Introduction
In recent years, flavonoids have been investigated
intensively for the treatment of various types of cancer and have
been proven to be potent anticancer agents (1,2).
The therapeutic efficacy of many natural plants has already been
described by practitioners of traditional medicine for several
disorders. Centipedegrass [Eremochloa ophiuroides (Munro)
Hack], which is native to China and Southeast Asia, is one of the
most versatile medicinal herbs (3). Composition analysis by liquid
chromatography-mass spectrometry (LC-MS)/MS has helped to identify
several C‑glycosidic flavones and phenolic constituents from
centipe-degrass, including maysin and its derivatives.
Centipedegrass extract (CGE), used in the present study, is mainly
composed of orientin, isoorientin, rhamnosylisoorientin,
derhamnosyl-maysin, maysin, luteolin and
luteolin-6-C-bovinopyranose at the concentrations of 1.4, 28.1,
63.1, 49.1, 97.3, 34 and 85 μg/mg DW, respectively. Maysin
and its derivatives have thus far been found only in
centipedegrass, maize silk and teosinte. Centipedegrass is known to
possess a wide spectrum of antibacterial, antifungal and
insecticidal properties (4,5),
and a recent study reported that CGE exhibits anti-adipogenic
activity (6). However, the
anticancer effects of flavonoid-rich centipedegrass against acute
lymphoblastic leukemia (ALL) are not yet known.
ALL, which involves the blood and bone marrow, is
the most common type of cancer in children younger than 5 years of
age (7). ALL is composed of many
subtypes that represent different clinical behaviors and require
different therapy schemes (8,9).
Family history and exposure to radiation may affect the risk of
developing ALL (10). Treatment
includes chemotherapy, steroids, radiation therapy, intensive
combined treatments (including bone marrow or stem cell
transplants) and growth factors. The cost for the complete
treatment of a child with ALL is approximately US $100,000 per
patient (11). Therefore, the
development of an effective chemotherapeutic regimen that
selectively induces apoptosis in cancer cells is of great
importance. Signaling pathways, such as the phosphatidylinositol
3-kinase (PI3K)/AKT and mitogen-activated protein kinase
(MAPK)/extracellular signal-regulated kinase (ERK) pathways are
frequently upregulated in ALL (12–14).
To maintain tissue homeostasis in multicellular
organisms, cell proliferation and cell death must be regulated.
This regulation may be achieved, in part, by coupling the process
of cell cycle progression and apoptosis (15). Apoptosis is a highly regulated
process and some of its important characteristics are DNA
fragmentation, cell shrinkage, nuclear condensation,
phosphatidylserine (PS) flipping from the inner to the outer
leaflet of the plasma membrane and the alteration of mitochondrial
membrane potential (∆Ψm) (16–19). The timing and order of cell cycle
events are monitored during cell cycle checkpoints that occur at
the G1/S boundary, in the S phase and during the G2/M phase
(20).
The current study was designed to demonstrate the
effects of CGE on human leukemia cell lines. To the best of our
knowledge, our data provide the first evidence that CGE functions
as a broad-range anticancer agent by effectively triggering
apoptosis in human leukemia cells through a mechanism involving the
regulation of PI3K/AKT and MAPKs, which in turn leads to caspase
activation and apoptosis.
Materials and methods
Preparation of the CGE
The preparation of CGE was carried out as previously
described (21). The dried
centipedegrass (5 kg) leaves were ground in a Wiley mill and passed
through a 420-μm sieve. The ground sample (1 kg) was
extracted 3 times with 80% methanol (MeOH, 100 l; Merck & Co,
Inc., Whitehouse Station, NJ, USA) for 24 h with constant shaking
at ambient temperature in the dark. The extracts were filtered
using No. 2 filter paper (Advantec Mfs Inc., Dublin, CA, USA) and
concentrated in vacuo. The MeOH extracts were fractionated
with n-hexane and ethyl acetate (EA) (Merck & Co, Inc.). The EA
extracts were concentrated in vacuo and the dried compounds
were dissolved in MeOH. The dissolved extracts in MeOH were diluted
in 20% MeOH and chromatographed over a Toyopearl HW-40C resin
(Tosoh Co., Tokyo, Japan) column using 70% MeOH (elution volume,
700 ml). The fraction was evaporated and freeze dried. The dried
extracts were reconstituted in dimethylsulfoxide (DMSO) for the
treatment of the cells.
Reagents and antibodies
Unless otherwise specified, all chemicals were
purchased from Sigma Chemical Co. (St. Louis, MO, USA), including
thiazolyl blue tetrazolium blue (MTT), Annexin V-FITC, protease
inhibitor cocktail, propidium iodide (PI) and DMSO. Antibodies to
Bcl-2 (no. 2870), BAX (no. 5023), Bid (no. 2002), cytochrome
c (no. 4247), poly(ADP-ribose) polymerase (PARP; no. 9542),
caspase-3 (no. 9662), caspase-7 (no. 9492), caspase-9 (no. 9502),
p-PI3K (no. 4228), p-AKT (Ser 473; no. 4058), p-AKT (Thr 308; no.
9275), AKT (no. 4691), p-BAD (Ser 136; no. 9291), HSP-60 (no. 4870)
and GAPDH (no. 2118), as well as a horseradish peroxidase
(HRP)-conjugated secondary antibody (no. 7074), the PI3K inhibitor,
LY294002, and the ERK inhibitor, U0126, were obtained from Cell
Signaling Technology (Beverly, MA, USA). Pan-caspase inhibitors
(Z-IETD-FMK and Z-VAD-FMK) were purchased from R&D Systems
(Minneapolis, MN, USA). The ECL plus chemiluminescence kit was
obtained from Amersham Pharmacia Biotech (Piscataway, NJ, USA). The
mitochondrial dye, 3,3′-dihexyloxacarbocyanine iodide
[DiOC6(3)], was
obtained from Molecular Probes (Carlsbad, CA, USA).
Cell culture and viability assay
The Jurkat (TIB-152), CEM-CM3 (TIB-195), CCRF-CEM
(CCL-119) and MOLT-4 (CRL-1582) cell lines were obtained from the
American Type Culture Collection (ATCC, Rockville, MD, USA) and
cultured in RPMI-1640 medium supplemented with 10% heat-inactivated
FBS and 100 U/ml penicillin at 37°C in a humidified 5%
CO2 incubator.
MTT was used to evaluate the viability of the cells.
Briefly, the leukemia cells were plated at a density
1×105 cells/well in 96-well plates. The cells were
treated with a series of concentrations (2.5, 5, 10, 20, 40, 80 and
160 μg/ml) of CGE. CGE was dissolved in DMSO and the final
concentration of DMSO in the culture medium was <0.05%. After 24
h of incubation with CGE, 10 μl of MTT solution (5 mg/ml in
PBS as a stock solution) were added to each well of a 96-well plate
followed by incubation for an additional 1 h at 37°C. The
MTT‑reducing activity of the cells was measured by treating them
with acidic isopropanol prior to reading at 570 nm using a
microplate reader (Tecan Systems Inc., San Jose, CA, USA). The
IC50 value was calculated using SigmaPlot 10.0 software
(Systat Software Inc., San Jose, CA, USA) with the 4-parameter
logistic function standard curve analysis for dose response.
