Introduction
The incidence of kidney cancer has been rising
steadily. Native Americans have the highest incidence of kidney
cancer and highest mortality rates for males and females (1,2).
Renal cell carcinoma (RCC) is the most common type of kidney cancer
in adults, responsible for approximately 80% of cases (3). At present, initial treatment is most
commonly a radical or partial nephrectomy and remains the mainstay
of curative treatment (1). RCC is
resistant to chemotherapy and radiotherapy in most cases, but
responds well to immunotherapy with interleukin-2 or interferon-α
(2).
Phosphodiesterase 5 (PDE5) is a member of several
cyclic nucleotide phoshodiesterase gene families, which hydrolyze
cyclic GMP (cGMP-PDEs) (4). The
abnormal expression of PDE5 and the increase in PDE5 activity has
been detected in a variety of tumor cell lines (5–10).
A sustained increase in intracellular cyclic GMP (cGMP) induced by
the inhibition of PDE5 is required to modify the process of
apoptosis and mitotic arrest in these carcinoma cells with an
enhanced PDE5 expression (5,6,8,9).
The inhibition of PDE5 induces sustained levels of intracellular
cGMP and activates its downstream protein kinase G (PKG), resulting
in the inhibition of proliferation and the induction of apoptosis
in colon cancer cells. Further studies exploring the mechanisms of
the PDE5 inhibitor-induced activation of the downstream cGMP
pathway found that the activated intracellular cGMP-PKG pathway
increased the degradation of β-catenin in SW480 colon cancer cells,
but not in SW620 cells (11,12). In the present study, to determine
the role of the PDE5 enzyme in controlling the inhibition and
apoptosis of renal carcinoma cells, we suppressed PDE5 expression
in OS-RC-2 human renal carcinoma cells by transfecting the cells
with PDE5 siRNA, and investigated the changes in cell
proliferation, apoptosis and the downstream molecules of PKG.
Materials and methods
MG132 (Z-leu-Leu-Leu-CHO) and
S-Nitroso-N-acetylpenicillamine (SNAP) were purchased from Biomol
Research Laboratories, Inc. (Plymouth Meeting, PA, USA) and Sigma
(St. Louis, MO, USA), respectively. 8-Br-cGMP was obtained from
Sigma. Anti-β-catenin, anti-phosphorylated (p)-β-catenin,
anti-p-c-Jun N-terminal kinase (JNK), anti-cyclin A, anti-cyclin
B1, KT5823, anti-PED5 and anti-PKG antibodies were purchased from
Abcam (Cambridge, MA, USA). Anti-actin (AC-74), anti-cyclin D1
anti-p21, anti-p27 antibodies and JNK inhibitor (SP600125) were
purchased from the Beyotime Institute of Biotechnology (Haimen,
China). Anti-Sp1 antibody was purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA).
Cell culture
The human renal carcinoma cell line, OS-RC-2, was
obtained from the Type Culture Collection of the Chinese Academy of
Sciences (Shanghai, China). The cells were cultured under 5%
CO2 at 37°C in RPMI-1640 medium with 5% FBS and 1%
penicillin/streptomycin (all from HyClone, Auckland, New
Zealand).
RNA interference
Target gene accession numbers NM_033437 and
NM_003109 were obtained from GenBank. According to the principles
of Ambion for the design of siRNAs, the sequences of 21 nucleotides
beginning with AA and containing 30–50% GC were selected and used
as target sites. The siRNA used were as follows: PDE5A siRNA,
5′-GGAAGAAACAAG AGAGCUAdTdT-3′ (sense) and 5′-UAGCUCUCUUGUUUCU
UCCdTdT-3′ (antisense); Sp1 siRNA, 5′-CAUCCAAGGCUGU GGGAAAdTdT-3′
(sense) and 5′-UUUCCCACAGCCUU GGAUG dTdT-3′ (antisense). The
control and negative control sequences were provide by the company,
and a report was sent to ensure the effects. No homology sequence
was found by Blast analysis. The siRNAs were purchased from Wuhan
Genesil Biotechnology Co., Ltd. (Wuhan, China), and 10 nmol/l of
each siRNA were transfected into the OS-RC-2 cells, using HiPerFect
transfection reagent (Qiagen, Valencia, CA, USA).
Assessment of cell apoptosis and
proliferation
The activity of caspase-3 was determined using the
caspase-3 activity kit (Beyotime Institute of Biotechnology). Cell
lysates were prepared after their respective treatment.
Subsequently, the OS-RC-2 cells were scraped, centrifuged,
resuspended and lysed in lysis buffer (RIPA buffer and protease
inhibitor PMSF; Solarbio, Beijing, China). Assays were performed in
96-well microtiter plates by incubating 10 μl protein of cell
lysate per sample in 80 μl reaction buffer (1% NP-40, 20 mM
Tris-HCl, pH 7.5, 137 mM Nad and 10% glycerol) containing 10 μl/mM
caspase-3 substrate (Ac-DEVD-pNA). The lysates were incubated at
37°C for 4 h. Samples were measured using a microplate reader
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) at an absorbance of
405 nm, according to the manufacturer’s instructions. Cell
proliferation was determined by MTT assay according to the
instructions provided with the MTT cell proliferation assay kit
(Beyotime Institute of Biotechnology). The absorbance was measured
at 570 nm on a microplate reader (Bio-Rad Laboratories, Inc.).
Quantitative RT-PCR (RT-qPCR)
Total RNA was extracted using TRIzol reagent
(Invitrogen, Carlsbad, CA, USA), reverse-transcribed into cDNA
templates and amplified using a ReverTra Ace qPCR RT kit (Toyobo,
Osaka, Japan). RT-qPCR was performed using a SYBR-Green Real-time
PCR Master Mix-Plus (Toyobo). The PCR primers (Table I) were used for gene
amplification. Target gene expression levels are presented as a
ratio of the levels in the treated cells and the control cells
using the ΔΔCt method, as previously described (13).
 | Table IPrimer sequences for real-time
RT-PCR. |
Table I
Primer sequences for real-time
RT-PCR.
| Genes | Primers sequences
(5′→3′) | Product length
(bp) |
|---|
| PDE5 | Forward:
CACACCGAATCTTGCTCTTG | 134 |
| Reverse:
TCAGAATCCTTGACAACAATGG | |
| Cyclin D1 | Forward:
TCTACACCGACAACTCCATCCG | 133 |
| Reverse:
TCTGGCATTTTGGAGAGGAAGTG | |
| p21 | Forward:
AGGTGGACCTGGAGACTCTCAG | 94 |
| Reverse:
TCCTCTTGGAGAAGATCAGCCG | |
| PKG | Iα Forward:
ACTCCACAAATGCCAGTCGG | 139 |
| Reverse:
GGTGAACTTCCGGAATGCCT | |
| PKG | Iβ Forward:
AGAGCGCGAGCACCTTG | 194 |
| Reverse:
GATCTGCGACAGCTCCAAGT | |
| PKG | II Forward:
AAAAGACATGCGAAGCGGTC | 166 |
| Reverse:
GCTCAACTCTTCCGAACCCA | |
| β-actin | Forward:
AATCGTGCGTGACATTAAG | 178 |
| Reverse:
GAAGGAAGGCTGGAAGAG | |
Western blot analysis
The OS-RC-2 cells were collected using the rubber
policeman, washed in phosphate-buffered saline (PBS) and lysed in
ice-cold PBS buffer [0.l M Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM
EDTA, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium
dodecylsulfate (SDS), and 0.1% protease inhibitor cocktail and 10
mM phenylmethylsulphonyl fluoride (PMSF)] for 30 min on ice. The
lysates were sonicated for 2 sec to shear the DNA to reduce its
viscosity, followed by centrifugation at 28,100 × g for 30 min at
4°C. The supernatant was collected, and protein concentrations were
measured using the Bradford method (Bio-Rad Laboratories, Inc.).
