Introduction
Osteoarthritis (OA), a degenerative joint disease,
is a multifactorial process in which mechanical factors play a
central role and is characterized by alterations in the structure
and function of the whole joint (1). OA involves the entire joint organ,
including the subchondral bone, meniscus, ligaments, periarticular
muscle, capsule and synovium, and is associated with risk factors,
such as age, gender, obesity, prior joint injury, genetic
predisposition and mechanical factors, including malalignment and
abnormal joint shape (2). During
skeletal development, chondrocytes differentiate from mesenchymal
progenitors to synthesize the cartilage (3). Differentiated chondrocytes express
cartilage-specific collagens II, IX and XI. Under normal
conditions, chondrocytes rest in a non-stimulated steady state and
maintain the synthesis of proteoglycans and other non-collagen
molecules (4).
Prostaglandins are produced by cyclooxygenases (COX)
from arachidonic acid and are induced in arthritic joints (5). COX has three forms: COX-1, COX-2 and
COX-3. Whereas COX-1 is constituvely expressed in various cell
types to maintain homeostasis, COX-2 is the inducible form of COX,
implicated in prostaglandin synthesis in the inflammatory response
and has been associated with osteoarthritic cartilage (6). COX-3 is a recently described variant
of COX-1 and is also known as COX-1 VI. However, to date, there is
not much conclusive evidence available regarding the existence of
COX-3 protein (7).
Reactive oxygen species (ROS), such as hydrogen
peroxide (H2O2), superoxide anion
(O2−). and hydroxyl radical (•OH)
are generally believed to be harmful to cells and tissues (8). ROS are generated by a variety of
endogenous and exogenous processes through several pathways and the
mitochondria are the major source of intracellular ROS (9,10).
ROS are destructive to DNA and proteins (11). ROS are involved in the regulation
of the production of biochemical factors involved in cartilage
degradation. They may cause damage to all matrix components, either
directly or indirectly by reducing matrix component synthesis
(12).
Thymoquinone (TQ) is the main active component of
Nigella sativa oil, traditionally used in the Middle East
(13). In this study, we
investigated the effects of TQ and the regulatory mechanisms of TQ
with respect to dedifferentiation and COX-2 expression in
chondrocytes, including alterations in the expression of various
signaling molecules in TQ-treated chondrocytes. Several signaling
cascades, including those involving phosphoinositide 3-kinase
(PI3K)/Akt and mitogen-activated protein kinases (MAPKs; p38, ERK)
and c-Jun N-terminal kinase (JNK), regulate the dedifferentiation
of chondrocytes and COX-2 expression by modulating the generation
of ROS (14,15). Although other investigators have
suggested that ROS inhibit differentiation and induce COX-2
expression, the mechanisms involved have not been fully elucidated.
In the present study, we investigated the molecular mechanisms
through which the TQ-induced generation of ROS affects
dedifferentiation and COX-2 expression in rabbit articular
chondrocytes. Our results suggest that TQ induces the generation of
ROS, which modulates the PI3K/Akt or MAPK signaling cascades,
leading to dedifferentiation and inflammation in rabbit
chondrocytes.
Materials and methods
Primary culture of rabbit articular
chondrocytes
Articular chondrocytes were isolated from cartilage
slices of 2-week-old New Zealand white rabbits (Koatech,
Pyeongtaek, Korea), as previously described (16). Briefly, the cartilage slices were
enzymatically dissociated in 0.2% collagenase in Dulbecco’s
modified Eagle’s medium (DMEM; Gibco, Carlsbad, CA, USA).
Individual cells were cultured in DMEM supplemented with 10% (v/v)
fetal bovine calf serum (Gibco). The chondrocytes were grown at
37°C in the DMEM in a humidified incubator containing 5%
CO2. Primary chondrocyte cultures at 3.5 days were
treated with 0.1 % DMSO (vehicle control) or with various
pharmacological reagents, including TQ (Sigma-Aldrich, St. Louis,
MO, USA). The cells were treated with various inhibitors
[N-acetyl-L-cysteine (NAC),
4,4′-diiso-thiocyano-2,2′-stilbenedisulphonic acid (DIDS),
SP600125, SB203580, PD98059 and LY294002] for 1 h prior to
treatment with TQ. NAC and DIDS were purchased from Sigma-Aldrich,
and SP600125 was obtained from Biomol (Plymouth Meeting, PA, USA).
The other chemicals used, SB203580 and PD98059, were purchased from
Calbiochem (San Diego, CA, USA). LY294002 was obtained from Tocris
Bioscience (Bristol, Avon, UK). The study was approved by the
Ethics Committee of Kongju National University, Gongju, Korea.
Western blot analysis
The cells were lysed in radioimmuno-precipitation
(RIPA) lysis buffer containing protease inhibitors [10 g/ml
leupeptin, 10 g/ml pepstatin A, 10 g/ml aprotinin and 1 mM
4-(2-aminoethyl)benzenesulfonyl fluoride] and phosphatase
inhibitors (1 mM NaF and 1 mM Na3VO4).
Protease inhibitors and phosphatase inhibitors were obtained from
Sigma-Aldrich. Equal amounts of protein were mixed with
electrophoresis sample buffer (Bio-Rad Laboratories, Hercules, CA,
USA) and boiled for 5 min before loading onto SDS-PAGE gels.
Proteins were fractionated by SDS-PAGE and transferred onto
nitrocellulose membranes (Millipore, Billerica, MA, USA). The
membranes were incubated with primary antibodies followed by
horseradish peroxidase-conjugated secondary antibodies
(Sigma-Aldrich). Primary antibodies were specific to phosphorylated
(p-)p38 (#9211; Cell Signaling Technology, Beverly, MA, USA),
p-ERK-1/2 (#9101; Cell Signaling Technology), p-JNK (#9251; Cell
Signaling Technology), p-Akt (#9271; Cell Signaling Technology),
type II collagen (MAB8887; Santa Cruz Biotechnology, Santa Cruz,
CA, USA), actin (sc-1615; Santa Cruz Biotechnology) and COX-2
(#160106; Cayman Chemical Co., Ann Arbor, MI, USA). Proteins were
visualized with ECL Plus reagent (Amersham Biosciences) on a
Chemilumino analyzer LAS 4000 mini (Fujifilm, Tokyo, Japan).