Detection of apoptosis by flow
cytometry
The extent of apoptosis was evaluated by flow
cytometry using Annexin V‑FITC. The cells were grown at a density
of 1×106 cells in 6-well plates and treated with various
concentrations of CGE [control (DMSO) and 2 × IC50] for
24 h. Following treatment with CGE, the cells were harvested,
washed with pre-chilled 1X PBS, and resuspended in 100 μl of
binding buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, and 2.5 mM
CaCl2). Subsequently, 100 ng/ml Annexin V-FITC were
added and the mixture was incubated for 10–15 min in the dark at
room temperature. PI (20 μg/ml) was added followed by incubation
for an additional 15 min in the dark. A total of 400 μl of
binding buffer was then added and fluorescence was monitored
immediately using an FC500 Flow Cytometer (Beckman Coulter,
Fullerton, CA, USA). A total of 10,000 events was collected per
sample. The analysis was carried out using CXP analysis software
version 2.2 (Beckman Coulter) and the percentage of apoptotic cells
was assessed.
Cell cycle distribution analysis
The cells were plated at a density of
1×106 cells/well in a 6-well plate. The cells were
treated with various concentrations of CGE [control (DMSO),
IC50 and 2 × IC50] for 24 h. After 24 h, the
cells were washed twice with PBS. The cells were fixed with 70%
ethanol overnight at 4°C. The fixed cells were washed and
resuspended in PBS containing 100 μg/ml RNase A and then
incubated for 1 h at 37°C. The cells were stained by the addition
of 20 μg/ml PI for 15–20 min at room temperature in the
dark. The DNA content of the stained cells was analyzed using an
FC500 Flow Cytometer (Beckman Coulter). The data were analyzed
using CXP analysis software version 2.2 (Beckman Coulter).
Determination of ∆Ψm by
DiOC6(3)
The ΔΨm was determined using the
DiOC6(3) dye. The
leukemia cells were treated with DMSO (controls) or with the
indicated concentrations of CGE for 24 h. Subsequently, the cells
were washed in cold PBS, resuspended in PBS supplemented with
DiOC6(3) (40 nM),
incubated in the dark at 37°C in an incubator with 5%
CO2 for 20 min, and then immediately analyzed using an
FC500 Flow Cytometer (Beckman Coulter). The data were analyzed
using CXP analysis software version 2.2 (Beckman Coulter).
Caspase activity assay
The activity of caspase-3/7 was determined using the
Caspase-Glo 3/7 Assay kit (Promega Corp., Madison, WI, USA)
according to the manufacturer’s instructions. The leukemia cells
(20,000 cells/well) were seeded in a white 96-well plate and
treated with CGE (2 × IC50 concentration) for 1, 3, 6,
12 and 24 h. Caspase-Glo 3/7 reagent (100 μl) was added to each
well, and the 96-well plate was incubated on a rotary shaker at
room temperature for 10 min, and the luminescence was measured
using a microplate reader (Tecan Systems Inc.). The quantification
of caspase activity was calculated as the fold increase over the
control sample. The caspase activity in the cells treated with CGE
was normalized to the caspase activity of the control cells. To
evaluate the caspase activity in the presence of various
inhibitors, some cells were pre-incubated for 1 h with 50 μM of
caspase-3 inhibitor (Z-DEVD-FMK), 50 μM of caspase-8 inhibitor
(Z-IEDT-FMK), PI3K inhibitor (LY294002) or ERK inhibitor
(U0126).
Preparation of the mitochondrial and
cytosolic fractions
The leukemia cells (1×106 cells) were
treated with CGE (2 × IC50 concentration) for 1, 3, 6,
12 and 24 h. The mitochondrial and cytosolic fractions were
prepared using the Cytosol/Mitochondria Fractionation kit
(Calbiochem, San Diego, CA, USA) according to the manufacturer’s
instructions. The cytosolic and mitochondrial fractions were stored
at −20°C for immunoblot analysis.
Immunoblot analysis
The protein concentration of the cytosolic and
mitochondrial fractions was determined using the BSA method. An
equal quantity of protein (10 μg) was subjected to SDS-PAGE and
transferred to PVDF membranes. The successful transfer of the
protein was assessed by Ponceau-red staining. The membranes were
blocked for 1 h at room temperature in Tris-buffered saline (pH
7.4) containing 0.1% Tween-20 and 5% skim milk. The blocked blots
were incubated with primary antibodies overnight at 4°C using
antibody dilutions recommended by the manufacturer. Further
incubation was performed with HRP-conjugated secondary antibody and
the protein expression was detected with the ECL Plus
Chemiluminescence kit (Amersham Pharmacia Biotech) according to the
manufacturer’s instructions.
Statistical analysis
Statistical analysis was performed by comparing the
mean of the CGE-treated groups with that of the control group using
an Student’s unpaired t-test (Sigma Plot 10.0). Values of
p<0.05, p<0.01, and p<0.001 were considered to indicate
statistically significant differences.