Twenty micrograms of protein determined by Bradford assay were
electrophoretically separated using a 12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel and
transferred onto polyvinylidene fluoride (PVDF) membranes, and then
immunoblotted with the corresponding antibodies. Immuno-detection
was performed with an enhanced chemiluminescence (ECL) detection
kit (Beyotime Institute of Biotechnology). The protein bands were
analyzed using Quantity One 4.4 software.
Construction and expression of PKG
wild-type and mutants
The wild-type and mutant constructs of PKG,
including PKG Iα and PKG Iβ were designed and purchased from
BioTech Co., Ltd. The PKG constructs were subcloned into the
expression vector pMIG upstream of the IRES-enhanced green
fluorescent protein (GFP) sequence. The cells were transfected with
pMIG as previously described (14).
Statistical analysis
All data are presented as the means ± standard
deviation (SD). Significant differences among the groups were
determined using the unpaired Student’s t-test. A value of
P<0.05 was accepted as an indication of statistical
significance. All data in the figures were obtained from at least 3
independent experiments.
Results
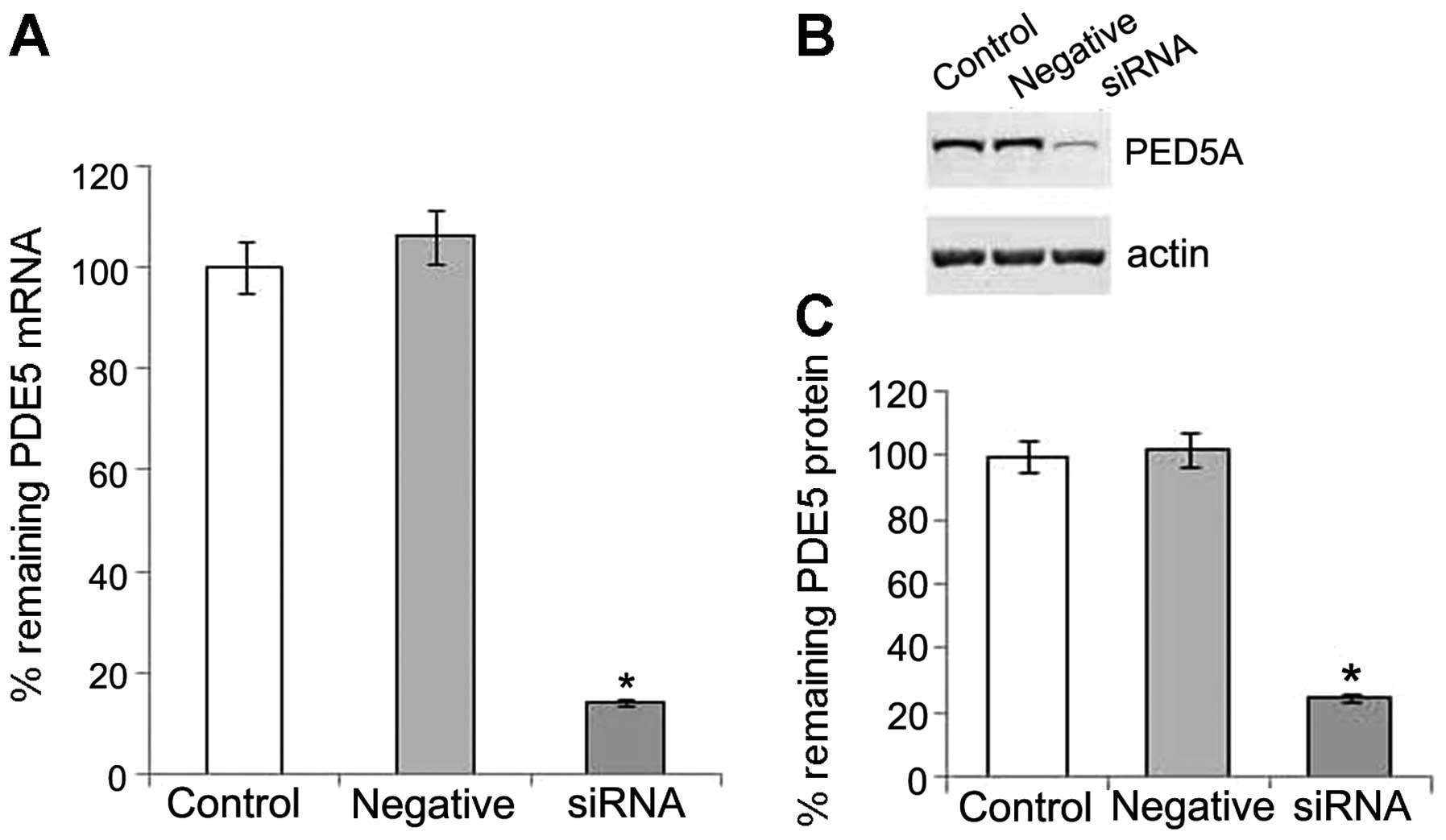
PDE5 siRNA suppresses PDE5 expression at
the post-transcriptional and protein level
We detected the mRNA and protein expression of PDE5
in the OS-RC-2 cells, and then used specific siRNA to silence PDE5
expression. Following transfection with PDE5 siRNA, real-time PCR
revealed that the level of PDE5 mRNA expression was significantly
reduced in the transfected cells when compared with the negative
control (Fig. 1A). Western blot
analysis also revealed a significant decrease in the levels of the
105 kDa PDE5 in the OS-RC-2 cells transfected with PDE5 siRNA, when
compared with the control (Fig. 1B
and C). Thus, our data indicated that treatment with PDE5 siRNA
successfully suppressed the expression of PDE5 in the OS-RC-2
cells.
