Lewy body disease is a heterogeneous group of
neurodegenerative disorders characterized by α-synuclein (α-syn)
accumulation that includes Lewy body dementia (LBD) and Parkinson’s
disease (PD) (1–3). While the progressive accumulation of
amyloid β oligomers has been identified as one of the important
toxic events in Alzheimer’s disease (AD) leading to synaptic
dysfunction, the accumulation of α-syn has been linked to the
pathogenesis of PD and LBD. Amyloid β also promotes α-syn
aggregation and toxicity in LBD (4,5).
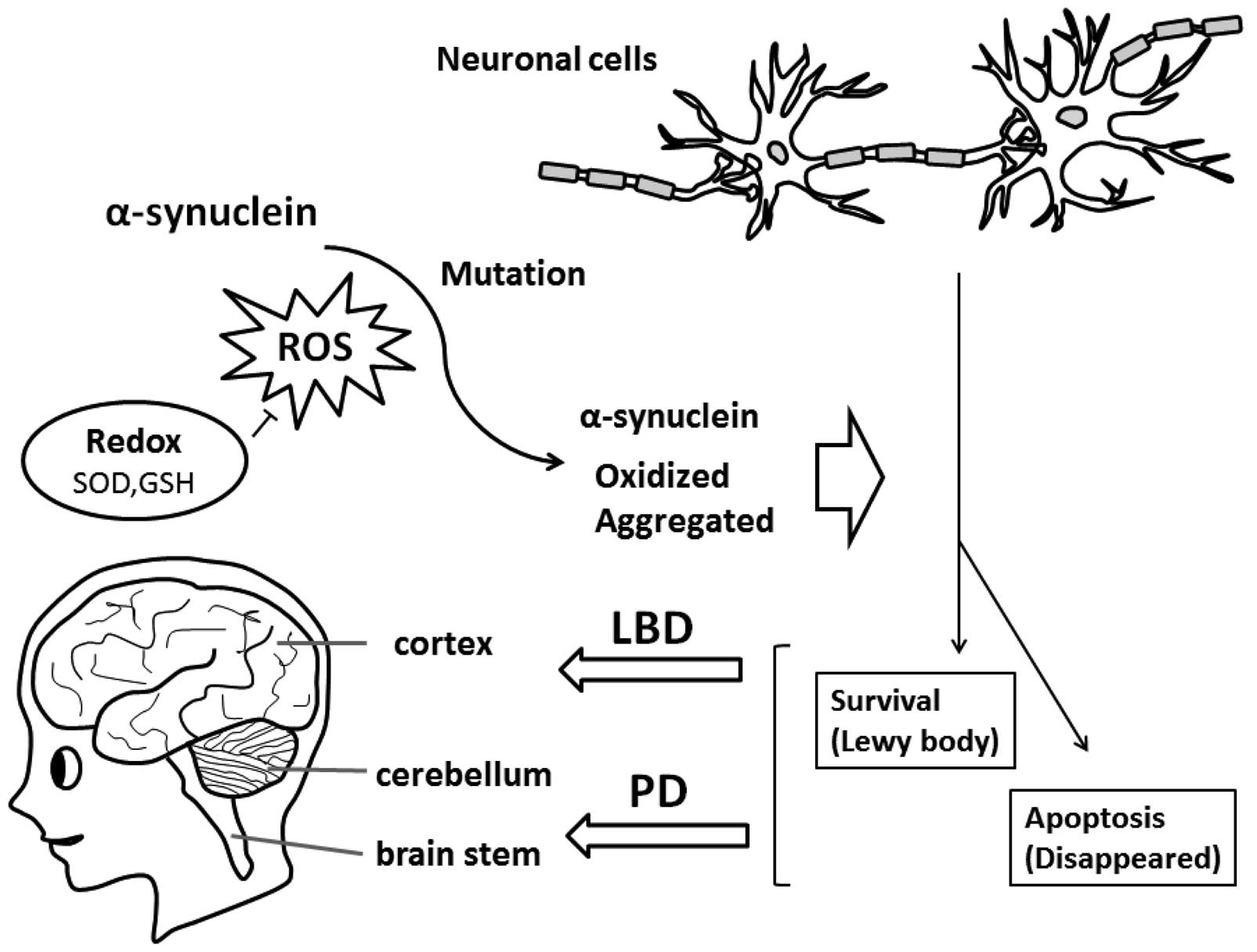
Aggregated α-syn is a major component of inclusions in PD and other
brains affected by synucleinopathy, indicating that α-syn
aggregation is associated with the key pathogenesis of
neurodegenerative disorders, such as LBD (Fig. 1). The impairment of lysosomal
pathways, including autophagy involved in α-syn clearance may play
an important role in LBD. Whereas in patients with LBD the clinical
presentation is of dementia followed by parkinsonism, in patients
with PD dementia the initial signs are of parkinsonism followed by
dementia (6,7). Although the mechanisms underlying
α-syn aggregation and toxicity are not fully elucidated, it is
clear that α-syn undergoes post-translational modifications, and
interacts with numerous proteins, hormones, neurotransmitters and
metals that can modulate its aggregation propensity (8,9).
Oxidized forms of α-syn found in sporadic PD and LBD
have been shown to block autophagy/mitophagy (10,11). The term mitophagy has been created
to describe the selective removal of mitochondria by autophagy.