Results
Inhibitory effects of CGE on cell growth
and the induction of apoptosis in leukemia cells
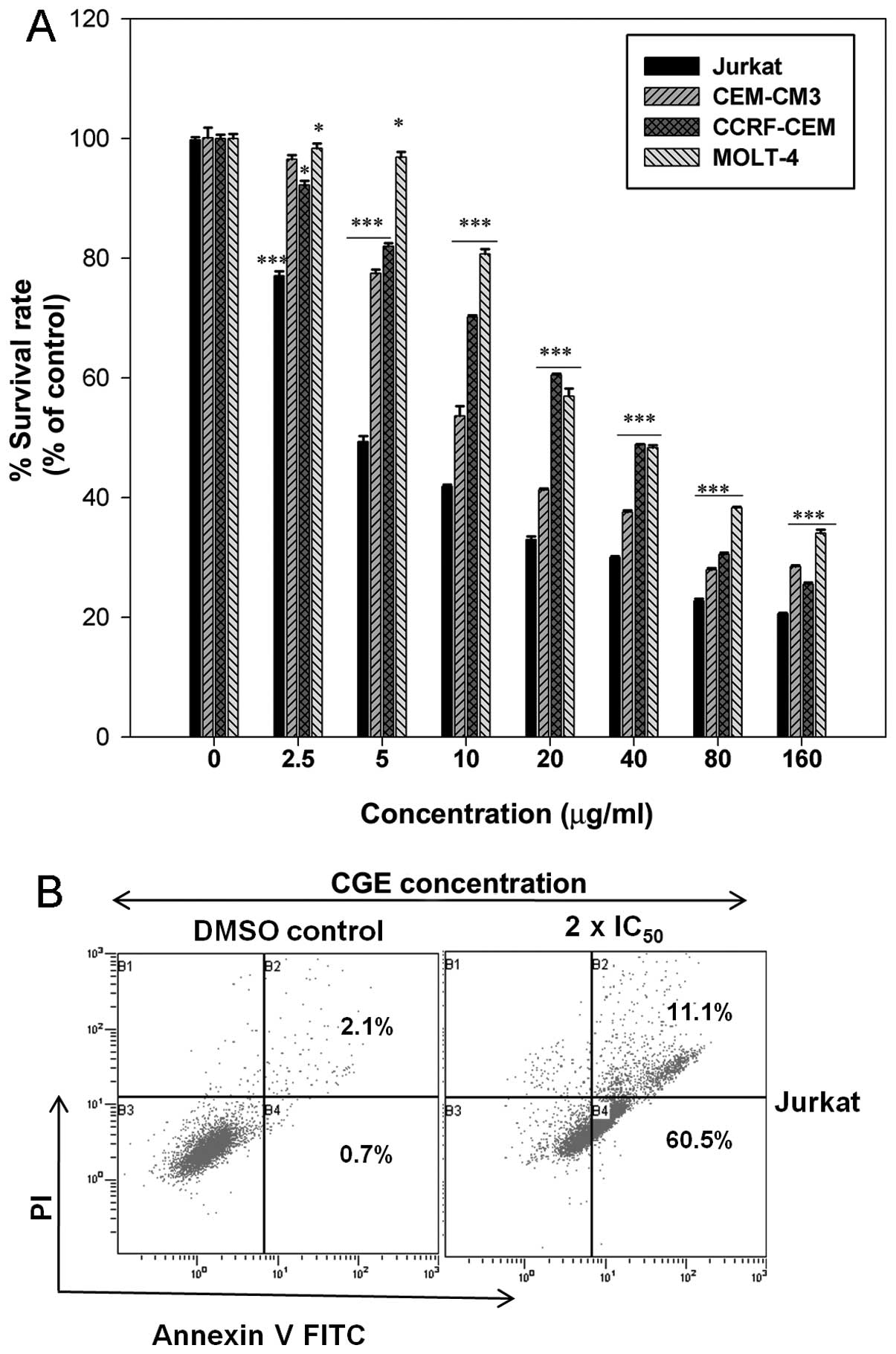
MTT assays were conducted to determine the growth
inhibitory effects of CGE on leukemia cells. Fig. 1A shows the cell survival rate of
the leukemia cells treated with the indicated concentrations of CGE
for 24 h. The cell survival (%) decreased with the increasing CGE
concentration. The IC50 values for CGE treatment were
estimated to be 3.91±0.66, 7.35±0.82, 43.16±2 and 51.17±1.57
μg/ml for the Jurkat, CEM-CM3, CCRF-CEM and MOLT-4 cell
lines, respectively, using SigmaPlot 10.0 software. These results
indicate that CGE exerts a significant cytotoxic effect upon
leukemia cells in a dose-dependent manner.
Due to the growth inhibitory effects observed,
further experiments to determine whether CGE induces apoptosis in
Jurkat cells were warranted (Fig.
1B). After treating the Jurkat cells with the 2 ×
IC50 concentration of CGE for 24 h, the number of early
apoptotic cells increased (60.5%) compared with those for the DMSO
control (0.7%). The total percentage of apoptotic cells was
directly related to the CGE concentration. This result is
consistent with the cytotoxicity assay, and it reveals that CGE
induced the apoptosis of Jurkat cells in a dose-dependent
manner.
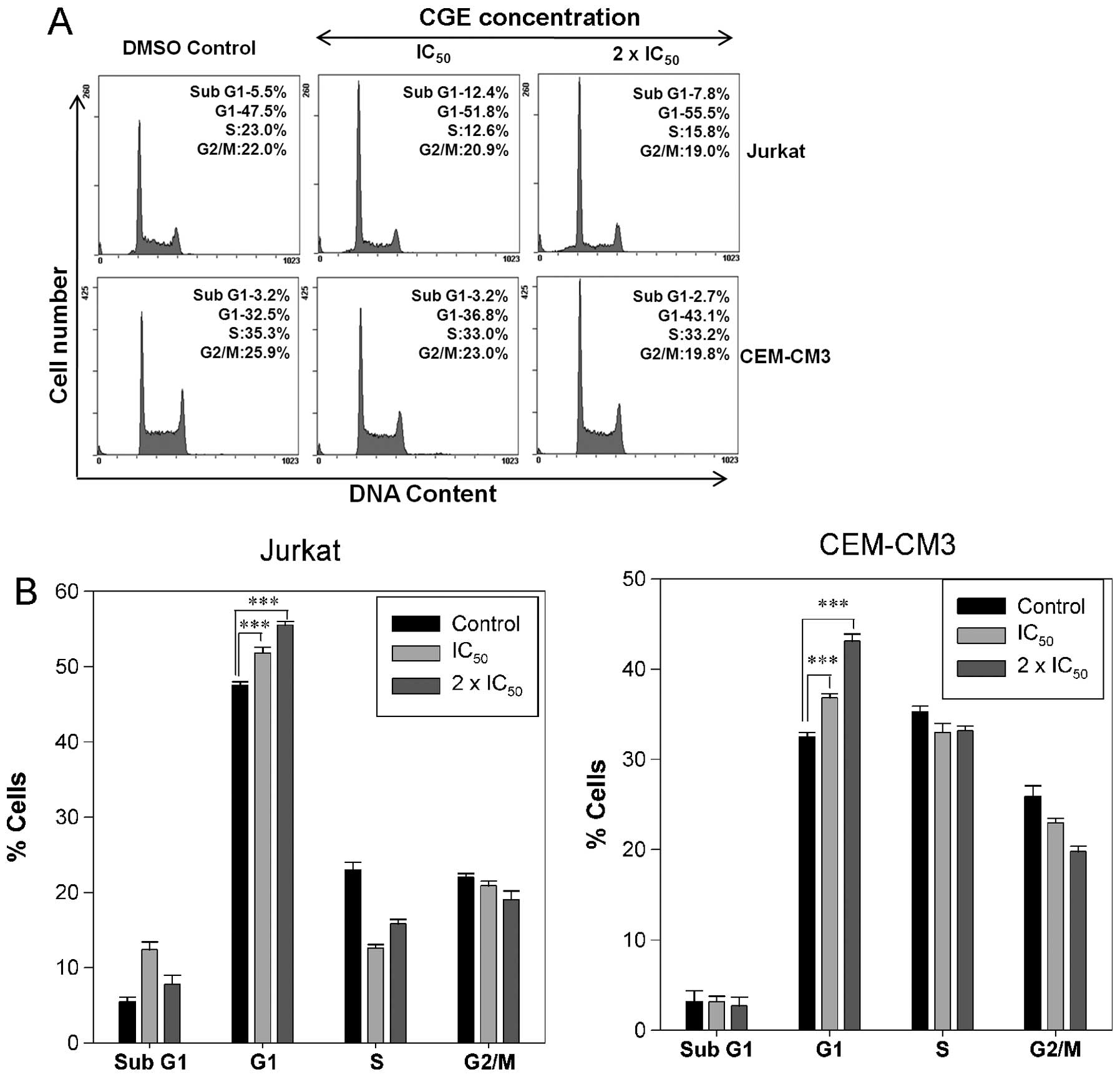
CGE induces cell cycle arrest in leukemia
cells
As the Jurkat and CEM-CM3 cell lines were highly
sensitive to the anti-proliferation effects of CGE, the ability of
CGE to interfere with the cell cycle was assessed. The Jurkat and
CEM-CM3 cells were incubated with various concentrations
(IC50, 2 × IC50) of CGE. Compared with the
DMSO-treated controls, treatment with CGE resulted in an
appreciable arrest of leukemia cells in the G1 phase of the cell
cycle. The G1 phase population of Jurkat cells significantly
increased from 47.5% in the control cells to 51.8 and 55.5% at the
IC50 and the 2 × IC50 concentrations of CGE,
respectively, whereas the G1 phase population in the CEM-CM3
control cells was 32.5% and this increased to 36.8 and 43.1% at the
IC50 and the 2 × IC50 concentrations of CGE,
respectively, after 24 h of treatment (Fig. 2A). This increase in the G1 cell
population was accompanied by a decrease in cell numbers in the S
phase and G2/M phase in the leukemia cell lines (Fig. 2B). These results indicate that the
cell cycle arrest and the induction of apoptosis may be the key
mechanisms behind the antitumor activity of CGE.