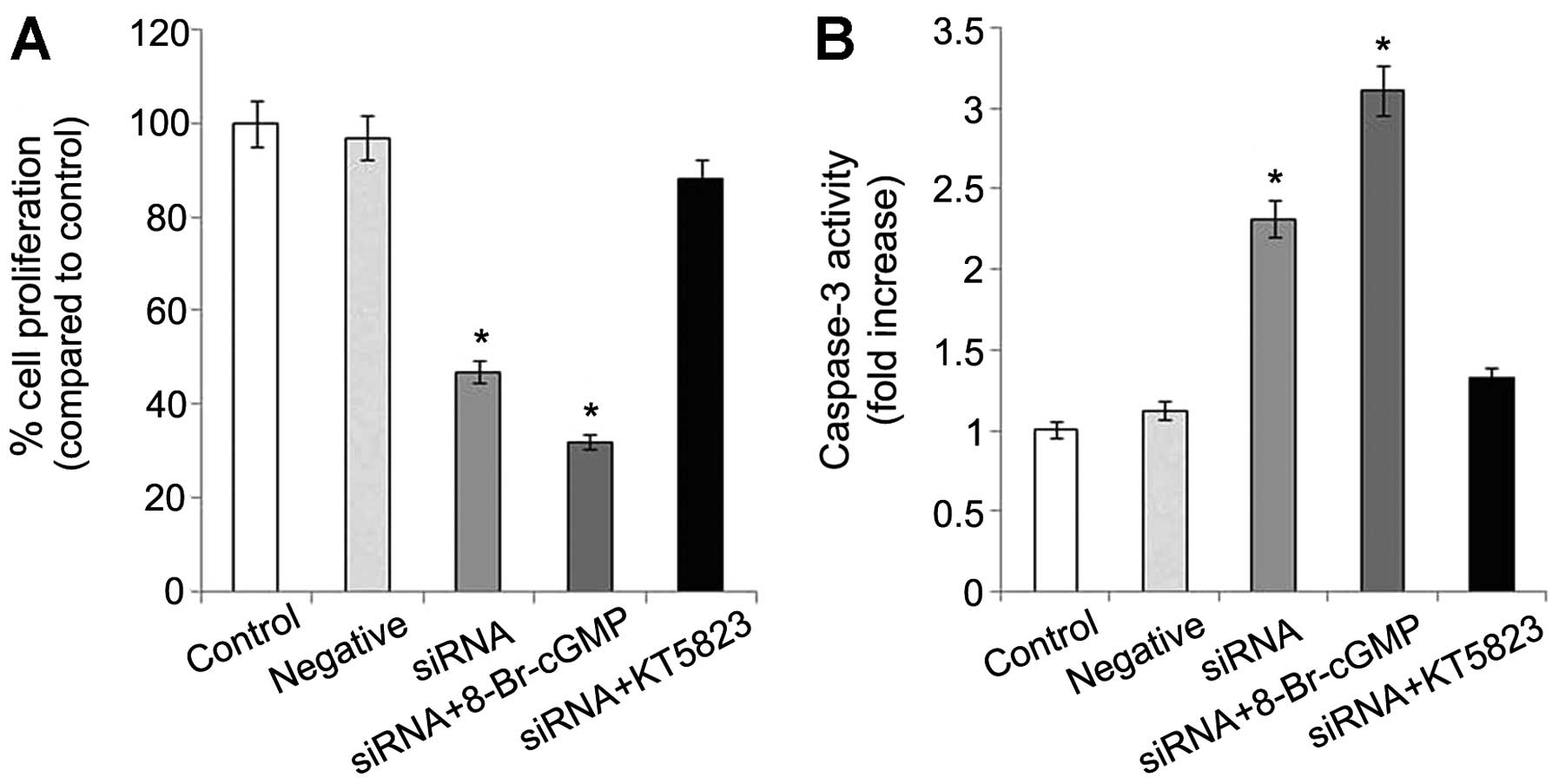
PDE5 suppression reduces proliferation
and increases apoptosis through cGMP-PKG
PDE5 siRNA-transfected OS-RC-2 cells had much lower
growth rates than the control and negative control cells. The
average rate of growth inhibition was approximately 50% in the PDE5
siRNA-transfected cells. When 8-Br-cGMP, a cell membrane permeable
cGMP derivative was added, a further inhibition was observed.
However, the inhibitory effect in the PDE5 siRNA-transfected
OS-RC-2 cells was blocked by KT5823, an inhibitor of PKG (Fig. 2A). Conversely, there was a 2-fold
increase in the relative level of caspase-3 in the transfected
cells compared to the control cells. Caspase-3 is an early
indicator of apoptosis. Caspase-3 activity was enhanced 3-fold
following treatment with 8-Br-cGMP and was inhibited following
treatment with KT5823 (Fig.
2B).
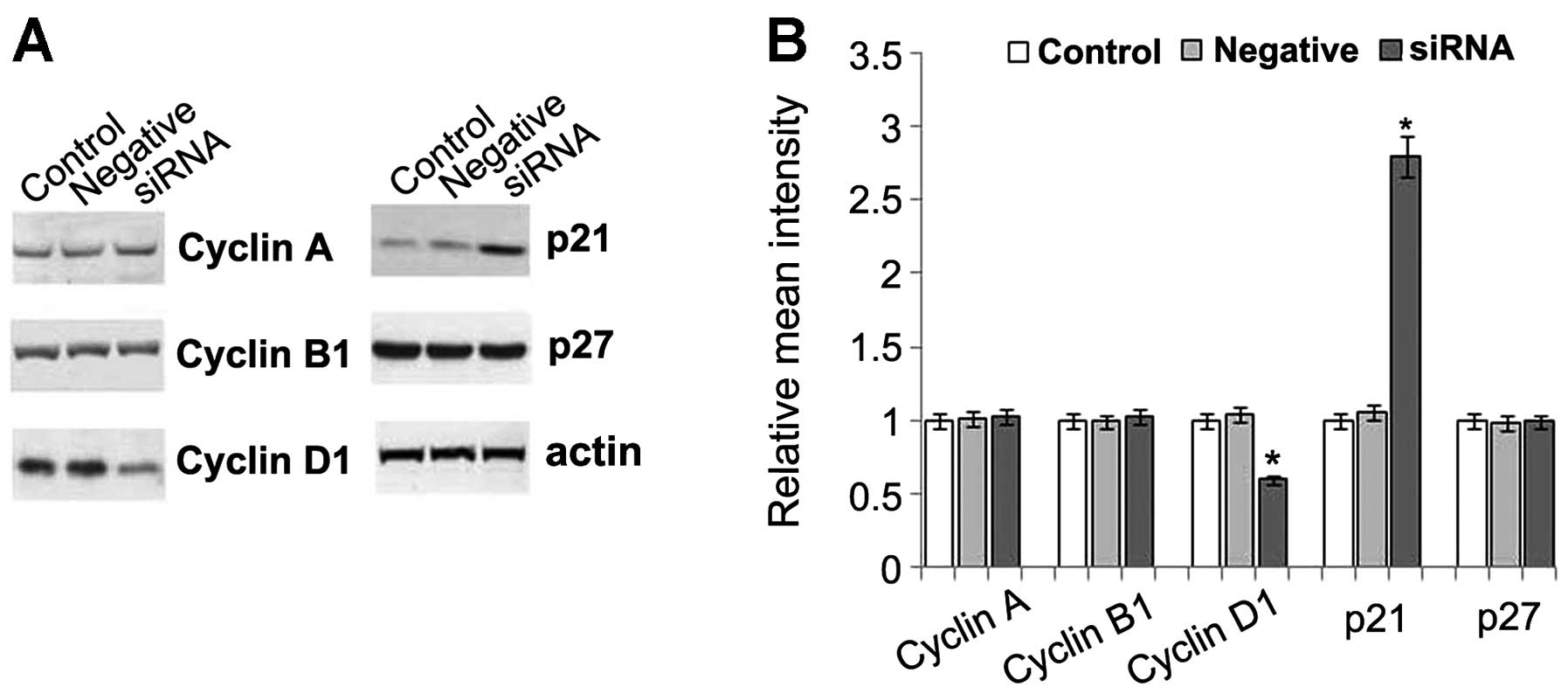
Effect of PDE5 siRNA on the molecules
related to cell cycle progression
To elucidate the mechanisms responsible for the
reduced proliferation and increased apoptosis in the PDE5
siRNA-transfected cells, the levels of cell cycle-specific
molecules, including cyclins (cyclin A, B and D1) and CDK
inhibitors (CKIs, p21 and p27) were determined. Our results
revealed that no changes in the levels of cyclin A and B1 were
detected in the PDE5 siRNA-transfected cells; however, a 50%
reduction in the levels of cyclin D1 was observed in these cells.
Further study indicated that both the mRNA and protein expression
of cyclin D1 was decreased in a time-dependent manner in these
cells. A 2- to 3-fold increase in the expression of p21, a mitotic
inhibitor, was observed in the PDE5 siRNA-transfected cells when
compared to the control cells. However, there were no changes
observed in p27 expression, another mitotic inhibitor involved in
the cyclin D1-CDK2, CDK4 and cyclin E-CDK2 inhibition at the G1
phase (Fig. 3). Therefore, cyclin
D1 and p21 may play an important role in the inhibition of
proliferation and the increase in apoptosis.