Mitophagy is constitutively active in healthy neurons. Evidence
indicates that alterations in mitophagy may participate in the
mechanisms of α-syn-mediated neurodegeneration (synucleinopathy)
(12,13). Modifications in the rate of
aggregation and clearance of α-syn may be responsible for the
formation of toxic amyloid β and α-syn oligomers in LBD. The
autophagic pathway is the major pathway involved in the degradation
of long-term proteins and organelles, cellular remodeling and
survival, which has been linked to cell death, and is markedly
activated in degenerative disorders (14,15). Alterations in the mitophagy
pathway in LBD/PD support the possibility that modulators of the
autophagic pathway may have potential therapeutic effects. For
example, activating autophagy by rapamycin treatment has been shown
to improve α-syn accumulation, related neuropathology and the
development of neurodegeneration (16). LBD represents the most common form
of dementia and movement disorders in the aging population
following AD, and displays widespread cortical and subcortical
pathology (17). A better
understanding of the mechanisms of α-syn dysfunction may help to
elucidate the molecular mechanisms of neurodegeneration and may be
the basis for the development of novel therapeutic strategies. The
present review discusses potential alterations in components of
mitophagy in LBD. We also provide an overview on the cellular
functions of the mitochondrial kinase phosphatase and tensin
homologue-induced putative kinase 1 (PINK1), with particular
emphasis on the mitochondrial damage response pathway and
mitochondrial quality control involved in LBD synucleinopathy.
LBD is characterized by the presence of
intra-neuronal cell inclusions termed Lewy bodies with α-syn as
their chief component (18).
Alterations of α-syn expression and impairment of its degradation
can lead to the formation of intra-cellular deposits of this
protein. α-syn aggregation is now accepted as a key step preceding
the formation of Lewy bodies. Whereas LBD is characterized by the
general neuronal loss of the dopaminergic system, a high percentage
of surviving neurons contain inclusions in the form of Lewy bodies
(19). Widespread distribution of
Lewy bodies through almost all brain areas is a characteristic
feature in LBD, while these are found mainly within the brainstem
in PD (20,21) (Fig.
1). Oligomerization and accumulation of fibrillar α-syn
aggregates are the molecular processes involved in the
pathophysiology of PD and LBD (22), in which the affectedness in PD
brainstem causes parkinsonian symptoms and the additional cortical
affectedness in LBD. The missense mutations in α-syn also promote
the aggregation process of this protein (23). Overexpressed or misfolded α-syn
can be secreted to the extracellular space (24). Although the precise sequence of
events responsible for α-syn fibrillation remains unknown,
aggregated α-syn species with altered solubility of α-syn species
with various molecular weights are found particularly in grey
matter (25,26). The mechanisms underlying cellular
α-syn aggregation are crucial to the understanding of the
pathogenic process of the diseases.