Chondrocyte differentiation
The chondrocytes were identified by staining for
sulfate proteoglycan with Alcian blue as previously described
(17). The cells were washed
twice with cold PBS, fixed with 95% methanol for 2 min (−20°C) and
stained overnight with 0.1% Alcian Blue 8GX (Wako Pure Chemical
Industries Ltd., Osaka, Japan) in 0.1 M HCl. After washing 3 times
with distilled water, the stain was extracted with 800 μl of
6 M guanidine-HCl for 6 h at room temperature; optical density was
measured at 595 nm.
Measurement of ROS production
The fluorogenic marker,
7′-dichlorodihydrofluorescein diacetate (DCFH-DA; Sigma-Aldrich),
was used to monitor the production of intracellular ROS. Following
treatment with various concentrations of TQ for 2 h, the cells were
washed twice with PBS and loaded for 30 min with DCFH-DA (10
μM; Sigma-Aldrich) in DMEM without phenol red. The
acetoxymethyl group on DCFH-DA is cleaved by non-specific esterases
within the cell, producing a non-fluorescent charged molecule that
does not cross the cell membrane. Intracellular ROS irreversibly
oxidizes DCFH-DA to dichlorofluorescein (DCF), which is a
fluorescent product. Following treatment, the medium was removed,
the chondrocytes were collected by centrifugation and fluorescence
was measured on an Flx8000 fluorometer (excitation, 485
nm/emission, 525 nm; Bio-Tek Instruments, Winooski, VT, USA). For
ROS visualization by fluorescence microscopy, the cells were
labeled for 30 min at 37°C in the dark with DCFH-DA (10 μM)
probe. The chondrocytes were washed twice with PBS. Fluorescence
was observed under an inverted Olympus BX50 microscope (Olympus,
Tokyo, Japan). DCF fluorescence intensity was quantified using
ImageJ software (Vector Laboratories, Burlingame, CA, USA).
Immunofluorescence (IF) staining
The chondrocytes cultured on glass coverslips were
fixed in 4% paraformaldehyde at 4°C for 10 min and permeabilized
with 0.1% Tween-20 in PBS for 15 min. For immunostaining, goat
polyclonal antibody to type II collagen (MAB8887; 1:50 dilution;
Santa Cruz Biotechnology) and anti-rabbit polyclonal antibody to
COX-2 (#160112; 1:50 dilution; Cayman Chemical Co.) were used as
primary and secondary antibodies, respectively. Counterstaining
with DAPI (Molecular Probe) enabled nuclear visualization. Images
of the cultured chondrocytes were acquired using a fluorescence
microscope (Olympus BX50; Olympus).
Measurement of prostaglandin
E2 (PGE2) levels
The chondrocytes were seeded in standard 96-well
microtiter plates at 1×104 cells/well. Following
treatment, COX-2 activity was determined by measuring
PGE2 levels in the culture medium. PGE2
concentrations were determined using a standardized enzyme
immunoassay (EIA) according to the manufacturer's instructions
(Assay Designs, Ann Arbor, MI, USA).
Statistical analysis
Data are expressed as the means ± SEM and analyzed
by one-way analysis of variance (ANOVA). Comparisons between groups
were performed by ANOVA followed by Turkey’s multiple comparison,
comparing all groups to the DMSO-treated group (control). Graphs
were generated using Microsoft Excel 2007. P-values <0.05 were
considered to indicate statistically significant differences.
Results
To the best of our knowledge, this is the first
study assessing the effects of TQ on normal rabbit articular
chondrocytes. Chondrocytes were treated with TQ (0, 5, 10 and 20
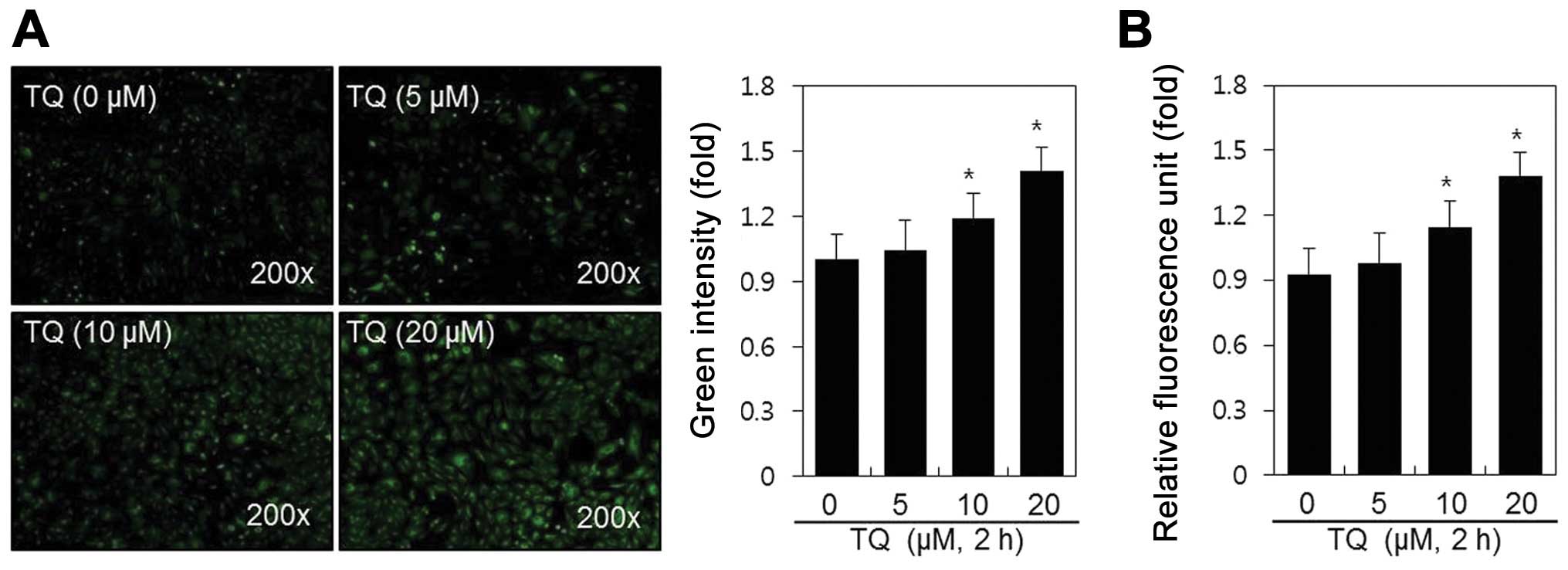
μM) for 2 h, after which we observed a marked induction of
ROS generation by fluorescence microscopy (Fig. 1A) and fluorometry (Fig. 1B). The dose-dependent increase in
the production of ROS increased 1.4-fold after 2 h of treatment
with TQ, as shown in Fig. 1B.