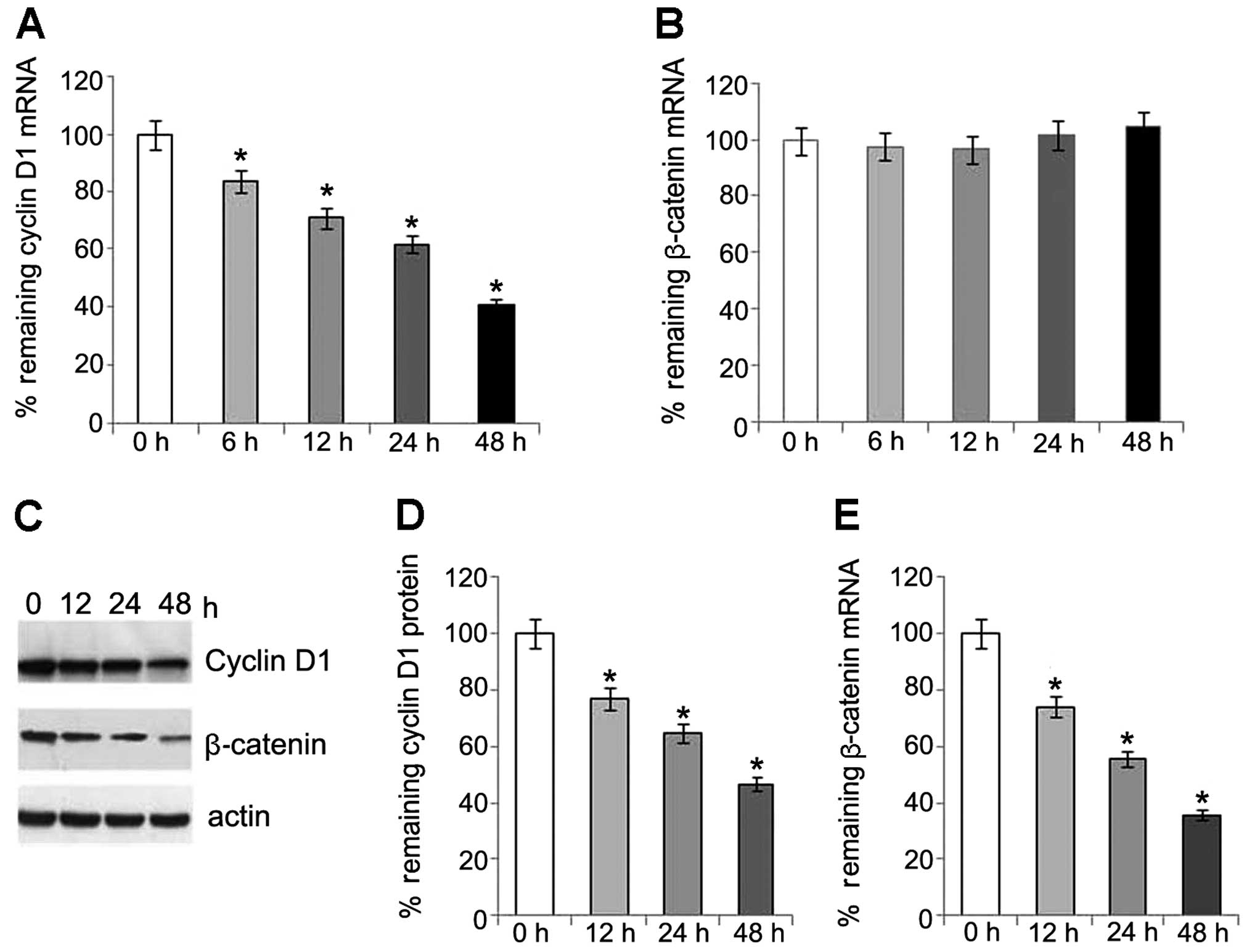
Involvement of β-catenin/TCF and JNK
pathways in regulation of cyclin D1 expression
To explore whether the time-dependent decrease in
the expression of cyclin D1 involves the regulation of β-catenin in
this study, we examined the mRNA and protein levels of β-catenin.
Of note, the protein levels of β-catenin also showed a
time-dependent reduction following transfection. However, no
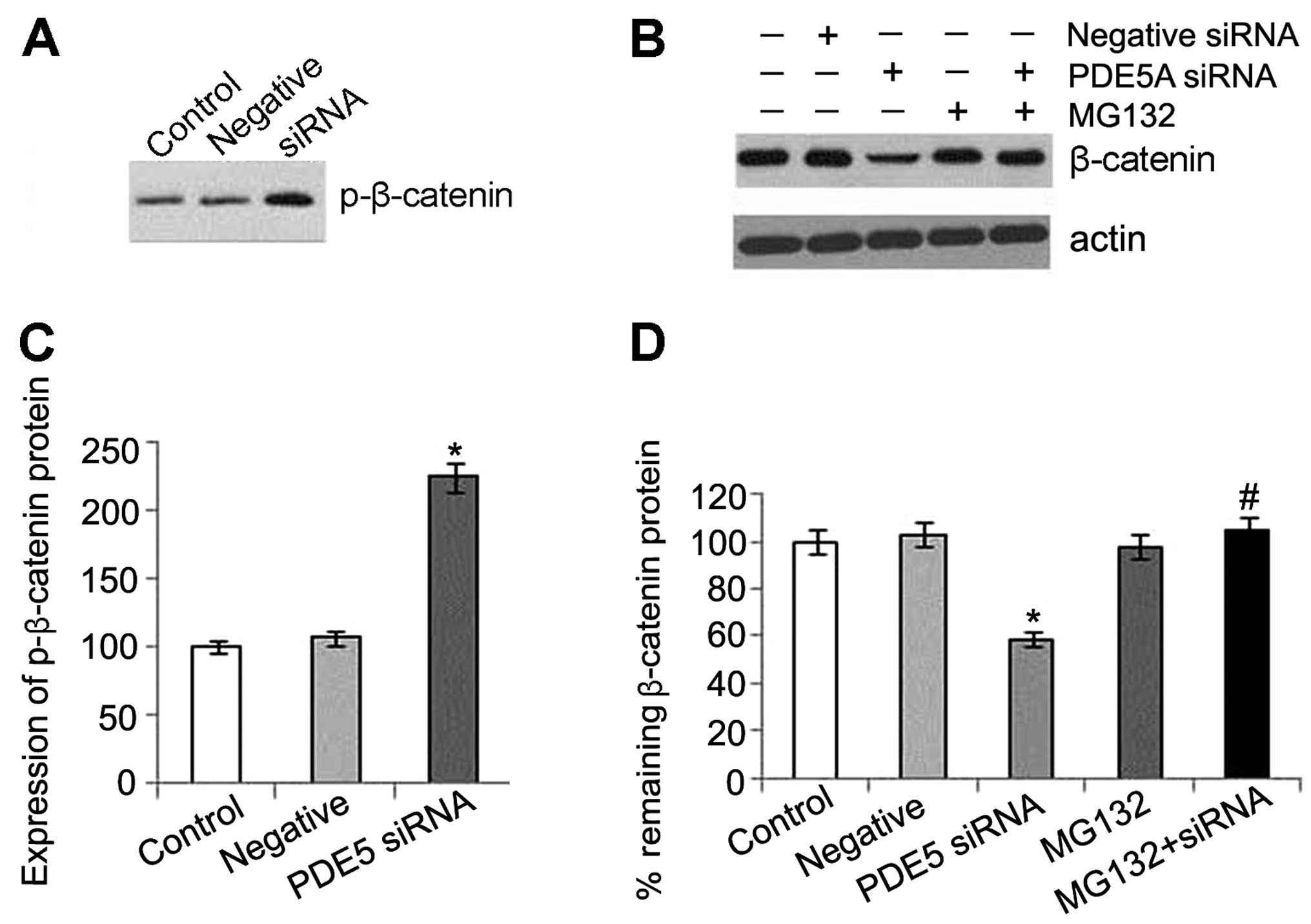
changes in the mRNA level of β-catenin were detected (Fig. 4). This may partly be explained by
the changes which occur in the expression of cyclin D1, as
previously described (15).