Protein misfolding and aggregation is a shared
feature of a number of neurodegenerative diseases, which can be
suppressed and promoted by several factors, such as protein
degradation systems, molecular chaperones and free radical
reactions. Several of these factors are under the control of normal
mitochondrial function. Thus, mitochondrial dysfunction may cause
the accumulation of protein aggregates, including the α-syn
protein. The toxic effects of α-syn have been linked to the
aggregated forms rather than the monomers (27). It has been demonstrated that
oxidative stress may play a key role in the spread of α-syn
pathology (28,29), therefore, α-syn-evoked protein
oxidation leads to disturbances in synaptic transmission and
neuronal cell death. The interaction of secreted α-syn with other
amyloidgenic proteins, such as amyloid β and its involvement in
irreversible mitochondrial disfunction with oxidative stress has
been previously reported (30),
which is an important factor involved in LBD neuronal cell loss. It
is well known that some of the genes that cause recessive PD are
associated with mitochondrial function, although recessive PD
phenotypically differs from LBD. α-syn accumulation is also caused
by the inhibition of the proteasome and autophagy-lysosome
(31). In general, α-syn
predominantly associates with the inner mitochondrial membrane,
where it can apparently interact with complex I, resulting in
reduced mitochondrial complex I activity and increased free radical
production (32). The failure of
the mitochondrial control eventually contributes to cell death.
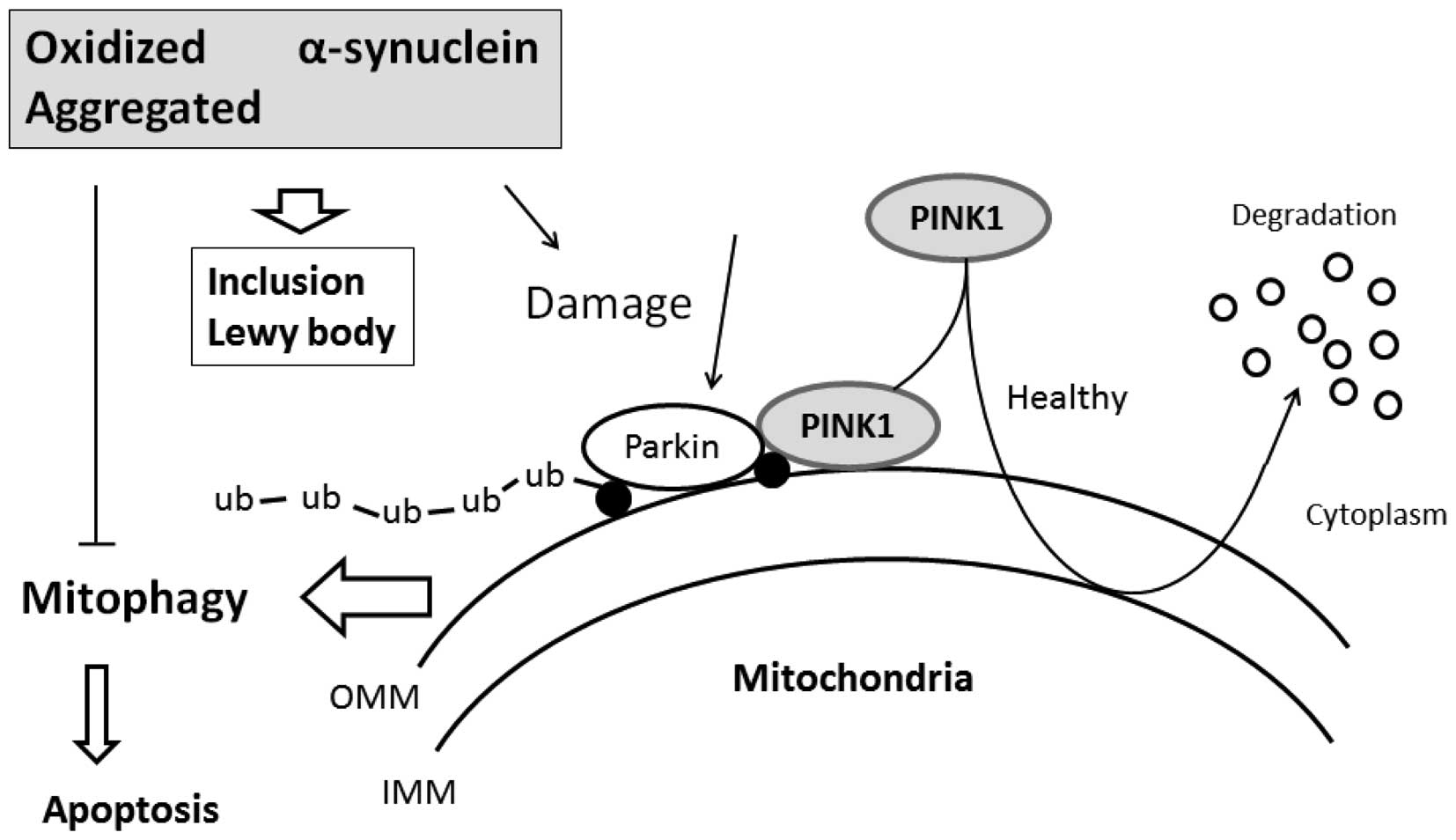
PINK1 may be a sensor of mitochondrial damage and marker of
mitophagy. The presence of LB in the brainstem and nigral neuronal
cells has been described in brains from PD carriers with
heterozygous PINK1 mutation (33).
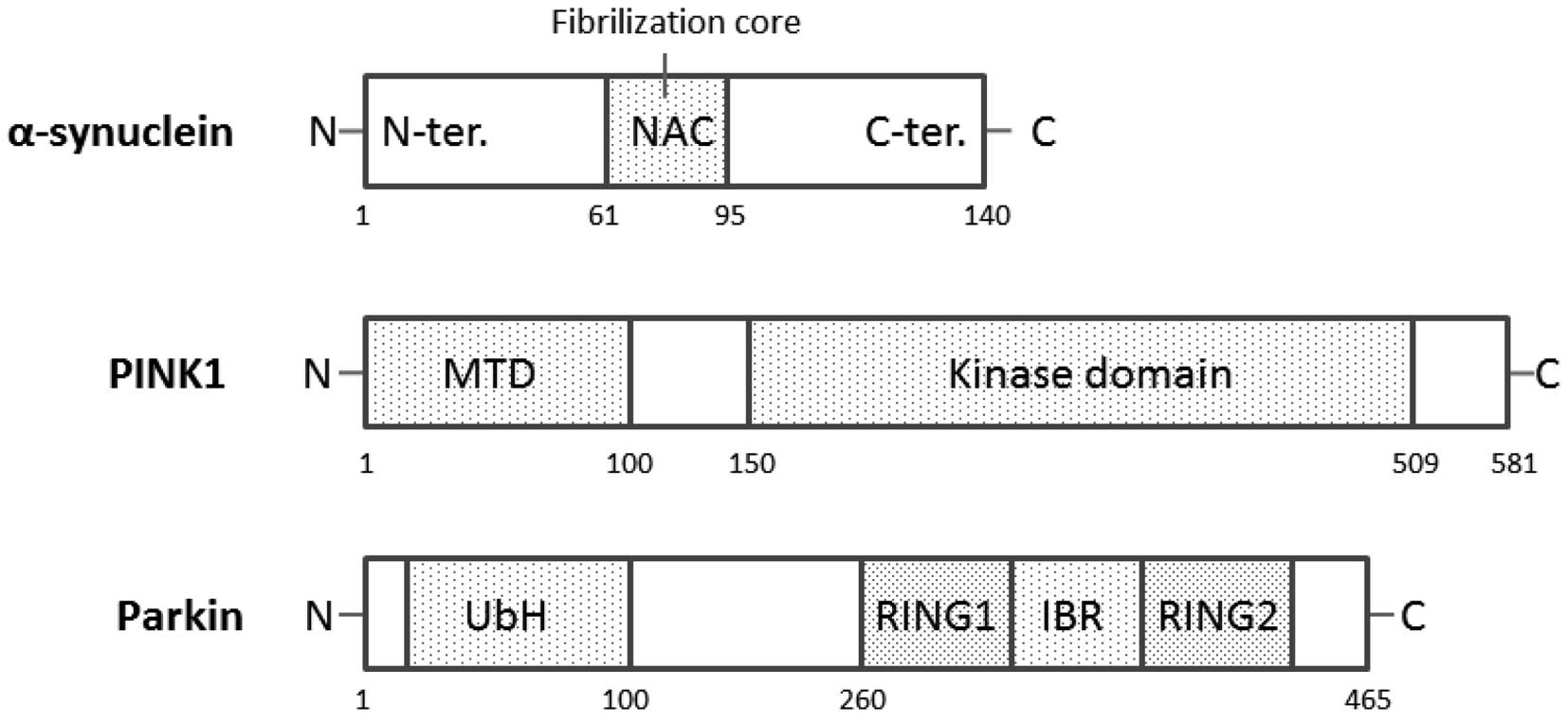
The α-syn cDNA encodes a 140-amino-acid protein,
which is predominantly expressed in the hippocampus, neocortex,
thalamus, cerebellum, olfactory bulb and substantia nigra. In these
neuronal tissue cells, α-syn is highly expressed in the
mitochondria, where the expression of cytosolic α-syn is also high
(34). α-syn is a 19-kDa protein
from which a 35-amino acid peptide [non-amyloid component (NAC)] is
derived. The schematic structure of human α-syn is shown in
Fig. 2. NAC is the second
component, after amyloid β, identified in the AD amyloid
preparation (35,36). Structure predictions indicate that
the NAC peptide sequence has a tendency to form β structures
consistent with its association with amyloid protein. α-syn has
also been shown to be linked to various aspects of mitochondrial
dysfunction (37). The
accumulation of α-syn in the mitochondria of dopaminergic neurons
leads to increased production of reactive oxygen species (ROS)
(38,39) that can be involved in a number of
pro-survival pathways, including the regulation of mitophagy via
the removal of defective mitochondria (40). The overexpression and/or mutation
of α-syn are associated with protein aggregation and inhibit a
number of cellular processes, including mitochondrial function.