These results indicate that TQ induces ROS production in rabbit
articular chondrocytes.
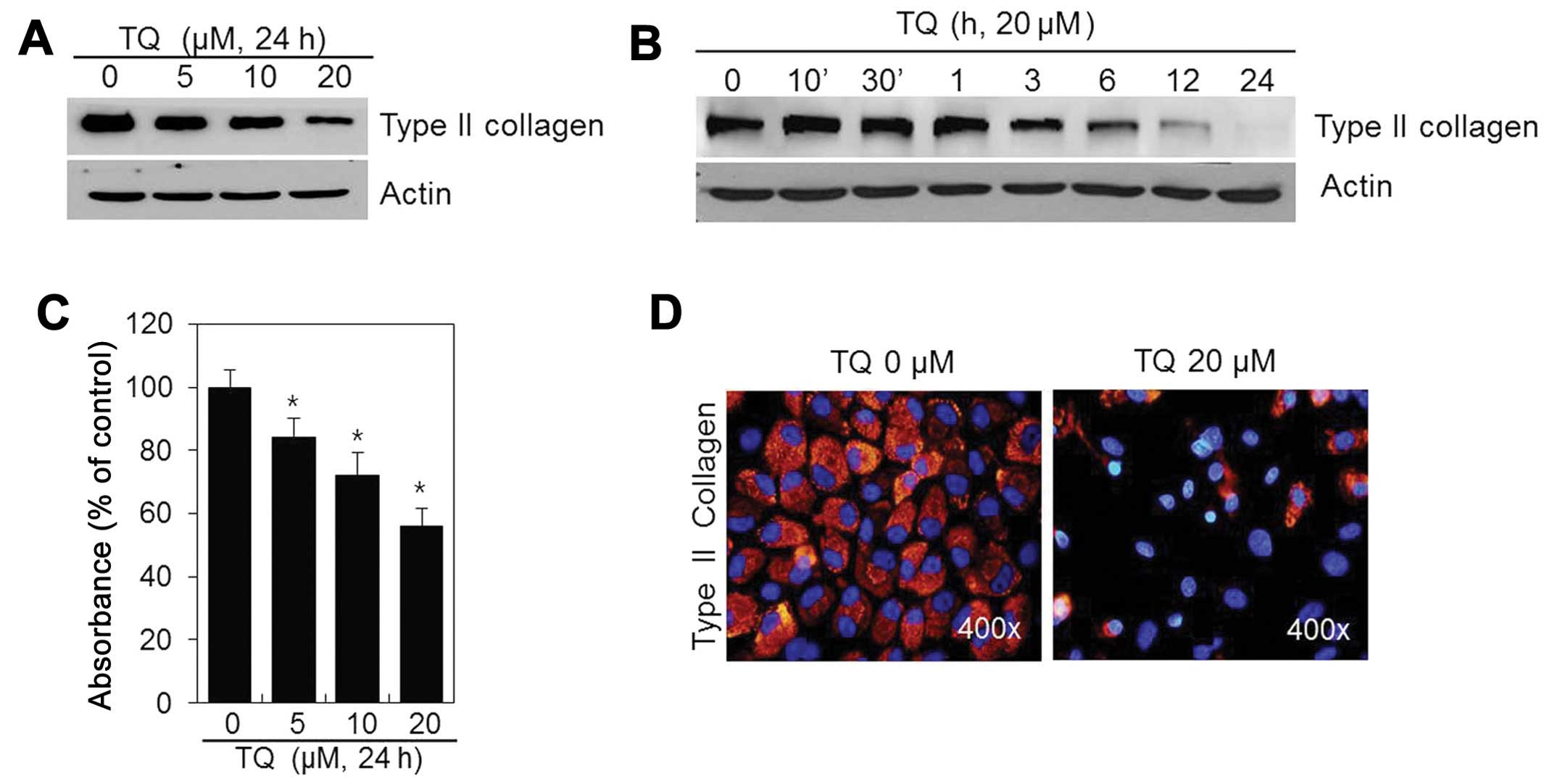
To determine whether TQ influences the chondrocyte
phenotype, the cells were treated with various concentrations of TQ
for 24 h or with 20 μM of TQ for various periods of time
(Fig. 2). The production of type
II collagen, a differentiation marker, was inhibited in a dose- and
time-dependent manner (Fig. 2A and
B) following treatment with TQ. Thus, TQ is capable of inducing
the dedifferentiation of chondrocytes.
We also examined the effects of TQ on the production
of chondroitin sulfate proteoglycan, which accumulates during
chondrocyte differentiation. The TQ-treated chondrocytes exhibited
a dose-dependent decrease in sulfate proteoglycan staining in
comparison to the controls (Fig.
2C). IF staining revealed that type II collagen was distributed
throughout the extracellular matrix of the control cells. However,
the type II collagen levels were decreased in the TQ-treated
chondrocytes (Fig. 2D). These
findings suggest that TQ induces the dedifferentiation of rabbit
articular chondrocytes.