Our results also indicate that a decrease in
β-catenin expression is caused by an effect at the protein level
rather than at the mRNA level in the activated PKG pathway.
Furthermore, a significant increase in the phosphorylation of
β-catenin was detected in the PDE5 siRNA-transfected cells, which
was also consistent with previous data indicating that PKG has a
similar role to GSK3β and directly phosphorylates β-catenin/TCF,
leading to proteasomal degradation (16). MG132, an ubiquitin-proteasome
inhibitor, was used to determine whether the decrease in β-catenin
expression induced by PKG activation could be blocked at the
proteasomal level. Our results revealed that MG132 prevented the
reduction in β-catenin expression which was caused by activated
PKG, and the inhibitor alone did not alter the level of β-catenin.
These results indicated that the protein was processed by the
ubiquitin-proteasome system (Fig.
5A). An increased expression of cyclin D1 levels was also
detected compared to the control cells, after MG132 was used to
inhibit the ubiquitin-proteasome system (Fig. 6A).
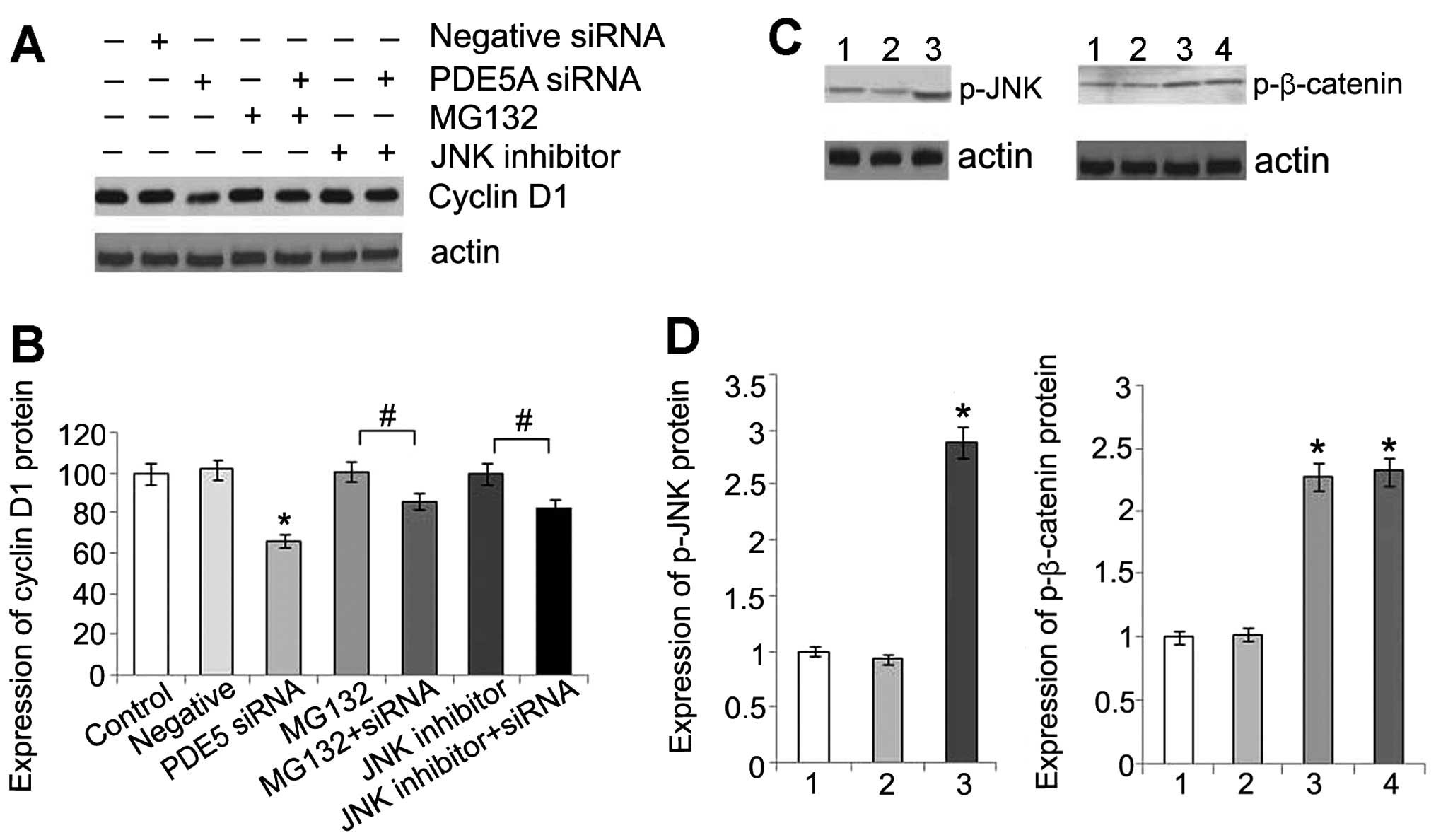 | Figure 6(A) Expression of cyclin D1 protein in
OS-RC-2 cells following treatment with phosphodiesterase type 5
(PDE5) siRNA, or MG132, or a combination of both, or c-Jun
N-terminal kinase (JNK) inhibitor, or a combination of PDE5 siRNA
and JNK inhibitor. (B) Statistical analysis of cyclin D1 protein
expression. Data are presented as the means ± standard deviation
(SD) (n=3). *P<0.05 compared with control cells.
#P<0.05. (C) Comparison of phosphorylated (p)-JNK in
OS-RC-2 cells in the different groups by western blot analysis
(lane 1, control group; lane 2, negative control group; lane 3,
PDE5 siRNA group). Comparison of p-β-catenin in OS-RC-2 cells in
the different groups by western blot analysis (lane 1, control
group; lane 2, negative control group; lane 3, PDE5 siRNA group;
lane 4, PDE5 siRNA + JNK inhibitor group). (D) Statistical analysis
of p-JNK and p-β-catenin protein. Data are presented as the means ±
SD (n=3). *P<0.05 compared with control. |
A previous study indicated that JNK activation was
required for the PKG-dependent regulation of TCF activity (17). Therefore, in this study, we
detected the level of phosphorylation of JNK and found that there
was a significant increase in the phosphorylation of JNK in the
transfected cells (Fig. 6B). This
result is consistent with that of a previous study showing that the
activation of PKG directly phosphorylates JNK (18). In addition, there was an increase
in the level of cyclin D1 and no change in the levels of
p-β-catenin following treatment with an inhibitor of JNK,
indicating that activated JNK may affect the downstream molecules
of β-catenin/TCF (Fig. 6A).
PKG activation increases the expression
of p21 through Sp1
To examine whether the activation of PKG alters the
transcription of p21, we treated the OS-RC-2 cells with PDE5 siRNA
or 8-Br-cGMP. Our results revealed that the mRNA levels of p21 were
increased in the cells treated with PDE5 siRNA or 8-Br-cGMP;
however, the increase was not time-dependent (data not shown).