α-syn and β-synuclein are homologous proteins implicated in
synucleinopathies (4). While
α-syn is neurotoxic and its aggregation and deposition are related
to neurodegeneration, β-synuclein is considered as a potent
inhibitor of α-syn aggregation and toxicity (41).
Increased mitophagy activity observed in cells
expressing α-syn is an important phenomenon linked to α-syn-induced
toxicity. When mitochondrial import is compromised by
depolarization, PINK1 accumulates on the mitochondrial surface,
which in turn mediates mitophagic destruction (52), which decreases α-syn toxicity and
promotes inclusion body formation. Therefore, PINK1 can protect
neuronal cells against possibly harmful proteins, such as α-syn
that would result in cellular stress (53). Consequently, PINK1 is involved in
α-synucleinopathy (54). The
mitochondrial chaperone protein TRAP1 which has been shown to be
phosphorylated by PINK1 also mitigates α-syn toxicity (55). However, the neuroprotective role
for the α-syn has been shown in attenuating manganese (Mn)-induced
toxicity in the background of PD (56). Evidence suggests that α-syn has an
effect on protein quality control systems, such as the
ubiquitin-proteasome system and autophagy, suggesting that
increased mitophagy activity is an important phenomenon linked to
α-syn toxicity during aging (57). PINK1 overexpression rescues the
α-syn-induced PD phenotype in D. melanogaster, apparently
through targeting of α-syn for degradation (58). A simultaneous increase in
α-syn and PINK1 may have a synergistic effect for cell
protection, which seems to be a result of the upregulation of
pro-survival mechanisms in response to an increase in ROS signaling
due to the effect of α-syn overexpression (59).
Therapeutic strategies exploit the observation that
defects in key processes required for cellular homeostasis produce
an alternative metabolic situation or diseases. ω-3 polyunsaturated
fatty acid (PUFA) induces autophagy. In general, several foods can
affect the cognitive processes in the central nervous system
neurons by ω-3 PUFA. It has been known that dietary ω-3 PUFA
improves memory and learning processes, and also affects gene
expression in neurons (60). A
long-term diet rich in ω-3 PUFA and/or docosahexaenoic acid (DHA)
leads to lower protein oxidative damage with no modifications in
the number of cortical astrocytes and microglial cells and with no
effects in α-syn expression. However, α-syn oligomerization is
markedly enhanced by ω-3 PUFA, while β-synuclein oligomerization is
not affected (41). PUFA-enriched
diets significantly alter the mRNA expression levels of several
genes in central nervous system neurons, and these effects may be
related to the balance of polyenoic fatty acids [(n-3)/(n-6)] in
the cell membrane. Diets enriched in saturated fatty acids and
simple carbohydrates are often deficient in ω-3 PUFA (61). Perilla frutescens is a good
source of ω-3 PUFA (>50% total fatty acid) which are
indispensable fatty acids that can be converted to DHA in the liver
and the developing brain, and contains one of the highest contents
of ω-3 fatty acid among edible plant seeds (62). The perilla diet supplementation
promotes neuronal signaling and alters synaptic plasticity for
improved learning and memory (63). A diet with spirulina, a natural
product from blue green algae, also provides neuroprotection, as
demonstrated in an α-syn model of PD (64). In other words, spirulina can
protect against the neuronal loss induced by α-syn.
Curcumin is a well-known polyphenol in commonly used
in the preparation of Asian food, known as the ingredient turmeric,
and has been shown to exhibit anti-inflammatory, anti-carcinogenic
and anti-microbial activities (65). Studies have suggested the
potential therapeutic role of curcumin in neurological disorders,
including PD and LBD. Curcumin has been shown to inhibit α-syn
aggregation and attenuate α-syn oligomer toxicity in neuronal cells
(66). Curcumin binds to the
preformed α-syn aggregates, and strongly reduces their cellular
toxicity by minimizing their hydrophobic surface exposure. In
addition, curcumin accelerates α-syn aggregation and reduces the
population of soluble oligomers which are cytotoxic. Of note,
curcumin does not bind to monomeric α-syn but binds specifically to
oligomeric intermediates (66).