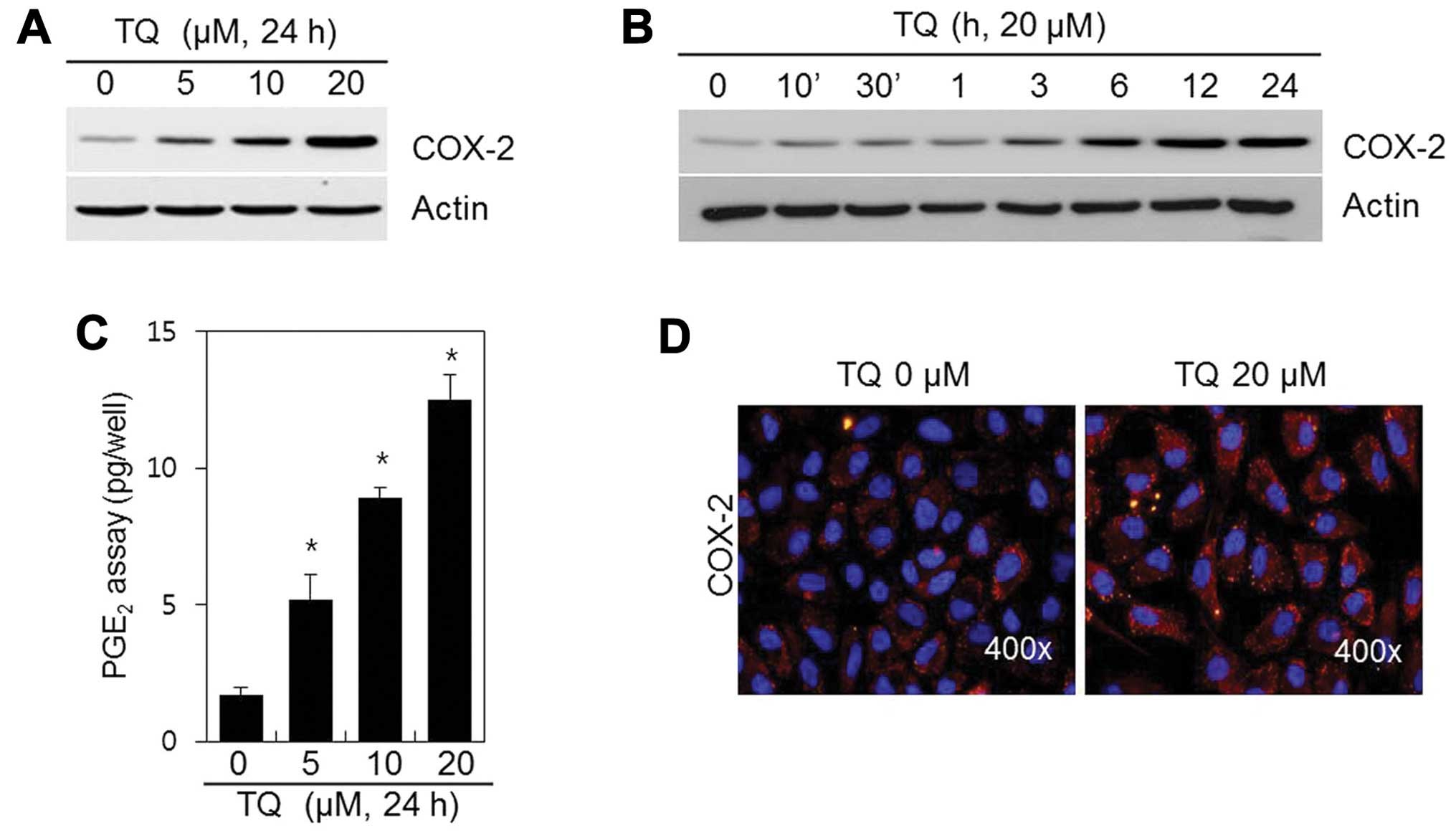
We sought to determine whether TQ affects COX-2
expression. The chondrocytes were treated with various
concentrations of TQ for 24 h or with 20 μM of TQ for
various periods of time (Fig. 3).
The expression of the inflammatory mediator, COX-2, was induced in
a dose- and time-dependent manner (Fig. 3A and B) following treatment with
TQ. We also found that TQ induced a marked dose-dependent induction
in PGE2 synthesis, which is known to mediate
inflammation (Fig. 3C). IF
staining revealed that COX-2 was distributed in low amounts
throughout the cytosol of the control cells; however, COX-2
expression was greater in the TQ-treated chondrocytes (Fig. 3D). These data suggest that TQ
induces COX-2 expression in rabbit articular chondrocytes.
After observing the prominent effects of TQ on
dedifferentiation and COX-2 expression in chondrocytes, we sought
to elucidate the mechanisms responsible for these effects. The
effects of TQ on dedifferentiation and COX-2 expression in
chondrocytes were determined after 24 h of treatment with 20
μM TQ. ROS play a key role in dedifferentiation and
inflammation (9,18). Thus, ROS function as critical
signaling molecules in various cell types, including
chondrocytes.
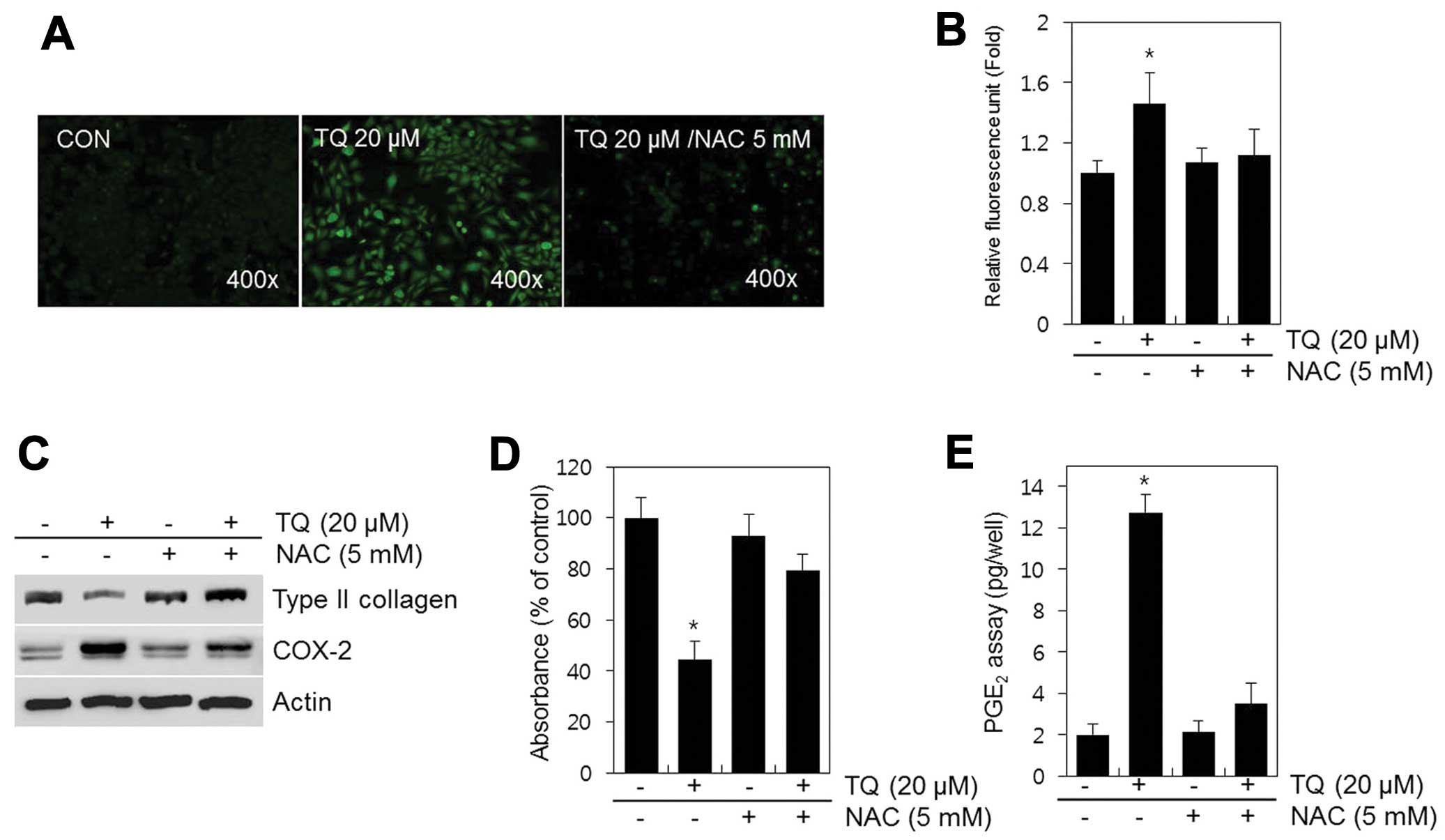
TQ induced the production of ROS (Fig. 4A); thus, this production of ROS
may mediate the TQ-induced dedifferentiation and expression of
COX-2 in chondrocytes. We examined this hypothesis by assessing the
effects of TQ in the presence of NAC, a ROS scavenger (Fig. 4). The TQ-treated chondrocytes were
incubated with NAC (5 mM) for 24 h and then analyzed by
fluorescence microscopy (Fig.
4A). In the TQ-treated cells, a 1.5-fold increase in ROS
production was observed; no change was observed in ROS production
in the control cells. Treatment of the chondrocytes with TQ in the
