28 frameshift
mutations (28/19.2%). The non-silent mutations included NADH
dehydrogenase (ND)2; 34/31.8%), ND4 (16/11.1%), ND5
(8/5.56%), cytochrome b (Cytb; 8/5.56%), cytochrome c
oxidase subunit 1 (COX1; 7/4.86%), ND6 (6/4.17%),
ND1 (6/4.17%), RNA, ribosomal cluster 1 (RNR1;
4/2.78%), ND3 (4/2.78%), RNA, ribosomal cluster 2
(RNR2; 3/2.08%), TRNE (3/2.08%), TRNG
(2/1.39%), ND4L (1/0.69%), cytochrome c oxidase
subunit 2 COX2; 1/0.69%), cytochrome c oxidase
subunit 3 (COX3; 1/0.69%), TRNY (1/0.69%) and
TRNA (1/0.69%). These data are presented in our previous
study (1). Mitochondrial
sequencing indicated that these mutations were found in the coding
region closely related to the mitochondrial oxidative respiratory
chain, covering ND1–6, ND4L, Cytb and other
related genes. We found that the mutation levels were particularly
high in ND2 and ND4.
mtDNA polymorphisms in patients with
AA
In this study, we detected 424 polymorphisms in 15
genomes. These polymorphisms were found in the D-loop region
(264/62.3%), RNR1 (47/11.08%), RNR2 (21/4.95%),
ND1 (5/1.18%), ND2 (16/3.77%), COX1
(16/3.77%), COX2 (1/0.24%), COX3 (2/0.47%),
ATP6 (2/0.47%), ND3 (6/1.42%), ND4L (6/1.42%),
ND4 (5/1.18%), ND5 (8/1.88%), ND6 (6/1.42%)
and Cytb (19/4.48%). Of note, the majority of these
polymorphisms were found in the D-loop region, where numerous
mutations are found to be involved in leukemia and MDS. However, in
this study, no novel polymorphisms were found.
Relative T/S value in patients with AA as
compared with the healthy individuals
We compared the relative T/S value in DNA obtained
from the healthy volunteers with those found in the patients with
AA (Tables I and II). The results presented herein and
those obtained from healthy volunteers indicated that there were
significant differences between the relative T/S values.
Additionally, whereas the relative T/S value in the normal
(healthy) group varied, overall, the relative T/S values were
shorter as a function of increasing age (P<0.05). In the
patients with AA, there was no obvious association between the
relative T/S value and age, and there was no obvious association
between the relative T/S value and gender in each group.
| Table IIPearson correlation coefficient
between the groups. |
Table II
Pearson correlation coefficient
between the groups.
| Age (mean ± SD,
years) | Male | Female |
|---|
| Relative T/S value
in normal group | −0.438a | 1.29±0.38 | 1.23±0.25 |
| Relative T/S value
in AA group | −0.204 | 1.13±0.42 | 0.96±0.31 |
Association between non-silent mutations,
relative T/S values and white blood cell (WBC), hemoglobin and
platelet count
We then compared the non-silent mutations, relative
T/S value, WBC, hemoglobin and platelet counts in the same patients
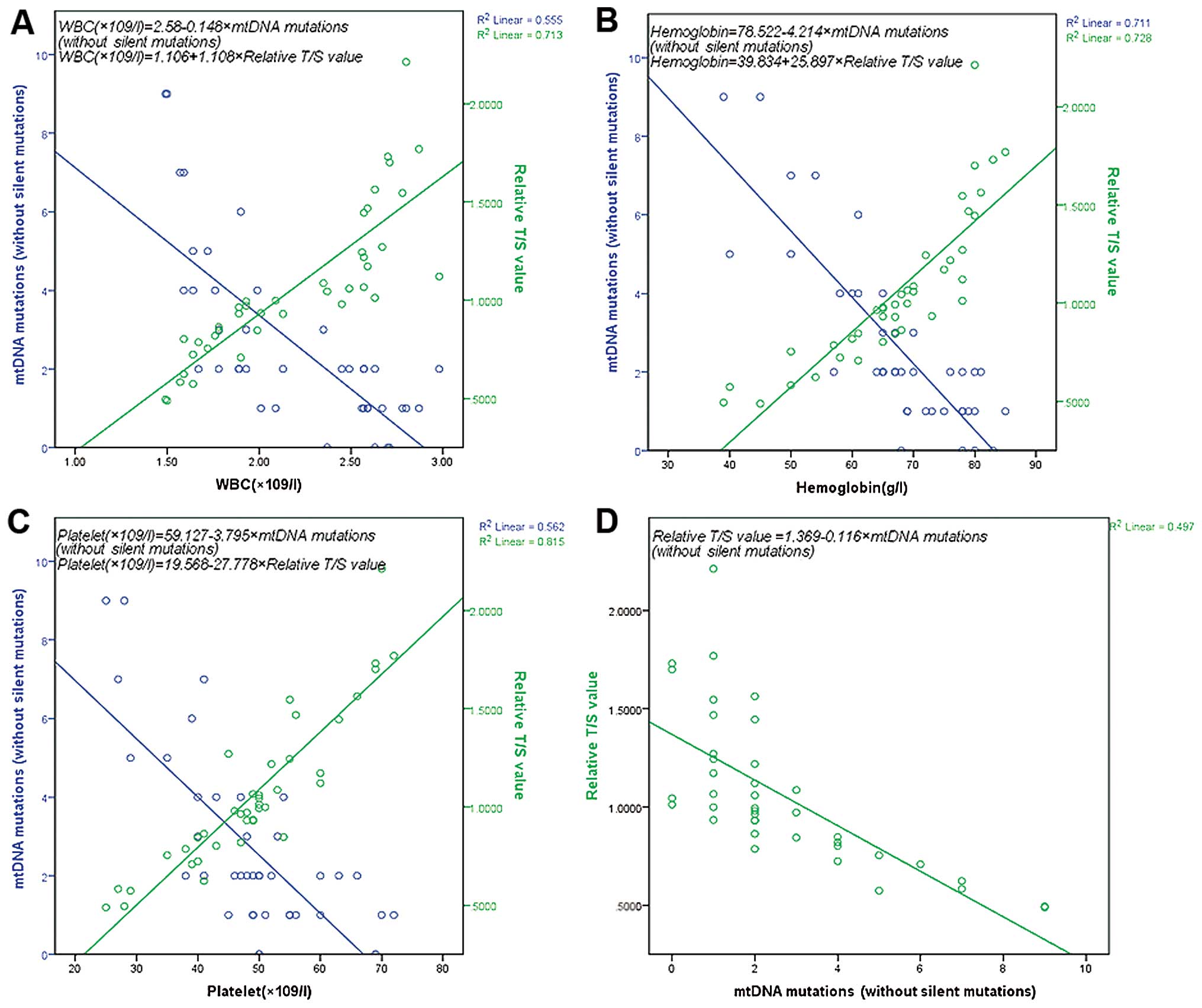