The degree of curcumin binding correlates with the extent of α-syn
oligomerization, suggesting that the oligomeric structure of α-syn
is required for effective curcumin binding. Curcumin also prevents
aggregation of α-syn by increasing the reconfiguration rate
(67), which may decrease the
population of toxic oligomeric intermediates of α-syn.
It is known that caloric restriction and the
polyphenolic antioxidant, resveratrol, may promote longevity.
Recently, several studies have shown that Sirt3 along with FoxO3 in
addition to Sirt1 are of importance in promoting the anti-aging
function of resveratrol (68).
Sirt3 in cooperation with Sirt1 activates FoxO3, and they contain
the initial mitochondrial signaling response to activate PINK-1,
thereby promoting mitophagy. By the way, PINK1 is overexpressed in
the mitochondria of hepatocytes of ethanol-treated rats, in which
ethanol treatment represents a possible protective mechanism
through the stimulation of mitophagy (69). Antioxidant vitamins, such as
vitamin E and the vitamin-like substance coenzyme Q10 have been
used in the treatment of LBD with some efficacy (70). With the potent anti-fibrillogenic,
as well as fibril-destabilizing activities of α-syn, these vitamin
compounds may prove to be useful in the prevention of LBD.
Treatment with rotenone, a toxic isoflavonoid, can reproduce
nigra-striatal cell loss and other features of PD in rodents
(71). Rotenone treatment results
in decreased spontaneous locomotor movement and increased
cytoplasmic α-syn expression. The mitochondrial PINK1 protein
levels are also increased following treatment with rotenone.
The present review was supported by Grants-in-Aid
from the Ministry of Education, Culture, Sports, Science and
Technology in Japan.
|
1
|
Overk CR and Masliah E: Pathogenesis of
synaptic degeneration in Alzheimer’s disease and Lewy body disease.
Biochem Pharmacol. 88:508–516. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Walker LC and LeVine H III: Corruption and
spread of pathogenic proteins in neurodegenerative diseases. J Biol
Chem. 287:33109–33115. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schwarz S, Froelich L and Burns A:
Pharmacological treatment of dementia. Curr Opin Psychiatry.
25:542–550. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Di Giovanni S, Eleuteri S, Paleologou KE,
Yin G, Zweckstetter M, Carrupt PA and Lashuel HA: Entacapone and
tolcapone, two catechol O-methyltransferase inhibitors, block
fibril formation of alpha-synuclein and beta-amyloid and protect
against amyloid-induced toxicity. J Biol Chem. 285:14941–14954.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Crews L, Tsigelny I, Hashimoto M and
Masliah E: Role of synucleins in Alzheimer’s disease. Neurotox Res.
16:306–317. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Goldstein DS, Holmes C, Kopin IJ and
Sharabi Y: Intra-neuronal vesicular uptake of catecholamines is
decreased in patients with Lewy body diseases. J Clin Invest.
121:3320–3330. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Onofrj M, Bonanni L, Manzoli L and Thomas
A: Cohort study on somatoform disorders in Parkinson disease and
dementia with Lewy bodies. Neurology. 74:1598–1606. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Paleologou KE and El-Agnaf OM: α-synuclein
aggregation and modulating factors. Subcell Biochem. 65:109–164.
2012. View Article : Google Scholar
|
|
9
|
Cheng F, Li X, Li Y, Wang C, Wang T, Liu
G, Baskys A, Uéda K, Chan P and Yu S: α-Synuclein promotes
clathrin-mediated NMDA receptor endocytosis and attenuates
NMDA-induced dopaminergic cell death. J Neurochem. 119:815–825.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Song JX, Lu JH, Liu LF, Chen LL,
Durairajan SS, Yue Z, Zhang HQ and Li M: HMGB1 is involved in
autophagy inhibition caused by SNCA/α-synuclein overexpression: a
process modulated by the natural autophagy inducer corynoxine B.
Autophagy. 10:144–154. 2014. View Article : Google Scholar
|
|
11
|
Settembre C, Fraldi A, Jahreiss L,
Spampanato C, Venturi C, Medina D, de Pablo R, Tacchetti C,
Rubinsztein DC and Ballabio A: A block of autophagy in lysosomal
storage disorders. Hum Mol Genet. 17:119–129. 2008. View Article : Google Scholar
|
|
12
|
Todde V, Veenhuis M and van der Klei IJ:
Autophagy: principles and significance in health and disease.
Biochim Biophys Acta. 1792:3–13. 2009. View Article : Google Scholar
|
|
13
|
Pan T, Kondo S, Le W and Jankovic J: The
role of autophagy-lysosome pathway in neurodegeneration associated
with Parkinson’s disease. Brain. 131:1969–1978. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Matus S, Valenzuela V and Hetz C: A new
method to measure autophagy flux in the nervous system. Autophagy.
10:710–714. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Giordano S, Darley-Usmar V and Zhang J:
Autophagy as an essential cellular antioxidant pathway in
neurodegenerative disease. Redox Biol. 2:82–90. 2013. View Article : Google Scholar
|
|
16
|
Spencer B, Potkar R, Trejo M, Rockenstein
E, Patrick C, Gindi R, Adame A, Wyss-Coray T and Masliah E: Beclin
1 gene transfer activates autophagy and ameliorates the
neurodegenerative pathology in alpha-synuclein models of
Parkinson’s and Lewy body diseases. J Neurosci. 29:13578–13588.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Isella V, Rucci F, Traficante D, Mapelli
C, Ferri F and Appollonio IM: The applause sign in cortical and
cortical-subcortical dementia. J Neurol. 260:1099–1103. 2013.