presence of NAC resulted in only a 1.1-fold increase in ROS
production (Fig. 4B). Blocking
the generation of ROS with NAC nearly abolished the TQ-induced loss
of type II collagen, as well as the increase in COX-2 expression
and PGE2 production in chondrocytes (Fig. 4C–E).
In order to gain further insight into the molecular
mechanisms underlying the induction of differentiation and COX-2
expression in chondrocytes, we investigated the activation of the
MAPK and PI3K pathways (Fig. 5).
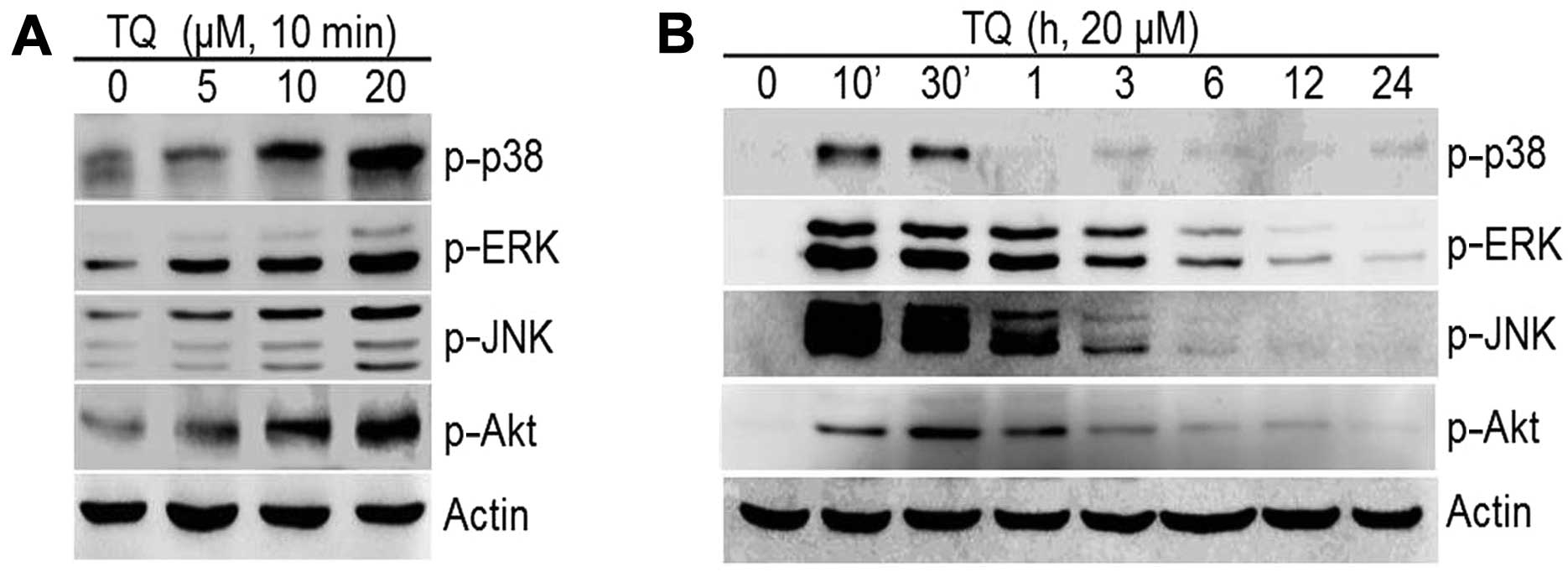
Our results revealed that TQ induced a dose-dependent increase in
the expression of the MAPKs, p-p38, p-ERK, p-JNK and PI3K/pAkt
(Fig. 5A). The TQ-induced
phosphorylation of MAPKs and PI3K was long-lasting and reached
maximum levels after 10 min of treatment for p-p38, p-ERK and p-JNK
and 30 min for p-Akt; the levels decreased thereafter (Fig. 5B). We then determined whether the
TQ-induced activation of MAPKs and PI3K is blocked by NAC (Fig. 6A). NAC inhibited the TQ-induced
phosphorylation of MAPKs and PI3K (Fig. 6A). To determine the association
between the TQ-induced generation of ROS, dedifferentiation and
COX-2 expression and the activation of MAPKs and PI3K, we inhibited
the phosphorylation of MAPKs and PI3K using specific inhibitors
(SB203580 for p38, PD98059 for ERK, LY294002 for PI3K/Akt and
SP600125 for JNK) prior to treatment with TQ (Fig. 6). None of these inhibitors blocked
the TQ-induced generation of ROS, but some slightly inhibited the
TQ-induced dedifferentiation and the expression of COX-2 (Fig. 6B and C). The inhibition of ERK by
PD98059 attenuated the TQ-induced loss in type II collagen
expression and proteoglycan synthesis (Fig. 6B and C). The inhibition of p38
with SB203580 or PI3K/Akt with LY294002 blocked the TQ-induced
expression of COX-2 and PGE2 synthesis (Fig. 6C and E).