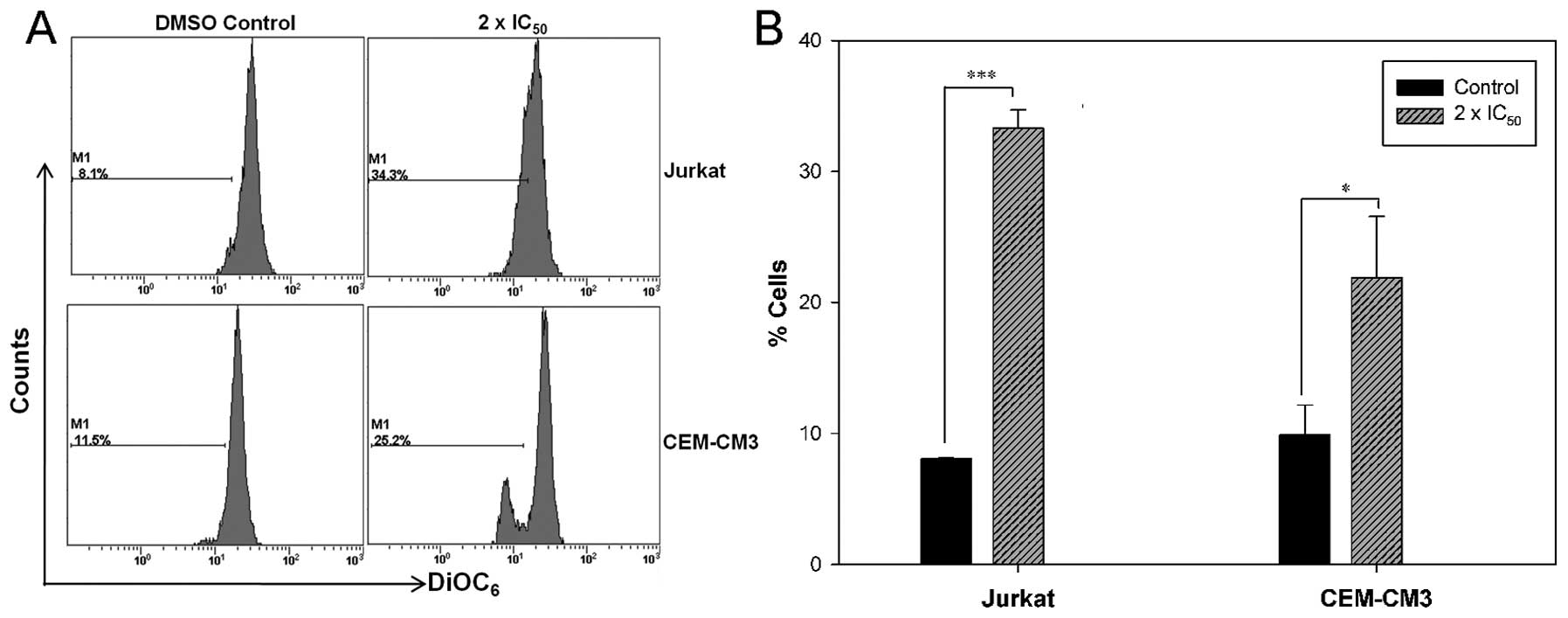
CGE induces mitochondrial
dysfunction
To determine whether the CGE-induced cell apoptosis
is mediated through mitochondrial dysfunction, we determined the
∆Ψm with the mitochondria-sensitive dye, DiOC6(3), by flow cytometry.
DiOC6(3) staining was
found to be increased, which indicated that CGE induced a decrease
in the ∆Ψm in the leukemia cells. A percentage increase from 8.1 to
34.3% was observed in the number of Jurkat cells with a loss of
∆Ψm, whereas a percentage increase from 11.5 to 25.2% was observed
in the number of CEM-CM3 cells with a loss of ∆Ψm (Fig. 3). These findings strongly suggest
that the CGE-induced apoptosis in leukemia cells is accompanied by
breakdown of the ∆Ψm.
CGE‑induced activation of effector
caspases and cyto‑ chrome c release in leukemia cells
In order to investigate the role of caspases in
apoptosis, caspase activity was analyzed after treating the
leukemia cells with the 2 x IC50 concentration of CGE
for 1, 3, 6, 12 and 24 h. We observed that caspase-3/7 activity
reached a maximum level (approximately 2-fold over the control)
after 12–24 h of incubation (Fig.
4A). In accordance with these results, the addition of cell
permeable-specific, irreversible inhibitors of caspase-8
(Z-IETD-FMK) or caspase-3 (Z-DEVD-FMK) enzymes to the leukemia
cells revealed that both the Z-IETD-FMK and Z-DEVD-FMK inhibitors
completely prevented the CGE-induced activation of caspase-3/7. The
CGE-induced apoptosis was inhibited in the presence of Z-VAD-FMK
and Z-IETD-FMK, thereby decreasing apoptosis (Fig. 4A and C). Conversely, the combined
treatment of CGE with the PI3K inhibitor, LY294002, and the ERK
inhibitor, U0126, resulted in enhanced caspase activity. CGE
treatment resulted in a significant increase in cleaved effector
caspases (caspase-3, -7, and -9) (Fig. 4B).