View Article : Google Scholar
|
|
18
|
Kahle PJ, Neumann M, Ozmen L, Müller V,
Odoy S, Okamoto N, Jacobsen H, Iwatsubo T, Trojanowski JQ,
Takahashi H, Wakabayashi K, Bogdanovic N, Riederer P, Kretzschmar
HA and Haass C: Selective insolubility of alpha-synuclein in human
Lewy body diseases is recapitulated in a transgenic mouse model. Am
J Patholx. 159:2215–2225. 2013. View Article : Google Scholar
|
|
19
|
Robinson PA: Protein stability and
aggregation in Parkinson’s disease. Biochem J. 413:1–13. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Olanow CW, Perl DP, DeMartino GN and
McNaught KS: Lewy-body formation is an aggresome-related process: a
hypothesis. Lancet Neurol. 3:496–503. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Braak H, Müller CM, Rüb U, Ackermann H,
Bratzke H, de Vos RA and Del Tredici K: Pathology associated with
sporadic Parkinson’s disease - where does it end? J Neural Transm
Suppl. 70:89–97. 2006.
|
|
22
|
Luk KC, Hyde EG, Trojanowski JQ and Lee
VM: Sensitive fluorescence polarization technique for rapid
screening of alpha-synuclein oligomerization/fibrillization
inhibitors. Biochemistry. 46:12522–12529. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ghosh D, Mondal M, Mohite GM, Singh PK,
Ranjan P, Anoop A, Ghosh S, Jha NN, Kumar A and Maji SK: The
Parkinson’s disease-associated H50Q mutation accelerates
α-Synuclein aggregation in vitro. Biochemistry. 52:6925–6927. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chai YJ, Kim D, Park J, Zhao H, Lee SJ and
Chang S: The secreted oligomeric form of α-synuclein affects
multiple steps of membrane trafficking. FEBS Lett. 587:452–459.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Campbell BC, McLean CA, Culvenor JG, Gai
WP, Blumbergs PC, Jäkälä P, Beyreuther K, Masters CL and Li QX: The
solubility of alpha-synuclein in multiple system atrophy differs
from that of dementia with Lewy bodies and Parkinson’s disease. J
Neurochem. 76:87–96. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wakabayashi K, Yoshimoto M, Fukushima T,
Koide R, Horikawa Y, Morita T and Takahashi H: Widespread
occurrence of alpha-synuclein/NACP-immunoreactive neuronal
inclusions in juvenile and adult-onset Hallervorden-Spatz disease
with Lewy bodies. Neuropathol Appl Neurobiol. 25:363–368. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sharon R, Goldberg MS, Bar-Josef I,
Betensky RA, Shen J and Selkoe DJ: alpha-synuclein occurs in
lipid-rich high molecular weight complexes, binds fatty acids, and
shows homology to the fatty acid-binding proteins. Proc Natl Acad
Sci USA. 98:9110–9115. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dryanovski DI, Guzman JN, Xie Z, Galteri
DJ, Volpicelli-Daley LA, Lee VM, Miller RJ, Schumacker PT and
Surmeier DJ: Calcium entry and α-synuclein inclusions elevate
dendritic mitochondrial oxidant stress in dopaminergic neurons. J
Neurosci. 33:10154–10164. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Müller SK, Bender A, Laub C, Högen T,
Schlaudraff F, Liss B, Klopstock T and Elstner M: Lewy body
pathology is associated with mitochondrial DNA damage in
Parkinson’s disease. Neurobiol Aging. 34:2231–2233. 2013.
View Article : Google Scholar
|
|
30
|
Wilkaniec A, Strosznajder JB and Adamczyk
A: Toxicity of extracellular secreted alpha-synuclein: its role in
nitrosative stress and neurodegeneration. Neurochem Int.
62:776–783. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xilouri M, Brekk OR and Stefanis L:
α-Synuclein and protein degradation systems: a reciprocal
relationship. Mol Neurobiol. 47:537–551. 2013. View Article : Google Scholar
|
|
32
|
Chinta SJ, Mallajosyula JK, Rane A and
Andersen JK: Mitochondrial α-synuclein accumulation impairs complex
I function in dopaminergic neurons and results in increased
mitophagy in vivo. Neurosci Lett. 486:235–239. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gandhi S, Muqit MM, Stanyer L, Healy DG,
Abou-Sleiman PM, Hargreaves I, Heales S, Ganguly M, Parsons L, Lees
AJ, Latchman DS, Holton JL, Wood NW and Revesz T: PINK1 protein in
normal human brain and Parkinson’s disease. Brain. 129:1720–1731.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hong L, Ko HW, Gwag BJ, Joe E, Lee S, Kim
YT and Suh YH: The cDNA cloning and ontogeny of mouse
alpha-synuclein. Neuroreport. 9:1239–1243. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
El-Agnaf OM, Jakes R, Curran MD, Middleton
D, Ingenito R, Bianchi E, Pessi A, Neill D and Wallace A:
Aggregates from mutant and wild-type alpha-synuclein proteins and
NAC peptide induce apoptotic cell death in human neuroblastoma
cells by formation of beta-sheet and amyloid-like filaments. FEBS
Lett. 440:71–75. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jensen PH, Hojrup P, Hager H, Nielsen MS,
Jacobsen L, Olesen OF, Gliemann J and Jakes R: Binding of Abeta to
alpha- and beta-synucleins: identification of segments in
alpha-synuclein/NAC precursor that bind Abeta and NAC. Biochem J.