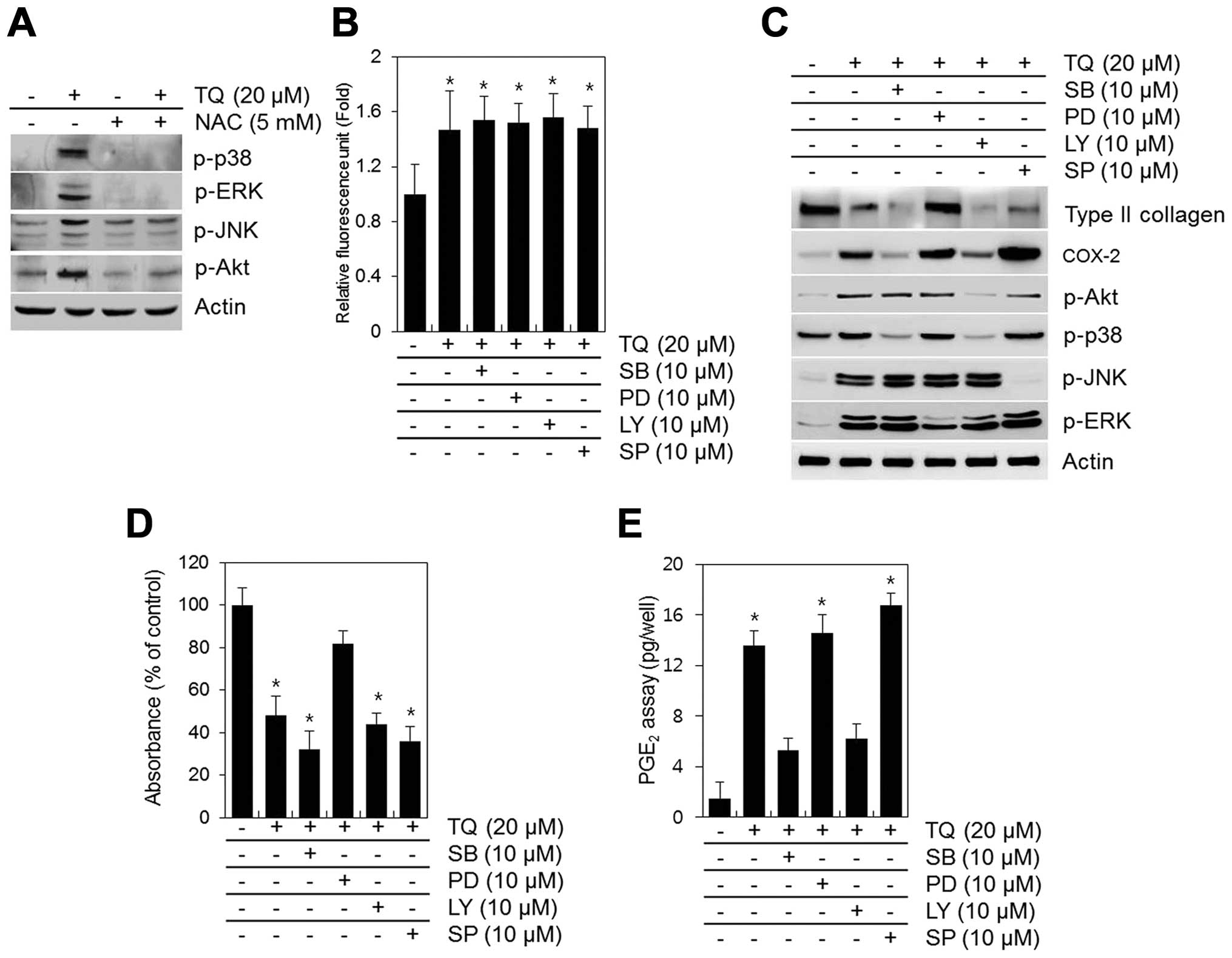 | Figure 6Thymoquinone (TQ)-induced reactive
oxygen species (ROS) generation regulates dedifferentiation through
p38 and cyclooxygenase-2 (COX-2) expression through PI3K/Akt and
ERK. (A) Chondrocytes were exposed to 20 μM TQ in the
absence or presence of 5 mM N-acetyl cysteine (NAC) for 24 h. (A)
The expression of p-p38, p-ERK, p-JNK, p-Akt and actin was
determined by western blot analysis with actin as a loading
control. Primary chondrocytes were exposed to 20 μM TQ in
the absence or presence of SB203580 (SB, PI3K inhibitor), PD98059
(PD, p38 inhibitor), LY294002 (LY, ERK inhibitor) or SP600125 (SP,
JNK inhibitor) (B) for 2 h or (C–E) for 24 h. (B) ROS fluorescence
was measured using an Flx 8000 fluorometer. (C) The expression of
type II collagen, COX-2, p-Akt, p-p38, p-JNK and p-ERK was
determined by western blot analysis with actin as a loading
control. (D) The production of sulfate proteoglycan was determined
by Alcian blue staining. (E) The synthesis of prostaglandin