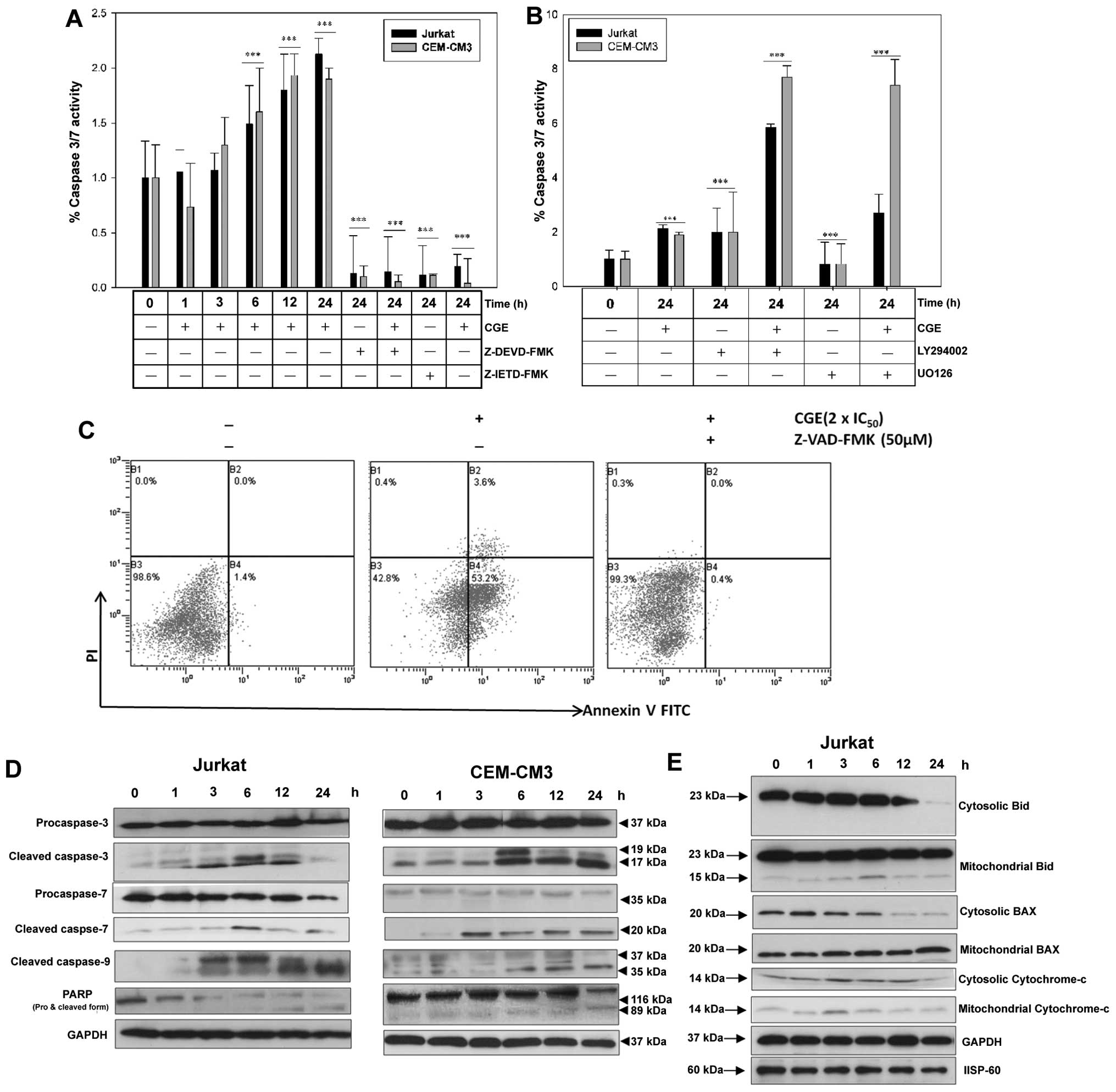 | Figure 4Jurkat and CEM-CM3 cells treated with
the 2 × IC50 concentration of centipedegrass extract
(CGE), for the indicated periods of time. (A) Time-dependent
detection of caspase-3/7 activity in the presence or absence of
specific caspase inhibitors, such as Z-IETD-FMK and Z-DEVD-FMK
(***p<0.001). (B) Time‑dependent detection of
caspase‑3/7 activity in the presence or absence of the specific
phosphatidylinositol 3‑kinase (PI3K) inhibitor, LY294002, and the
ERK inhibitor, U0126 (***p<0.001). (C) Apoptotic cell
death measured by Annexin V‑FITC and propidium iodide (PI) by flow
cytometry. Cells were treated with CGE (2 × IC50) for 24
h in the presence or absence of a general caspase inhibitor. (D)
Whole cell lysates were prepared after the cells were incubated
with CGE for the indicated periods of time. Lysates were used for
the detection of caspase-3, -7, -9, and poly(ADP-ribose) polymerase
(PARP) in CGE-treated Jurkat and CEM-CM3 cells. GAPDH was used as
an internal control. (E) The cytosolic and mitochondrial fractions
were prepared as described in the Materials and methods. Cell
lysates (10 μg) were subjected to 12% SDS-PAGE and
immunoblotted with the Bid, BAX, and cytochrome c
antibodies. A GAPDH antibody (for the cytosolic fraction) and an
HSP-60 antibody (for the mitochondrial fraction) were used as the
loading controls. |
In addition, we found that treatment with CGE
activated caspase-3, -7 and -9 and resulted in the cleavage of the
PARP substrate of caspase-3 (Fig.
4D). The effects of CGE treatment on BAX, Bid and cytochrome
c in the cytosolic and the mitochondrial fractions isolated
from the CGE-treated leukemia cells were evident based on the low
amounts of cytochrome c in the cytosol after 2 h; however,
the cytochrome c content markedly increased from 3 to 12 h.
A time-dependent decrease was observed in the mitochondrial
fraction together with a decrease in cytosolic BAX, whereas the
cleaved form of Bid and mitochondrial BAX increased, and cleaved
Bid decreased in a time-dependent manner (Fig. 4D), which indicates that BAX
translocation from the cytosol to the mitochondria induced
CGE-mediated apoptosis.