323:539–546. 1997.PubMed/NCBI
|
|
37
|
Schapira AH and Gegg M: Mitochondrial
contribution to Parkinson’s disease pathogenesis. Parkinsons Dis.
2011:1591602011.
|
|
38
|
Liu F, Hindupur J, Nguyen JL, Ruf KJ, Zhu
J, Schieler JL, Bonham CC, Wood KV, Davisson VJ and Rochet JC:
Methionine sulfoxide reductase A protects dopaminergic cells from
Parkinson’s disease-related insults. Free Radic Biol Med.
45:242–255. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dias V, Junn E and Mouradian MM: The role
of oxidative stress in Parkinson’s disease. J Parkinsons Dis.
3:461–491. 2013.
|
|
40
|
Weber TA and Reichert AS: Impaired quality
control of mitochondria: aging from a new perspective. Exp
Gerontol. 45:503–511. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Israeli E and Sharon R: Beta-synuclein
occurs in vivo in lipid-associated oligomers and forms
hetero-oligomers with alpha-synuclein. J Neurochem. 108:465–474.
2009. View Article : Google Scholar
|
|
42
|
Valente EM, Abou-Sleiman PM, Caputo V,
Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR,
Healy DG, Albanese A, Nussbaum R, González-Maldonado R, Deller T,
Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola
B, Auburger G and Wood NW: Hereditary early-onset Parkinson’s
disease caused by mutations in PINK1. Science. 304:1158–1160. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Blackinton JG, Anvret A, Beilina A, Olson
L, Cookson MR and Galter D: Expression of PINK1 mRNA in human and
rodent brain and in Parkinson’s disease. Brain Res. 1184:10–16.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Weihofen A, Thomas KJ, Ostaszewski BL,
Cookson MR and Selkoe DJ: Pink1 forms a multiprotein complex with
Miro and Milton, linking Pink1 function to mitochondrial
trafficking. Biochemistry. 48:2045–2052. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jin SM, Lazarou M, Wang C, Kane LA,
Narendra DP and Youle RJ: Mitochondrial membrane potential
regulates PINK1 import and proteolytic destabilization by PARL. J
Cell Biol. 191:933–942. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Beilina A, Van Der Brug M, Ahmad R,
Kesavapany S, Miller DW, Petsko GA and Cookson MR: Mutations in
PTEN-induced putative kinase 1 associated with recessive
parkinsonism have differential effects on protein stability. Proc
Natl Acad Sci USA. 102:5703–5708. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pridgeon JW, Olzmann JA, Chin LS and Li L:
PINK1 protects against oxidative stress by phosphorylating
mitochondrial chaperone TRAP1. PLoS Biol. 5:e1722007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Michiorri S, Gelmetti V, Giarda E,
Lombardi F, Romano F, Marongiu R, Nerini-Molteni S, Sale P, Vago R,
Arena G, Torosantucci L, Cassina L, Russo MA, Dallapiccola B,
Valente EM and Casari G: The Parkinson-associated protein PINK1
interacts with Beclin1 and promotes autophagy. Cell Death Differ.
17:962–974. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Matsuda S, Kitagishi Y and Kobayashi M:
Function and characteristics of PINK1 in mitochondria. Oxid Med
Cell Longev. 2013:6015872013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Rakovic A, Shurkewitsch K, Seibler P,
Grünewald A, Zanon A, Hagenah J, Krainc D and Klein C: Phosphatase
and tensin homolog (PTEN)-induced putative kinase 1
(PINK1)-dependent ubiquitination of endogenous Parkin attenuates
mitophagy: study in human primary fibroblasts and induced
pluripotent stem cell-derived neurons. J Biol Chem. 288:2223–2237.
2013. View Article : Google Scholar :
|
|
51
|
Chu CT: A pivotal role for PINK1 and
autophagy in mitochondrial quality control: implications for
Parkinson disease. Hum Mol Genet. 19:R28–R37. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Greene AW, Grenier K, Aguileta MA, Muise
S, Farazifard R, Haque ME, McBride HM, Park DS and Fon EA:
Mitochondrial processing peptidase regulates PINK1 processing,
import and Parkin recruitment. EMBO Rep. 13:378–385. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Dexter DT and Jenner P: Parkinson disease:
from pathology to molecular disease mechanisms. Free Radic Biol
Med. 62:132–144. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Murakami T, Moriwaki Y, Kawarabayashi T,
Nagai M, Ohta Y, Deguchi K, Kurata T, Morimoto N, Takehisa Y,
Matsubara E, Ikeda M, Harigaya Y, Shoji M, Takahashi R and Abe K:
PINK1, a gene product of PARK6, accumulates in
alpha-synucleinopathy brains. J Neurol Neurosurg Psychiatry.
78:653–654. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Butler EK, Voigt A, Lutz AK, Toegel JP,
Gerhardt E, Karsten P, Falkenburger B, Reinartz A, Winklhofer KF
and Schulz JB: The mitochondrial chaperone protein TRAP1 mitigates
α-Synuclein toxicity. PLoS Genet. 8:e10024882012. View Article : Google Scholar
|
|
56
|
Bornhorst J, Chakraborty S, Meyer S,
Lohren H, Brinkhaus SG, Knight AL, Caldwell KA, Caldwell GA, Karst
U, Schwerdtle T, Bowman A and Aschner M: The effects of pdr1,
djr1.1 and pink1 loss in manganese-induced toxicity and the role of
α-synuclein in C. elegans. Metallomics. 6:476–490. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Sampaio-Marques B, Felgueiras C, Silva A,
Rodrigues M, Tenreiro S, Franssens V, Reichert AS, Outeiro TF,
Winderickx J and Ludovico P: SNCA (α-synuclein)-induced toxicity in
yeast cells is dependent on sirtuin 2 (Sir2)-mediated mitophagy.