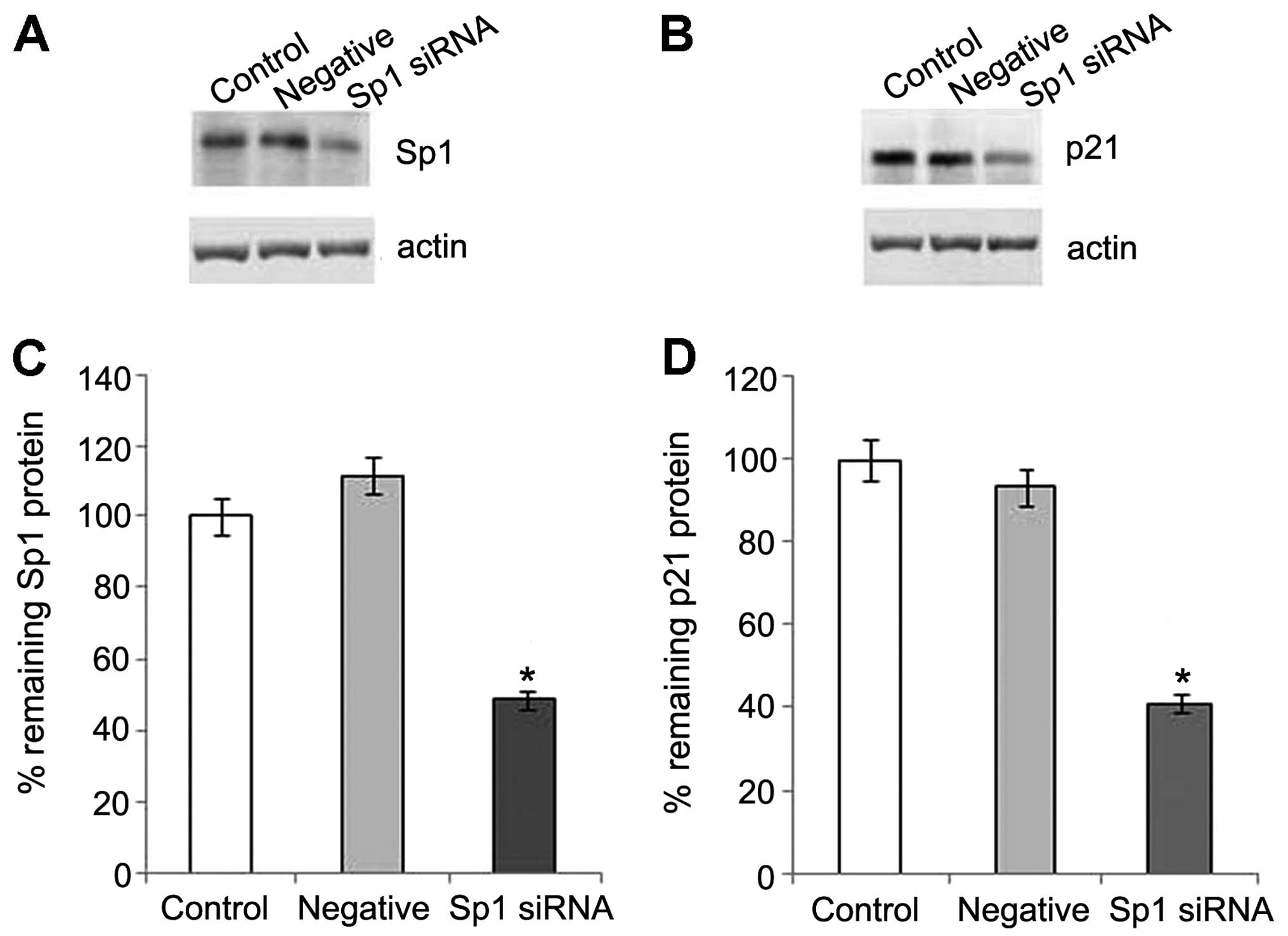
Previous studies have confirmed that Sp1 is required for the
PKG-induced upregulation of p21, p27 and HINT1 in SW480 cells, and
can be phosphorylated by activating PKG on serine residues
(11,12). To further investigate the
requirement of Sp1 in the increase in p21 expression induced by PKG
in OS-RC-2 cells, we knocked down the expression of endogenous Sp1
by transfecting the cells with Sp1 siRNA. The level of Sp1 in the
OS-RC-2 cells was markedly reduced by Sp1 siRNA. The activation of
p21 protein expression was clearly observed in the control group
following treatment with 8-Br-cGMP, and was inhibited by the
suppression of Sp1 (Fig. 7).
Thus, our data indicate that the increased expression of p21 may be
regulated through Sp1 by activating PKG.
PKG Iβ plays a vital role in anticancer
activities in OS-RC-2 cells
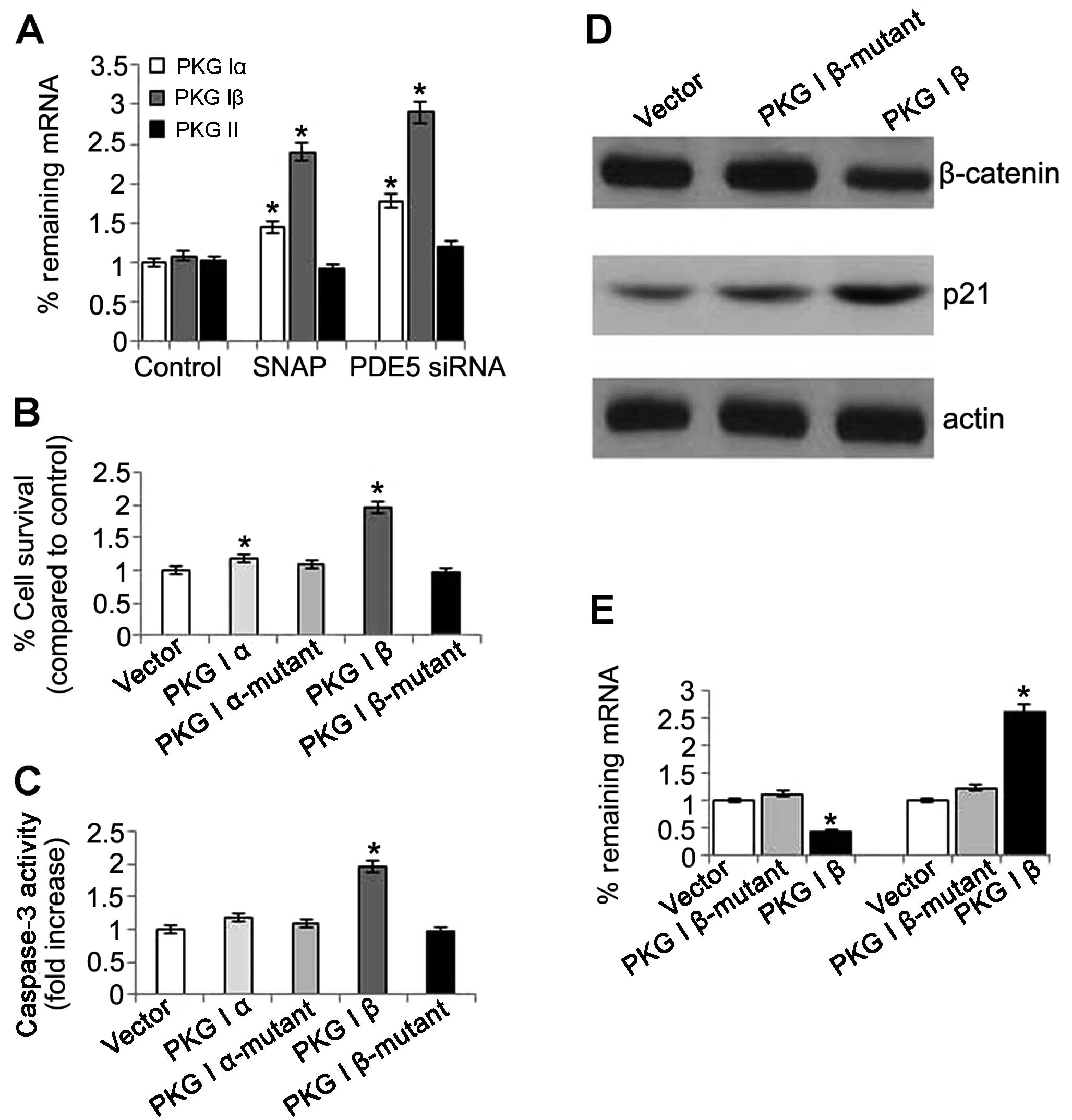
PKG has various isozymes, including PKG I and PKG
II; the N terminus of PKG I is encoded by 2 alternatively spliced
exons that produce the isoforms α and β (19). In this study, we investigate which
type of PKG is responsible for the anticancer activities in OS-RC-2
cells. The mRNA expression of PKG I and PKG II was detected in the
cells treated with PDE5 siRNA. The results revealed that the mRNA
expression of PKG Iα and PKG Iβ was significantly increased. The
same result was also observed in the nitric oxide (NO) donor SNAP
group, which indicated that the NO-cGMP pathway was involved in the
anticancer activities induced by the activation of PKG.