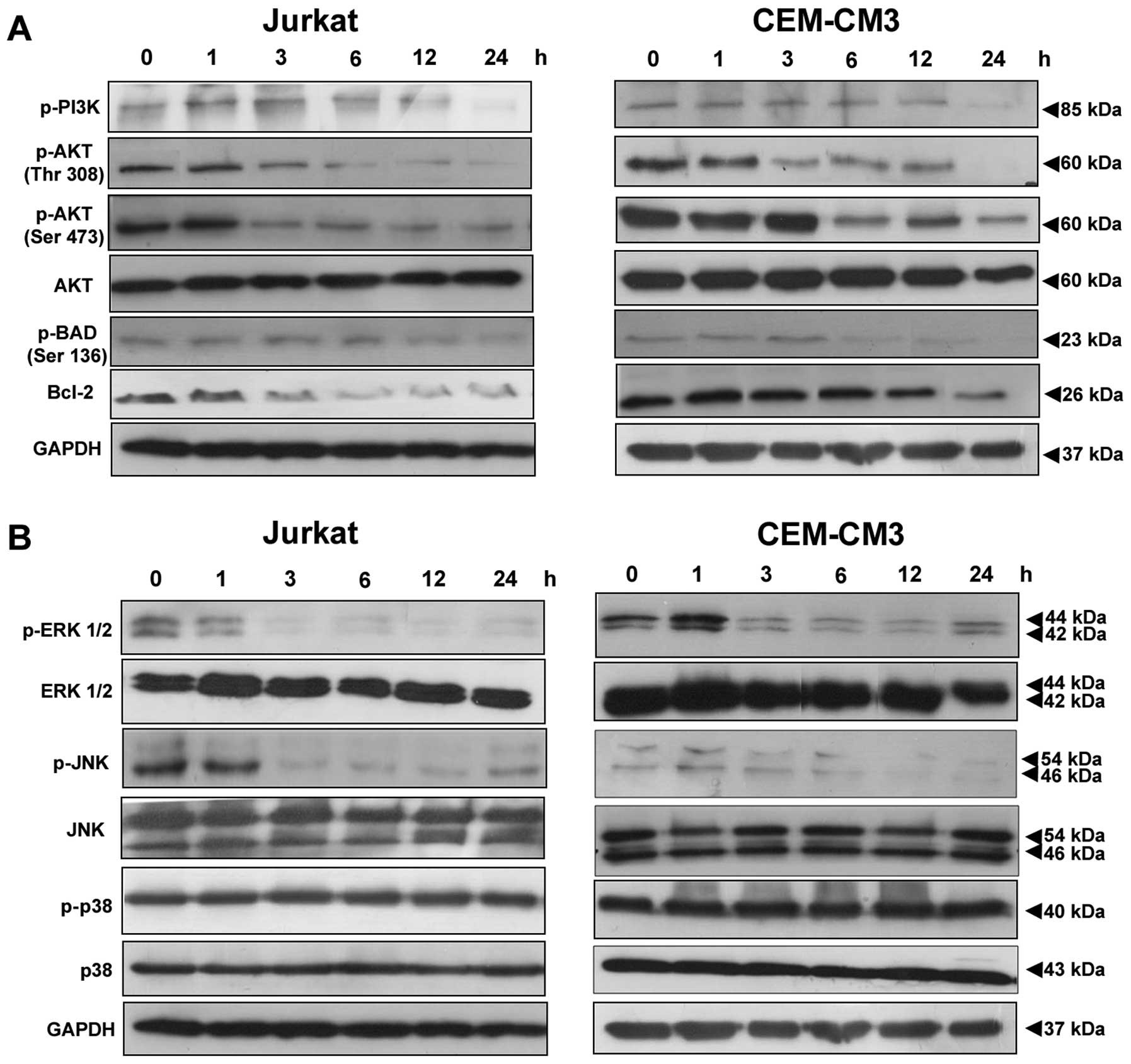
Decreased phosphorylation of PI3K/AKT and
MAPK pathway‑related proteins
CGE downregulated the phosphorylation of AKT and
MAPKs in the leukemia cells. We examined CGE-induced cell death by
measuring the activation of apoptotic proteins in leukemia cells.
The cells were treated with CGE (2 × IC50 concentration)
for 0, 1, 3, 6, 12 and 24 h. CGE induced a decrease in the levels
of p-PI3K, p-AKT (Ser 473), p-AKT (Thr 308), p-BAD, p-ERK and
p-JNK, but the p-p38 protein levels were not altered (Fig. 5).
Discussion
The compositional analysis of CGE has demonstrated
that it is composed of various C-glycosidic flavonoids, such as
maysin and its derivatives, including luteolin, orientin,
isoorientin, rhamnosyl isoorientin, derhamnosly maysin and
luteoin-6-C-boivinopyranose (17). CGE has been reported to exhibit
several biological activities, such as pancreatic lipase inhibitory
activity, antioxidant activity (4) and insecticidal activity (5). The presence of C-glycosidic
flavonoids, such as maysin and its derivatives, in CGE may
attribute to the anticancer properties of CGE. Our results
demonstrated that CGE inhibited cell proliferation in 4 human
leukemia cell lines in a dose-dependent manner. The biochemical
mechanisms through which CGE exerts its effects remain unclear, and
the present study aimed to identify the molecular signaling
pathways involved in the induction of apoptosis by CGE. Our results
clearly demonstrated that CGE inhibited the growth of leukemia
cells by inducing apoptotic cell death as determined by Annexin
V-FITC analysis (Fig. 1).
Generally, the early apoptotic translocation of the phospholipid PS
from the inner to outer leaflet of the plasma membrane occurs due
to the loss of membrane asymmetry, which can be detected by the
binding of Annexin V-FITC to PS (22,23).
Cell cycle control has been proven to be a major
event in cellular division. The disruption of the normal cell cycle
plays a vital role in the development of cancer. A large number of
anticancer natural compounds have been shown to induce cell death
and apoptosis in close association with cell cycle arrest at the G1
phase 9 (24–26). In this study, Jurkat and CEM-CM3
cells treated with CGE showed a significant accumulation of cells
in the G1 phase; there was a concurrent reduction of the cell
population in S and G2/M phase in a dose-dependent manner (Fig. 2), which suggests that the
anti-proliferative effects of CGE are mainly due to the induction
of apoptosis.
There are two types of apoptotic cysteine-dependent
aspartate-directed caspases: initiator and effector (executioner)
caspases. Initiator caspases (e.g., caspase-8 and -9) cleave and
activate effector caspases (e.g., caspase-3, -6 and -7), which in
turn triggers the apoptotic process (27). Caspase-3 has been shown to
inactivate or cleave nuclear proteins, such as PARP, which plays an
important role in DNA repair (28). The initiation of this cascade
reaction is regulated by caspase inhibitors, which can originate
from a number of natural compounds (29,30). In this study, caspase activation
was examined by treating the leukemia cells with CGE. Caspase-3/7
activity increased in a time-dependent manner to approximately
2-fold over the controls after 12–24 h (Fig. 4A). Blocking caspase activation
using Z-VAD-FMK and Z-IETD-FMK significantly suppressed CGE‑induced
apoptosis, whereas the PI3K inhibitor, LY294002, and the ERK
inhibitor, U0126, enhanced the CGE-induced apoptosis (Fig. 4B) and further activated the
effector caspases (caspase-3, -7, and -9) with the concomitant
induction of apoptosis (Fig.