Autophagy. 8:1494–1509. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Todd AM and Staveley BE: Pink1 suppresses
alpha-synuclein-induced phenotypes in a Drosophila model of
Parkinson’s disease. Genome. 51:1040–1046. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Todd AM and Staveley BE: Expression of
Pink1 with α-synuclein in the dopaminergic neurons of Drosophila
leads to increases in both lifespan and healthspan. Genet Mol Res.
11:1497–1502. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Hajjar T, Meng GY, Rajion MA, Vidyadaran
S, Othman F, Farjam AS, Li TA and Ebrahimi M: Omega 3
polyunsaturated fatty acid improves spatial learning and
hippocampal peroxisome proliferator activated receptors (PPARα and
PPARγ) gene expression in rats. BMC Neurosci. 13:1092012.
View Article : Google Scholar
|
|
61
|
Galland L: Diet and inflammation. Nutr
Clin Pract. 25:234–241. 2010. View Article : Google Scholar
|
|
62
|
Eckert GP, Franke C, Nöldner M, Rau O,
Wurglics M, Schubert-Zsilavecz M and Müller WE: Plant derived
omega-3-fatty acids protect mitochondrial function in the brain.
Pharmacol Res. 61:234–241. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lee J, Park S, Lee JY, Yeo YK, Kim JS and
Lim J: Improved spatial learning and memory by perilla diet is
correlated with immunoreactivities to neurofilament and α-synuclein
in hilus of dentate gyrus. Proteome Sci. 10:722012. View Article : Google Scholar
|
|
64
|
Pabon MM, Jernberg JN, Morganti J,
Contreras J, Hudson CE, Klein RL and Bickford PC: A
spirulina-enhanced diet provides neuroprotection in an α-synuclein
model of Parkinson’s disease. PLoS One. 7:e452562012. View Article : Google Scholar
|
|
65
|
Villegas I, Sánchez-Fidalgo S and Alarcón
de la Lastra C: New mechanisms and therapeutic potential of
curcumin for colorectal cancer. Mol Nutr Food Res. 52:1040–1061.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Singh PK, Kotia V, Ghosh D, Mohite GM,
Kumar A and Maji SK: Curcumin modulates α-synuclein aggregation and
toxicity. ACS Chem Neurosci. 4:393–407. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Ahmad B and Lapidus LJ: Curcumin prevents
aggregation in α-synuclein by increasing reconfiguration rate. J
Biol Chem. 287:9193–9199. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Das S, Mitrovsky G, Vasanthi HR and Das
DK: Antiaging properties of a grape-derived antioxidant are
regulated by mitochondrial balance of fusion and fission leading to
mitophagy triggered by a signaling network of
Sirt1-Sirt3-Foxo3-PINK1-PARKIN. Oxid Med Cell Longev.
2014:3451052014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Eid N, Ito Y, Maemura K and Otsuki Y:
Elevated autophagic sequestration of mitochondria and lipid
droplets in steatotic hepatocytes of chronic ethanol-treated rats:
an immunohistochemical and electron microscopic study. J Mol
Histol. 44:311–326. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kones R: Parkinson’s disease:
mitochondrial molecular pathology, inflammation, statins, and
therapeutic neuroprotective nutrition. Nutr Clin Pract. 25:371–389.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Girish C and Muralidhara: Propensity of
Selaginella delicatula aqueous extract to offset rotenone-induced
oxidative dysfunctions and neurotoxicity in Drosophila
melanogaster: implications for Parkinson’s disease.
Neurotoxicology. 33:444–456. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Chen L, Thiruchelvam MJ, Madura K and
Richfield EK: Proteasome dysfunction in aged human alpha-synuclein
transgenic mice. Neurobiol Dis. 23:120–126. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Liu W, Vives-Bauza C, Acín-Peréz- R,
Yamamoto A, Tan Y, Li Y, Magrané J, Stavarache MA, Shaffer S, Chang
S, Kaplitt MG, Huang XY, Beal MF, Manfredi G and Li C: PINK1 defect
causes mitochondrial dysfunction, proteasomal deficit and
alpha-synuclein aggregation in cell culture models of Parkinson’s
disease. PLoS One. 4:e45972009. View Article : Google Scholar
|
|
74
|
Kamp F, Exner N, Lutz AK, Wender N,
Hegermann J, Brunner B, Nuscher B, Bartels T, Giese A, Beyer K,
Eimer S, Winklhofer KF and Haass C: Inhibition of mitochondrial
fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J.
29:3571–3589. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wilhelmus MM, Nijland PG, Drukarch B, de
Vries HE and van Horssen J: Involvement and interplay of Parkin,
PINK1, and DJ1 in neurodegenerative and neuroinflammatory
disorders. Free Radic Biol Med. 53:983–992. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Tamura T, Yoshida M, Hashizume Y and Sobue
G: Lewy body-related α-synucleinopathy in the spinal cord of cases
with incidental Lewy body disease. Neuropathology. 32:13–22. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Funabe S, Takao M, Saito Y, Hatsuta H,
Sugiyama M, Ito S, Kanemaru K, Sawabe M, Arai T, Mochizuki H,
Hattori N and Murayama S: Neuropathologic analysis of Lewy-related
α-synucleinopathy in olfactory mucosa. Neuropathology. 33:47–58.
2013. View Article : Google Scholar
|