E2 (PGE2) was analyzed by PGE2
assay. Data are presented as the means ± SD from 3 independent
experiments performed in triplicate. *P<0.01,
compared with the control group. |
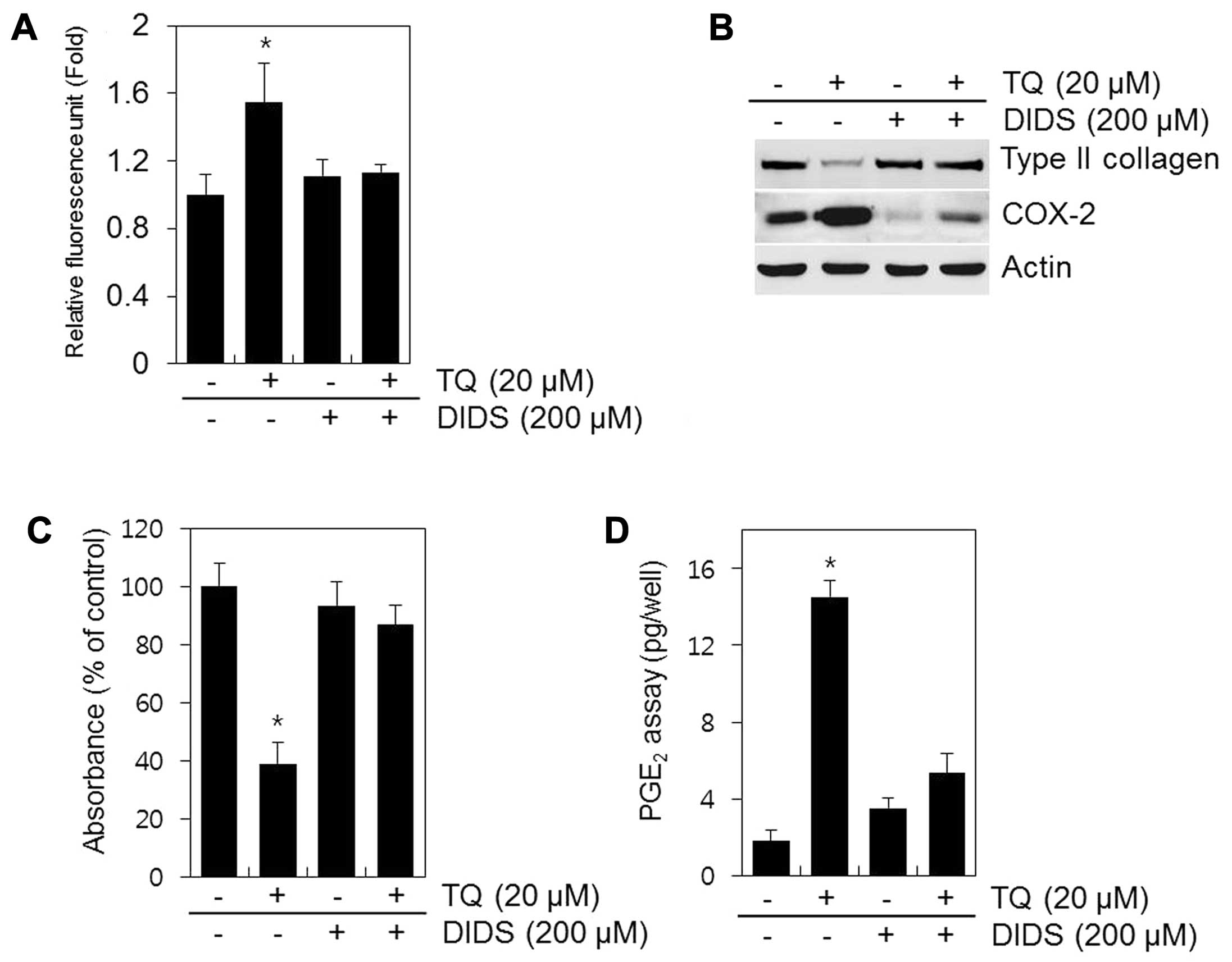
A recent study demonstrated that DIDS, a selective
inhibitor of mitochondrial electron transport, prevents the
production of ROS (19). In the
present study, the cells treated with DIDS no longer produced ROS
in response to TQ (Fig. 7A).
Treatment with DIDS restored type II collagen expression and
sulfate proteoglycan synthesis and decreased the expression of
COX-2 and PGE2 production in the TQ-treated chondrocytes
(Fig. 7B–D). These findings
suggest that the inhibition of ROS production from the mitochondria
by DIDS inhibits dedifferentiation and COX-2 expression and that
ROS is the key source of cartilage destruction (Fig. 7).
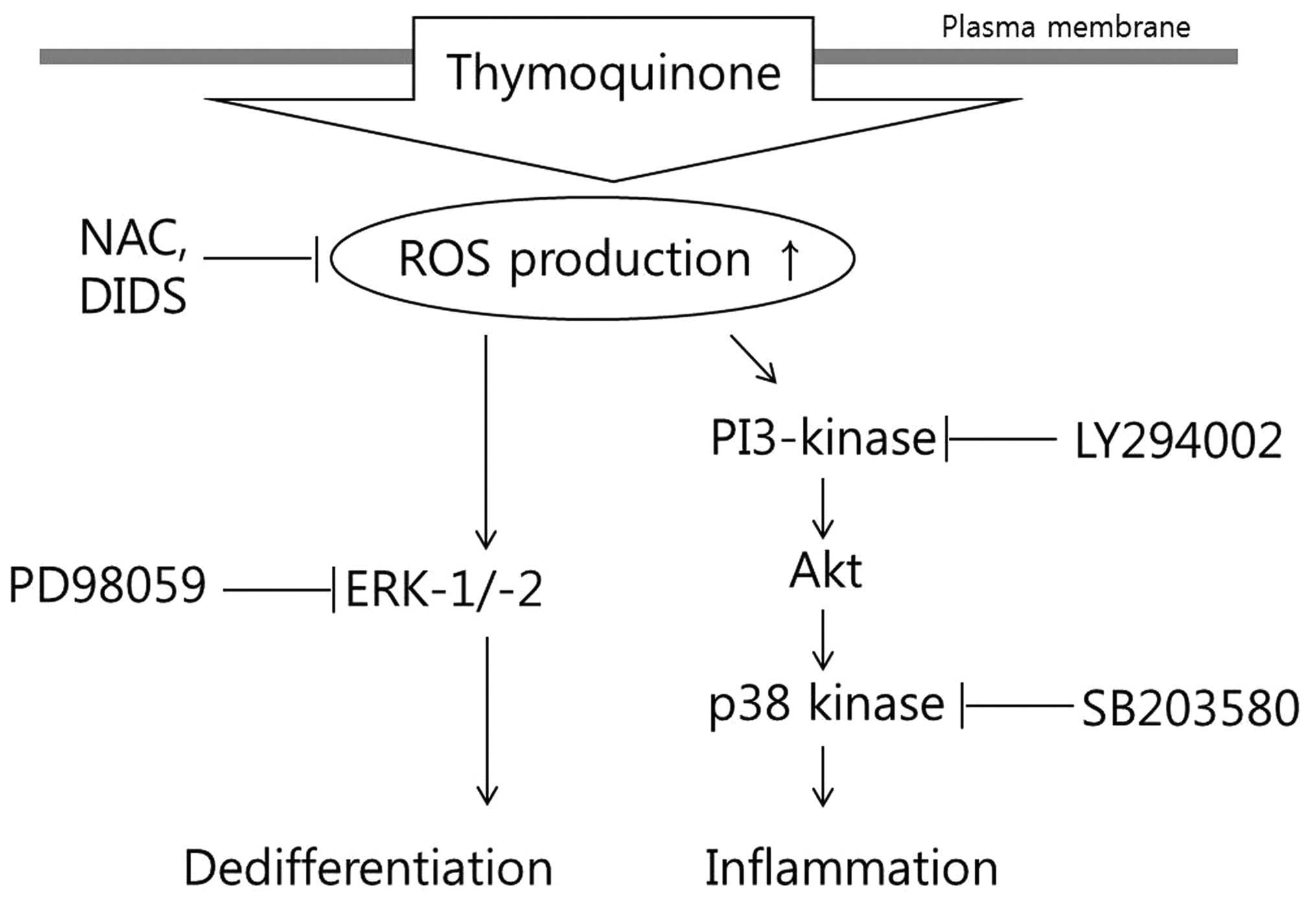
A schematic diagram displaying the cascade of TQ
-induced dedifferentiation and inflammation and the mechanisms
involved is presented in Fig.