Unexpectedly, there were no changes observed in the PKG II levels
in the cells treated with PDE5 siRNA of SNAP when compared with the
control (Fig. 8A). These results
suggested that not PKG II, but PKG I was involved in the anticancer
activities. Next, we investigated the effects of PKG Iβ
overexpression on cell proliferation and apoptosis. The results
revealed that the cells with an overexpression of PKG had
significantly increased growth rates, whereas this increase was
abolished by treatment with the mutants (Fig. 8B). Relative caspase-3 expression
was significantly increased in the cells with PKG Iβ
overexpression, but there was no obvious change in the cells with
PKG Iα overexpression (Fig. 8C).
However, the role of PKG I was according to its intact structure as
no changes in cell proliferation and apoptosis were observed in the
cells overexpressing mutant PKG Iα and Iβ (Fig. 8B and C). Futhermore, immunoblot
analysis revealed that the protein expression of β-catenin was
reduced and the protein expression of p21 was increased in the PKG
Iβ group (Fig. 8D). These results
suggest that PKG Iβ plays a vital role in anticancer activities in
OS-RC-2 cells.
Discussion
PDE5 inhibition induces sustained levels of
intracellular cGMP and activates its downstream PKG, resulting in
the inhibition of proliferation and the induction of apoptosis in
colon cancer cells (11,12). In this study, we demonstrated that
suppressing PDE5 gene expression by PDE5 siRNA transfection
inhibited proliferation and induced apoptosis in OS-RC-2 human
renal carcinoma cells. The effects were enhanced by 8-Br-cGMP, a
cell membrane permeable cGMP derivative, and blocked by KT5823, a
PKG inhibitor. Our data support previous findings showing that
inhibiting PDE5 by exisulind and its higher affinity analogues,
such as CP461 induces apoptosis in colon tumor cells throug the
cGMP-PKG signaling pathway (4,5,12,16). The main activated PKG isoform in
OS-RC-2 cells is PKG Iβ, which is responsible for the anticancer
activities, such as the inhibition of cell proliferation and the
increase in the cell apoptotic rate. It has been reported that the
activation of soluble guanylyl cyclase by NO leads to an increase
in cGMP synthesis and the activation of PKG (20). In this study, the PKG Iα and PKG
Iβ were significantly increased in the SNAP group, indicating that
NO was involved in the cGMP-PKG pathway.
Activated cGMP-PKG may play a key role in
determining the levels of cellular β-catenin that has been well
known for its role in carcinogenesis. It has been found that
various types of human cancer, such as kidney (21), liver (22), prostate (23) and colon cancer (24) involved an abnormal Wnt signaling
pathway characterized by an accumulation of β-catenin. Moreover,
β-catenin plays a central role in oncogenesis through tge
regulation of cell adhesion and the Wnt signaling pathways
(25). Along with TCF/Lef,
accumulated β-catenin facilitates the expression of multiple genes
involved in proliferation and apoptosis. However, mutations of
adenomatous polyposis coli (APC) in tumor cells prevent the
formation of the APC/axin/GSK3β/PP2A complex, leading to a
defective phosphorylation of β-catenin by GSK3b kinase (26–28). Finally, β-catenin is unable to be
degraded by the ubiquitin-proteasome pathway, resulting in an
accumulation of a cellular β-catenin. We found that the reduction
of β-catenin was followed by an increased phosphorylation of
β-catenin in the OS-RC-2 cells treated with PDE5 siRNA. Our data
confirmed previous findings that the attenuation of cellular
β-catenin accumulation through an activated cGMP-PKG pathway and
the increased phosphorylation of β-catenin occur in
exisulind-treated colonic tumor cells. Moreover, it has been
suggested that PKG can phosphorylate β-catenin directly, leading to
increased proteasomal degradation in a manner mimicking the effect
of GSK3β (16).
The reduction in β-catenin expression suggested that
either an activated proteolysis pathway or an inhibited
transcription was initiated in the OS-RC-2 cell transfected with
PDE5 siRNA. The levels of β-catenin mRNA did not decrease within 48
h following transfection with PDE5 siRNA, indicating an involvement
of proteolysis. In this study, we confirmed directly that the
levels of β-catenin could not be reduced by MG132, an
ubiquitin-proteasome inhibitor in OS-RC-2 cells transfected with
PDE5 siRNA. Furthermore, the proteolysis of β-catenin was induced
not only by the ubiquitin-proteasomal pathway, but also by
caspases. A previous study demonstrated that the inhibition of
caspase-3 did not block the degradation of β-catenin in
exisulind-treated cells (29),
indicating the necessity of the ubiquitin-proteasome pathway in the
proteolysis of β-catenin. Further studies are required to determine
the function of caspases in OS-RC-2 cells transfected with PDE5
siRNA.
β-catenin/TCF/Lef controls the expression of a
number of oncogenes, including cyclin D1, c-myc and peroxisome
proliferator-activated receptor (PPAR)β. The transcription of
cyclin D1 can be initiated by the accumulation of β-catenin
(11,15). It has been found that the levels
of β-catenin are related to the activated pathway of cGMP-PKG in
various tumor cells treated with exisulind and its higher affinity
analogues (5,12). This study demonstrated that
β-catenin protein and both cyclin D1 mRNA and protein levels
decreased in a time-dependent manner, indicating that the decrease
in β-catenin levels promoted the downregulation of TCF/Lef-mediated
promoter transcription. The suppression of cyclin D1 by antisense
oligonucleotide can induce apoptosis in squamous carcinoma cells
(30), suggesting that the
reduction in cyclin D1 levels may play a direct role in promoting
the progression of apoptosis in PDE5 siRNA-transfected cells.
However, we found that the reduction in cyclin D1 levels was not
blocked by MG132. This may be explained by the fact that cyclin D1
protein can be degraded through an ubiquitin-proteasome pathway
triggered by GSK3β phosphorylation. The reduction in cyclin D1
expression can be blocked by an inhibitor of JNK. This supports
previous findings that the phosphorylation and activation of the
MEKK1-SEK1-JNK1 pathway triggered by either exisulind-mediated PKG
activation or a constitutively active mutant PKG construct promotes
cGMP-mediated apoptosis (18).
Therefore, the reduction in cyclin D1 expression is mainly affected
by the β-catenin and JNK pathways, increasing the apoptosis of
OS-RC-2 cells.