with AA. We found that there was a negative correlation between the
non-silent mutations and the WBC, hemoglobin and platelet counts
obtained. Additionally, there was a positive correlation between
the relative T/S value and the WBC, hemoglobin and platelet counts
obtained. Finally, there was also a negative correlation observed
between the non-silent mutations and the relative T/S value
(Fig. 1).
Discussion
In this study, we analyzed entire mtDNA nucleotide
sequences and telomere lengths of patients with AA. To the best of
our knowledge, this is the first study indicating that there is an
association between telomere length and mtDNA mutations in patients
with AA. We also found that there was a negative correlation
between the non-silent mutations and the WBC, hemoglobin, platelet
counts obtained and the relative T/S value.
Acquired mtDNA deletions in hematopoietic diseases
have been observed in association with severe pancytopenia and
reticulocytopenia (17). In this
study, we detected 146 mutations in 18 genes, including 39 silent
mutations (39/26.7%) and 28 frameshift mutations (28/19.2%). The
mutation rate was particularly high in ND2 and ND4 of
the mtDNA genome of the patients with AA, with the notable
exception of these silent mutations.
These genes are closely linked to oxidation in the
respiratory chain. We also found that there was a negative
correlation between the non-silent mutations and the WBC,
hemoglobin and platelet counts obtained. ND1–6, ND4L
and Cytb are important components of the NADH-ubiquinone
oxidoreductase (complex I) system, and the ubiquinone cytochrome
c oxidoreductase (complex III) system. Mitochondrial injury
is reflected by mtDNA damage and by a decrease in the levels of
mtRNA transcripts, protein synthesis and mitochondrial function. A
decrease in these complex activities may result in decreased
cellular energy, disruption of cell signaling and interference with
cellular differentiation and programmed cell death or apoptosis.
Furthermore, deficient mitochondrial ATP production due to
mutations in mtDNA may promote chromosomal instability (18). Cells with an inadequate ATP supply
may also have difficulty in correctly segregating their chromosomes
during mitosis. These factors may also result in decreased energy
metabolism, which will affect the self-renewal and differentiation
of hematopoietic stem cells. Of note, the genes affected were
involved in oxidative phosphorylation. In contrast to the mutations
involved in hematologic malignancies, such as acute leukemia (AL)
and MDS, these mutations were mainly found in the D-loop (19,20). In these studies, the majority of
nucleotide alterations were detected in the D-loop region in
patients with AL and MDS, suggesting that the D-loop is a
mutational hotspot in human cancer.