4C).
Apoptotic signaling occurs through the mitochondria
and involves the cellular redistribution of BAX and cytochrome
c followed by the activation of multiple caspases, which
manifest the apoptotic phenotype. During this process, decreased
cytosolic levels of BAX, increased cytosolic levels of cytochrome
c and cleaved Bid are observed (31,32). In this study, to better understand
the cellular redistribution of proteins in CGE-induced apoptosis,
we examined changes in the levels of proteins by western blot
analysis. The levels of BAX decreased gradually, whereas the levels
of cytochrome c and cleaved Bid increased in the cytosolic
fraction (Fig. 4D).
The PI3K/AKT and MAPK signaling pathways are known
to function as crucial components of cell proliferation in cancer
cells. Activated AKT and ERK can function as key effectors of the
PI3K and MAPK signaling pathways to promote cell survival and
proliferation (33,34). Among the wide spectrum of proteins
and genes involved in apoptosis, members of the Bcl-2 family play a
central role in this process (35). The phosphorylation of Bad can
block its apoptotic function (36). In addition, in this study, we
observed the loss of AKT and PI3K activity and the decreased
expression of p-BAD and Bcl-2, which led to apoptosis
eventually.
To the best of our knowledge, this study is the
first to demonstrate the anticancer effects of C‑glycosidic
flavonoids, such as maysin and its derivatives, in CGE, which
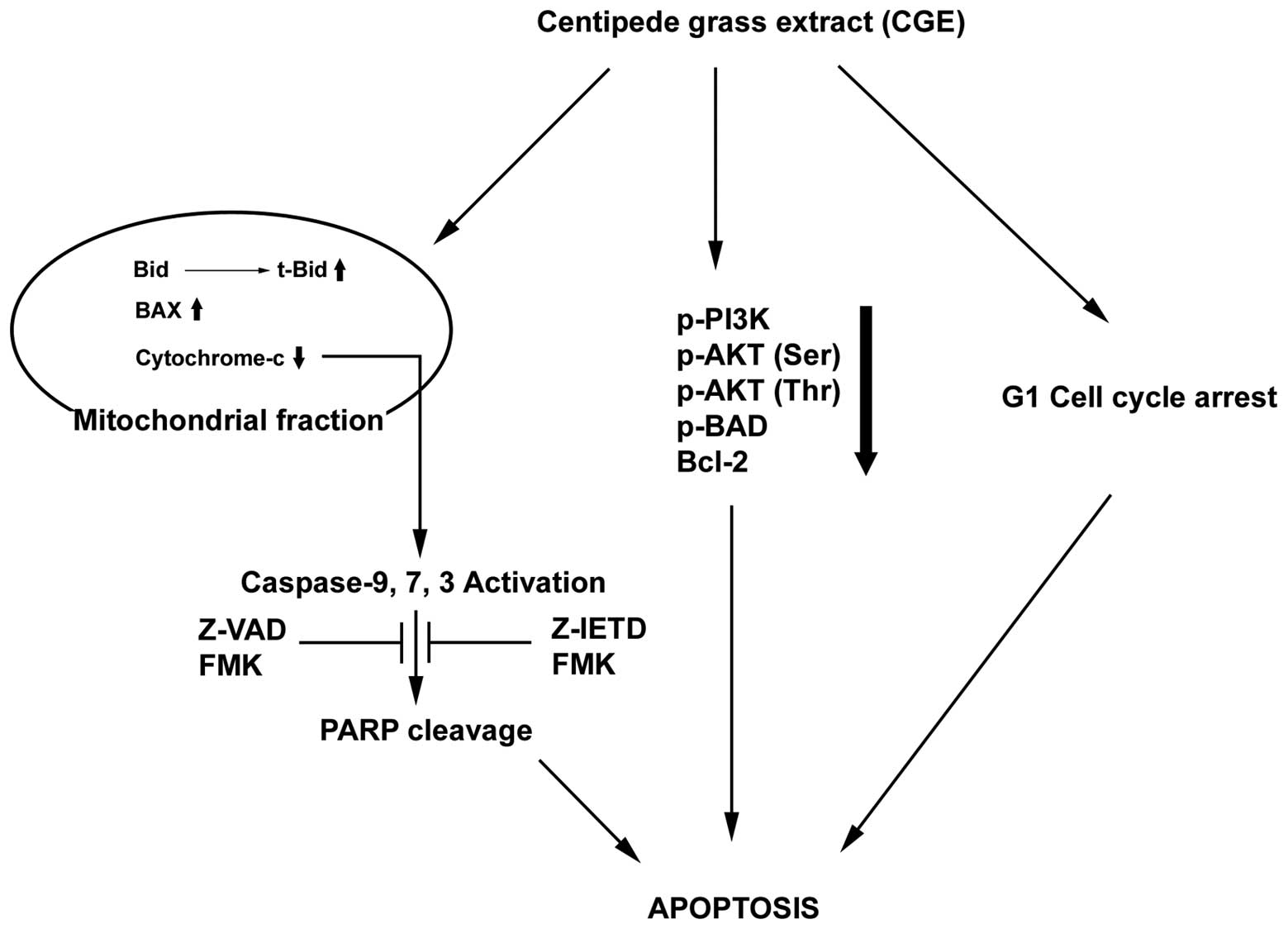
inhibit the growth of leukemia cells. Our results are summarized in
Fig. 6. The selective inhibition
of cancer cell growth, apoptosis induction via the significant G1
cell cycle arrest and BAX translocation from the cytosol to the
mitochondria induced the disruption of the ∆Ψm, thereby increasing
the caspase-3/7 activity and activation of effector caspase-3, -7
and -9. In addition, CGE inhibited PI3K activity by reducing p-AKT,
p-BAD, and Bcl-2 levels and inhibited MAPKs, including p-ERK and
p-JNK. Taken together, CGE may prove to be a potential
chemopreventive agent in the treatment of ALL.
Acknowledgments
This study was supported by the National Research
Foundation of Korea (NRF) grant funded by the Korean government
(MSIP) (No. 2012M2A2A6036022).
References
|
1
|
Prasad S, Phromnoi K, Yadav VR, Chaturvedi
MM and Aggarwal BB: Targeting inflammatory pathways by flavonoids
for prevention and treatment of cancer. Planta Med. 76:1044–1063.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chahar MK, Sharma N, Dobhal MP and Joshi
YC: Flavonoids: a versatile source of anticancer drugs. Pharmacogn
Rev. 5:1–12. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wiseman BR, Gueldner RC, Lynch RE and
Severson RF: Biochemical activity of centipedegrass against fall
armyworm larvae. J Chem Ecol. 16:2677–2690. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee EM, Lee SS, Chung BY, et al:
Pancreatic lipase inhibition by C‑glycosidic flavones isolated from
Eremochloa ophiuroides. Molecules. 15:8251–8259. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Johnson AW: Influence of organic
pesticides on nematode populations and seed production of centipede
grass. J Nematol. 2:252–254. 1970.PubMed/NCBI
|
|
6
|
Park HJ, Chung BY, Lee MK, et al:
Centipede grass exerts anti-adipogenic activity through inhibition
of C/EBPβ, C/EBPα, and PPARγ expression and the AKT signaling
pathway in 3T3-L1 adipocytes. BMC Complement Altern Med.