The results for the levels of p21 and p27 are not
similar in different colon cancer cell lines treated with
exisulind; i.e., the levels of both were increased in SW480 cells
(12) and only the level of p21
protein was increased in HT29 cells (4). However, all the changes were
reversed by KT5823, showing that both p21 and p27 are downstream
targets of activated PKG. In this study, the increased levels of
p21 protein were blocked by Sp1 siRNA in 8-Br-cGMP-treated OS-RC-2
cells. Therefore, the mechanism involved in the changes of p21 may
be related to the activation of Sp1 by PKG in 8-Br-cGMP-treated
OS-RC-2 cells. Further study supports the hypothesis that the
activation of PKG can lead to the serine phosphorylation of the
transcription factor, Sp (30).
In conclusion, decreasing levels of cyclin D1 and increasing levels
of p21 can inhibit proliferation and promote apoptosis through the
cGMP-PKG pathway in OS-RC-2 cells transfected with PDE5 siRNA.
Acknowledgements
This study was supported by a grant from the Natural
Foundation of Ningbo Science and Technology Bureau, China,
(2010A610047). This study also was supported by a grant from the
Technology foundation of Yinzhou Science and Technology Bureau,
China.
References
|
1
|
Rini BI, Rathmell WK and Godley P: Renal
cell carcinoma. Curr Opin Oncol. 20:300–306. 2008. View Article : Google Scholar
|
|
2
|
Escudier B: Chemo-immunotherapy in RCC:
the end of a story. Lancet. 375:613–614. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mulders PF, Brouwers AH, Hulsbergen-van
der Kaa CA, et al: Guideline ‘Renal cell carcinoma’. Ned Tijdschr
Geneeskd. 152:376–380. 2008.(In Dutch).
|
|
4
|
Zhu B and Strada SJ: The novel functions
of cGMP-specific phosphodiesterase 5 and its inhibitors in
carcinoma cells and pulmonary/cardiovascular vessels. Curr Top Med
Chem. 7:437–454. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thompson WJ, Piazza GA, Li H, et al:
Exisulind induction of apoptosis involves guanosine 3′,5′-cyclic
monophosphate phosphodiesterase inhibition, protein kinase G
activation, and attenuated beta-catenin. Cancer Res. 60:3338–3342.
2000.PubMed/NCBI
|
|
6
|
Piazza GA, Thompson WJ, Pamukcu R, et al:
Exisulind, a novel proapoptotic drug, inhibits rat urinary bladder
tumorigenesis. Cancer Res. 61:3961–3968. 2001.PubMed/NCBI
|
|
7
|
Zhu B, Vemavarapu L, Thompson WJ and
Strada SJ: Suppression of cyclic GMP-specific phosphodiesterase 5
promotes apoptosis and inhibits growth in HT29 cells. J Cell
Biochem. 94:336–350. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lim JT, Piazza GA, Han EK, et al: Sulindac
derivatives inhibit growth and induce apoptosis in human prostate
cancer cell lines. Biochem Pharmacol. 58:1097–1107. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tinsley HN, Gary BD, Keeton AB, et al:
Sulindac sulfide selectively inhibits growth and induces apoptosis
of human breast tumor cells by phosphodiesterase 5 inhibition,
elevation of cyclic GMP, and activation of protein kinase G. Mol
Cancer Ther. 8:3331–3340. 2009. View Article : Google Scholar
|
|
10
|
Tinsley HN, Gary BD, Keeton AB, et al:
Inhibition of PDE5 by sulindac sulfide selectively induces
apoptosis and attenuates oncogenic Wnt/beta-catenin-mediated
transcription in human breast tumor cells. Cancer Prev Res (Phila).
4:1275–1284. 2011. View Article : Google Scholar
|
|
11
|
Tetsu O and McCormick F: Beta-catenin
regulates expression of cyclin D1 in colon carcinoma cells. Nature.
398:422–426. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li H, Liu L, David ML, et al:
Pro-apoptotic actions of exisulind and CP461 in SW480 colon tumor
cells involve beta-catenin and cyclin D1 downregulation. Biochem
Pharmacol. 64:1325–1336. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Deguchi A, Thompson WJ and Weinstein IB:
Activation of protein kinase G is sufficient to induce apoptosis
and inhibit cell migration in colon cancer cells. Cancer Res.
64:3966–3973. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Carlson B, Lahusen T, Singh S, et al:
Downregulation of cyclin D1 by transcriptional repression in MCF-7
human breast carcinoma cells induced by flavopiridol. Cancer Res.
59:4634–4641. 1999.PubMed/NCBI
|
|
16
|
Liu L, Li H, Underwood T, et al: Cyclic
GMP-dependent protein kinase activation and induction by exisulind
and CP461 in colon tumor cells. J Pharmacol Exp Ther. 299:583–592.
2001.PubMed/NCBI
|
|
17
|
Kwon IK, Wang R, Thangaraju M, et al: PKG
inhibits TCF signaling in colon cancer cells by blocking
beta-catenin expression and activating FOXO4. Oncogene.
29:3423–3434. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Soh JW, Mao Y, Kim MG, et al: Cyclic GMP
mediates apoptosis induced by sulindac derivatives via activation
of c-Jun NH2-terminal kinase 1. Clin Cancer Res. 6:4136–4141.
2000.PubMed/NCBI
|
|
19
|
Hofmann F: The biology of cyclic
GMP-dependent protein kinases. J Biol Chem. 280:1–4. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Das A, Xi L and Kukreja RC: Protein kinase
G-dependent cardioprotective mechanism of phosphodiesterase-5
inhibition involves phosphorylation of ERK and GSK3beta. J Biol
Chem. 283:29572–29585. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Peruzzi B and Bottaro DP: Beta-catenin
signaling: linking renal cell carcinoma and polycystic kidney
disease. Cell Cycle. 5:2839–2841. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Thompson MD and Monga SP: WNT/beta-catenin
signaling in liver health and disease. Hepatology. 45:1298–1305.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chesire DR and Isaacs WB: Beta-catenin
signaling in prostate cancer: an early perspective. Endocr Relat
Cancer. 10:537–560. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stein U, Arlt F, Smith J, et al:
Intervening in beta-catenin signaling by sulindac inhibits
S100A4-dependent colon cancer metastasis. Neoplasia. 13:131–144.
2011.PubMed/NCBI
|
|
25
|
Li H, Pamukcu R and Thompson WJ:
beta-Catenin signaling: therapeutic strategies in oncology. Cancer
Biol Ther. 1:621–625. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ueda M, Gemmill RM, West J, et al:
Mutations of the beta-and gamma-catenin genes are uncommon in human
lung, breast, kidney, cervical and ovarian carcinomas. Br J Cancer.
85:64–68. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tarmin L, Yin J, Harpaz N, et al:
Adenomatous polyposis coli gene mutations in ulcerative
colitis-associated dysplasias and cancers versus sporadic colon
neoplasms. Cancer Res. 55:2035–2038. 1995.PubMed/NCBI
|
|
28
|
Lewis A, Davis H, Deheragoda M, et al: The
C-terminus of Apc does not influence intestinal adenoma development
or progression. J Pathol. 226:73–83. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Whitehead CM, Earle KA, Fetter J, et al:
Exisulind-induced apoptosis in a non-small cell lung cancer
orthotopic lung tumor model augments docetaxel treatment and
contributes to increased survival. Mol Cancer Ther. 2:479–488.
2003.PubMed/NCBI
|
|
30
|
Saikawa Y, Kubota T, Otani Y, et al:
Cyclin D1 antisense oligonucleotide inhibits cell growth stimulated
by epidermal growth factor and induces apoptosis of gastric cancer
cells. Jpn J Cancer Res. 92:1102–1109. 2001. View Article : Google Scholar : PubMed/NCBI
|