It was previously found that the mitochondria of
leukemic cells utilize glycolysis more vigorously than oxidative
phosphorylation (5,17,20–22). This may be caused by the
differences observed between benign and malignant hematologic
disorders.
In this study, we used HBG as the control gene in
real-time PCR analysis to survey relative telomere length
measurement in patients with AA and healthy volunteers. In patients
with AA, there was no obvious association between the relative T/S
value and age. We then compared the relative T/S values and WBC
count, hemoglobin and platelet counts in the same patients with AA.
We found that there was a positive association between the context
of the relative T/S value and WBC, hemoglobin and platelet counts.
In addition, a negative association was found between the
non-silent mutation and relative T/S value. Critically, short
telomeres promote apoptosis, cell senescence and chromosomal
instability in both tissue culture and animal models (23–25).
The major reverse transcriptase-incompetent splice
variant of the human telomerase protein inhibits telomerase
activity but protects cells from apoptosis. It was also found that
the telomere lengths in granulocytes from patients with AA were
significantly shorter than those in age-adjusted controls (12). In a large series of patients
undergoing immunosuppressive therapy (n=183), patients with shorter
telomeres (mostly without any telomerase mutation) had a higher
possibility of relapse, an increased risk to evolve to MDS
(particularly the most feared monosomy 7) and AML, coupled with a
poorer overall survival in comparison to patients with longer
telomeres (26). Bone marrow
cells of short-telomere patients with AA also present increased
chromosomal instability in vitro (27). In addition to absolute
reticulocyte and lymphocyte counts (27), telomere length is likely to be
critical in therapy decision making (28). Moreover, telomere length has been
found to be related to the risk of relapse, clonal evolution and
overall survival in severe AA (29).
In this study, we also compared non-silent mutations
and the relative T/S value, and found that there was a negative
correlation by this comparison. In conclusion, the mutations
occurred in the region that influences the replication and
transcription of mtDNA, and thus are likely to have significant
effects on function, increasing the permeability of the
mitochondrial inner membrane and destroying the mitochondrial
membrane potential, which would then trigger caspase activity and,
eventually, cell death by apoptosis. Therefore, functional defects
caused by mtDNA mutations may be the primary cause of bone marrow
failure in AA. In addition, since there was a negative correlation
between the non-silent mutations and the relative T/S value, we
suggest that mtDNA mutations and the shortening of telomere length
may influence each other and cause the failure of the bone marrow.
It is formally possible that our previously described supposition
may represent an important pathogenic mechanism in AA (Fig. 2). In future studies, we aim to
increase the number of cases to prevent bias and then survey the
related indicators of the oxidative respiratory chain and
telomerase in an attempt to determine the pathogenesis of AA.
Acknowledgements
This study was supported by grants from the Chinese
Postdoctoral Science Foundation (no. 2012M521356), the National
Natural Science Foundation of China (no. 81202839/H2902), the
Natural Science Foundation of Shandong Province, China (no.
ZR2012HQ023), the Shandong Province Technology Development Program
of Traditional Chinese Medicine (no. 2011-063) and the Affiliated
Hospital of Shandong University of Traditional Chinese Medicine and
Shandong University, China. We thank Sangon Biotech Co., Ltd.
(Shanghai, China), and SinoGenoMax Co., Ltd. for sequencing the
mtDNA.
References
|
1
|
Cui X, Liu F, Wang JQ, Zhang WJ, Wang JY,
Liu K, Cui SY, Zhang J and Xu RR: Complete sequence analysis of
mitochondrial DNA of aplastic anemia patients. Genet Mol Res.
11:2130–2137. 2012. View Article : Google Scholar
|
|
2
|
Chinnery PF and Schon EA: Mitochondria. J
Neurol Neurosurg Psychiatry. 74:1188–1199. 2003. View Article : Google Scholar
|
|
3
|
Richter C, Park JW and Ames BN: Normal
oxidative damage to mitochondrial and nuclear DNA is extensive.
Proc Natl Acad Sci USA. 85:6465–6467. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Penta JS, Johnson FM, Wachsman JT and
Copeland WC: Mitochondrial DNA in human malignancy. Mutat Res.
488:119–133. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Grist SA, Lu XJ and Morley AA:
Mitochondrial mutations in acute leukaemia. Leukemia. 18:1313–1316.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Linnartz B, Anglmayer R and Zanssen S:
Comprehensive scanning of somatic mitochondrial DNA alterations in
acute leukemia developing from myelodysplastic syndromes. Cancer
Res. 64:1966–1971. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wulfert M, Küpper AC, Tapprich C, et al:
Analysis of mitochondrial DNA in 104 patients with myelodysplastic
syndromes. Exp Hematol. 36:577–586. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hatfill SJ, La Cock CJ, Laubscher R,
Downing TG and Kirby R: A role for mitochondrial DNA in the
pathogenesis of radiation-induced myelodysplasia and secondary
leukemia. Leuk Res. 17:907–913. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rowe TC, Weissig V and Lawrence JW:
Mitochondrial DNA metabolism targeting drugs. Adv Drug Deliv Rev.