12:2302012. View Article : Google Scholar
|
|
7
|
Hjalgrim LL, Rostgaard K, Schmiegelow K,
et al: Age- and sex-specific incidence of childhood leukemia by
immuno phenotype in the Nordic countries. J Natl Cancer Inst.
95:1539–1544. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yeoh EJ, Ross ME, Shurtleff SA, et al:
Classification, subtype discovery, and prediction of outcome in
pediatric acute lympho-blastic leukemia by gene expression
profiling. Cancer Cell. 1:133–143. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Downing JR: Acute leukemia: subtype
discovery and prediction of outcome by gene expression profiling.
Verh Dtsch Ges Pathol. 87:66–71. 2003.
|
|
10
|
Chow EJ, Simmons JH, Roth CL, et al:
Increased cardio-metabolic traits in pediatric survivors of acute
lymphoblastic leukemia treated with total body irradiation. Biol
Blood Marrow Transplant. 16:1674–1681. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rahiala J, Riikonen P, Kekäläinen L and
Perkkiö M: Cost analysis of the treatment of acute childhood
lymphocytic leukemia according to Nordic protocols. Acta Paediatr.
89:482–487. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Silva A, Yunes JA, Cardoso BA, et al: PTEN
posttranslational inactivation and hyperactivation of the PI3K/Akt
pathway sustain primary T cell leukemia viability. J Clin Invest.
118:3762–3774. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Paganin M and Ferrando A: Molecular
pathogenesis and targeted therapies for NOTCH1-induced T-cell acute
lymphoblastic leukemia. Blood Rev. 25:83–90. 2011. View Article : Google Scholar :
|
|
14
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pucci B, Kasten M and Giordano A: Cell
cycle and apoptosis. Neoplasia. 2:291–299. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Krajarng A, Nilwarankoon S, Suksamrarn S
and Watanapokasin R: Antiproliferative effect of α-mangostin on
canine osteosarcoma cells. Res Vet Sci. 93:788–794. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Elmore S: Apoptosis: a review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sun C, Guo XX, Zhu D, et al: Apoptosis is
induced in cancer cells via the mitochondrial pathway by the novel
xylocydine-derived compound JRS-15. Int J Mol Sci. 14:850–870.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bejarano I, Redondo PC, Espino J, et al:
Melatonin induces mitochondrial-mediated apoptosis in human myeloid
HL-60 cells. J Pineal Res. 46:392–400. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Foster SS, De S, Johnson LK, Petrini JH
and Stracker TH: Cell cycle- and DNA repair pathway-specific
effects of apoptosis on tumor suppression. Proc Natl Acad Sci USA.
109:9953–9958. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Badaboina S, Bai HW, Park CH, et al:
Molecular mechanism of apoptosis induction in skin cancer cells by
the centipedegrass extract. BMC Complementary Altern Med.
13:3502013. View Article : Google Scholar
|
|
22
|
Demchenko AP: Beyond Annexin V:
fluorescence response of cellular membranes to apoptosis.
Cytotechnology. 65:157–172. 2013. View Article : Google Scholar :
|
|
23
|
Arur S, Uche UE, Rezaul K, et al: Annexin
I is an endogenous ligand that mediates apoptotic cell engulfment.
Dev Cell. 4:587–598. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li H, Wang P, Liu Q, Cheng X, Zhou Y and
Xiao Y: Cell cycle arrest and cell apoptosis induced by Equisetum
hyemale extract in murine leukemia L1210 cells. J Ethnopharmacol.
144:322–327. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mohan S, Abdul AB, Abdelwahab SI, et al:
Typhonium flagelliforme induces apoptosis in CEMss cells via
activation of caspase-9, PARP cleavage and cytochrome c release:
its activation coupled with G0/G1 phase cell cycle arrest. J
Ethnopharmacol. 131:592–600. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Abubakar MB, Abdullah WZ, Sulaiman SA and
Suen AB: A review of molecular mechanisms of the anti-leukemic
effects of phenolic compounds in honey. Int J Mol Sci.
13:15054–15073. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Grütter MG: Caspases: key players in
programmed cell death. Curr Opin Struct Biol. 10:649–655. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rodriguez-Hernandez A, Brea-Calvo G,
Fernandez-Ayala DJ, Cordero M, Navas P and Sanchez-Alcazar JA:
Nuclear caspase-3 and caspase-7 activation, and Poly(ADP-ribose)
polymerase cleavage are early events in camptothecin-induced
apoptosis. Apoptosis. 11:131–139. 2006. View Article : Google Scholar
|
|
29
|
El-Mahdy MA, Zhu Q, Wang QE, Wani G and
Wani AA: Thymoquinone induces apoptosis through activation of
caspase-8 and mitochondrial events in p53-null myeloblastic
leukemia HL-60 cells. Int J Cancer. 117:409–417. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tao Z, Zhou Y, Lu J, et al: Caspase-8
preferentially senses the apoptosis-inducing action of NG-18, a
Gambogic acid derivative, in human leukemia HL-60 cells. Cancer
Biol Ther. 6:691–696. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Granville DJ, Shaw JR, Leong S, et al:
Release of cytochrome c, Bax migration, Bid cleavage, and
activation of caspases 2, 3, 6, 7, 8, and 9 during endothelial cell
apoptosis. Am J Pathol. 155:1021–1025. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Martinou JC and Youle RJ: Mitochondria in
Apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev
Cell. 21:92–101. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shin DY, Kim GY, Lee JH, Choi BT, Yoo YH
and Choi YH: Apoptosis induction of human prostate carcinoma DU145
cells by diallyl disulfide via modulation of JNK and PI3K/AKT
signaling pathways. Int J Mol Sci. 13:14158–14171. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chang L and Karin M: Mammalian MAP kinase
signalling cascades. Nature. 410:38–40. 2001.
|
|
35
|
Levine B, Sinha S and Kroemer G: Bcl-2
family members: dual regulators of apoptosis and autophagy.
Autophagy. 4:600–606. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Khor TO, Gul YA, Ithnin h and Seow HF:
Positive correlation between overexpression of phospho-BAD with
phosphorylated Akt at serine 473 but not threonine 308 in
colorectal carcinoma. Cancer Lett. 210:139–150. 2004. View Article : Google Scholar : PubMed/NCBI
|