49:175–187. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee JJ, Kook H, Chung IJ, et al: Telomere
length changes in patients with aplastic anaemia. Br J Haematol.
112:1025–1030. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ball SE, Gibson FM, Rizzo S, Tooze JA,
Marsh JC and Gordon-Smith EC: Progressive telomere shortening in
aplastic anemia. Blood. 91:3582–3592. 1998.PubMed/NCBI
|
|
12
|
Brümmendorf TH, Maciejewski JP, Mak J,
Young NS and Lansdorp PM: Telomere length in leukocyte
subpopulations of patients with aplastic anemia. Blood. 97:895–900.
2001.PubMed/NCBI
|
|
13
|
Kaufman DW, Kelly JP, Levy M and Shapiro
S: The Drug Etiology of Agranulocytosis and Aplastic Anemia. Oxford
University Press; New York: 1991
|
|
14
|
Andrews RM, Kubacka I, Chinnery PF,
Lightowlers RN, Turnbull DM and Howell N: Reanalysis and revision
of the Cambridge reference sequence for human mitochondrial DNA.
Nat Genet. 23:1471999. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee MS and Levin BC: MitoAnalyzer, a
computer program and interactive web site to determine the effects
of single nucleotide polymorphisms and mutations in human
mitochondrial DNA. Mitochondrion. 1:321–326. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cawthon RM: Telomere measurement by
quantitative PCR. Nucleic Acids Res. 30:e472002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Clayton DA: Transcription of the mammalian
mitochondrial genome. Annu Rev Biochem. 53:573–594. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gattermann N: Mitochondrial DNA mutations
in the hematopoietic system. Leukemia. 18:18–22. 2004. View Article : Google Scholar
|
|
19
|
Kwok CS, Quah TC, Ariffin H, Tay SK and
Yeoh AE: Mitochondrial D-loop polymorphisms and mitochondrial DNA
content in childhood acute lymphoblastic leukemia. J Pediatr
Hematol Oncol. 33:e239–e244. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shin MG, Kajigaya S, Levin BC and Young
NS: Mitochondrial DNA mutations in patients with myelodysplastic
syndromes. Blood. 101:3118–3125. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gattermann N: From sideroblastic anemia to
the role of mitochondrial DNA mutations in myelodysplastic
syndromes. Leuk Res. 24:141–151. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Suganuma K, Miwa H, Imai N, et al: Energy
metabolism of leukemia cells: glycolysis versus oxidative
phosphorylation. Leuk Lymphoma. 51:2112–2119. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Listerman I, Sun J, Gazzaniga FS, Lukas JL
and Blackburn EH: The major reverse transcriptase-incompetent
splice variant of the human telomerase protein inhibits telomerase
activity but protects from apoptosis. Cancer Res. 73:2817–2828.
2013. View Article : Google Scholar
|
|
24
|
Lu R, Pal J, Buon L, et al: Targeting
homologous recombination and telomerase in Barrett’s
adenocarcinoma: impact on telomere maintenance, genomic instability
and tumor growth. Oncogene. 33:1495–1505. 2014.PubMed/NCBI
|
|
25
|
Begus-Nahrmann Y, Hartmann D, Kraus J, et
al: Transient telomere dysfunction induces chromosomal instability
and promotes carcinogenesis. J Clin Invest. 122:2283–2288. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Calado RT, Yewdell WT, Wilkerson KL, et
al: Sex hormones, acting on the TERT gene, increase telomerase
activity in human primary hematopoietic cells. Blood.
114:2236–2243. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cooper JN, Calado R, Wu C, Scheinberg P
and Young NS: Telomere length of peripheral blood leukocytes
predicts relapse and clonal evolution after immunosuppressive
therapy in severe aplastic anemia. Blood. 112:4422008.
|
|
28
|
Scheinberg P, Wu CO, Nunez O and Young NS:
Predicting response to immunosuppressive therapy and survival in
severe aplastic anaemia. Br J Haematol. 144:206–216. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Scheinberg P, Cooper JN, Sloand EM, Wu CO,
Calado RT and Young NS: Association of telomere length of
peripheral blood leukocytes with hematopoietic relapse, malignant
transformation, and survival in severe aplastic anemia. JAMA.
304:1358–1364. 2010. View Article : Google Scholar
|