8.
Discussion
The pathogenesis of OA is associated with risk
factors, such as oxidative stress and free radicals (20,21). Oxidative stress is caused by
abnormal cell metabolism exceeding the physiologicalbuffering
capacity. Oxidative stress has been described to increase cellular
aging, thus weakening organ function (22). Previous studies have demonstrated
that OA cartilage has high oxidative activity (23,24). ROS overproduction in cartilage
originates in the mitochondria and results in chondrocyte
destruction, which in turn causes OA (25). ROS serve as second messengers that
mediate gene transcription, cell proliferation, necrosis, apoptosis
and differentiation in a variety of cell types (26). In this study, we found that TQ
induced intracellular ROS production and induced dedifferentiation
and COX-2 expression in chondrocytes. TQ induced a dose-dependent
increase in ROS production (Fig.
1). TQ also induced the loss of type II collagen and an
increase in COX-2 expression (Figs.
1 and 2), while NAC inhibited
the TQ-induced dedifferentiation and inflammation (Fig. 3). These findings demonstrate that
TQ is an effective inducer of ROS generation in chondrocytes,
indicating that TQ may play a role in the process of cartilage
destruction through ROS-mediated pathways. Since treatment of the
chondrocytes with low concentrations of TQ (<5 μM) than
those used in the present study had no effect on ROS accumulation,
type II collagen and COX-2 expression (data not shown), we used
concentrations of 5–20 μM in this study. In our previous
study, we demonstrated that the treatment of chondrocytes with TQ
(5–20 μM) resulted in apoptosis, suggesting that TQ may be
effectively used to elucidate the pathways or mechanisms
responsible for apoptosis in chondrocytes (27). TQ may thus be a suitable reagent
for determining the mechanisms responsible for dedifferentiation
and inflammation.
Increasing evidence has attributed cellular damage
in a variety of disorders in humans to oxidative stress that leads
to ROS production, and these effects are mediated by the
interaction with matrix metalloproteinases (MMPs) (28,29). Therefore, in this study, we
investigated whether ROS leads to the destruction of matrix
components, such as type II collagen, by activating MMPs. However,
our results indicated that TQ did not affect MMP production (data
not shown).
Our findings also suggested that TQ increased the
expression and production of the pro-inflammatory mediators, COX-2
and PGE2 (Fig. 3).
Dedifferentiation and inflammation are supported by an
intracellular signaling network involving the PI3K/Akt and MAPKs
pathways (30). Phosphorated Akt
translocates to the nucleus and phosphorylates numerous target
molecules to mediate signals (31). MAPKs are a family of proteins
promoting a phosphorylative signaling cascade, leading to the
activation of transcription factors involved either in cellular
dedifferentiation and inflammation (32). It has also been reported that ROS
induces dedifferentiation, inflammation and proliferation in a
variety of cell types through the temporal activation of the PI3K
and MAPKs pathways (31,32). In addition, several studies have
linked dedifferentiation and COX-2 expression with MAPKs, p38,
ERK-1/2 and JNK and PI3K/Akt. (21,33,34).
In the present study, TQ induced the activation of
MAPKs and PI3K (Fig. 5) and the
inhibition of TQ-induced dedifferentiation by PD98059 was due to
the inhibition of ERK activation (Fig. 6C). The inhibition of p38 and PI3K
decreased the TQ-induced expression of COX-2, but did not influence
dedifferentiation (Fig. 6C).
As demonstrated in our study, DIDS inhibits anion
channels in the mitochondrial inner membrane, thus, inhibiting ROS
release from the organelle. Pre-treatment of the TQ-treated cells
with DIDS abolished dedifferentiation and COX-2 expression,
suggesting that the transition of ROS through anion channels may be
required for the activation of the MAPK and PI3K pathways (Fig. 7). Thus, our results indicate that
the TQ-induced production of ROS triggers dedifferentiation through
ERK and COX-2 expression through the p38 and PI3K pathways.
Acknowledgments
The present study was supported by the Korean Health
Technology R&D Project, Ministry of Health and Welfare,
Republic of Korea (A120960-1201-0000300) and the National Research
Foundation of Korea (NRF) (MEST) (NRF-2012R1A1A2043276).
References
|
1
|
Hunter DJ and Felson DT: Osteoarthritis.
BMJ. 332:639–642. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Blagojevic M, Jinks C, Jeffery A and
Jordan KP: Risk factors for onset of osteoarthritis of the knee in
older adults: a systematic review and meta-analysis. Osteoarthritis
Cartilage. 18:24–33. 2010. View Article : Google Scholar
|
|
3
|
Goldring MB, Tsuchimochi K and Ijiri K:
The control of chondrogenesis. J Cell Biochem. 97:33–44. 2006.
View Article : Google Scholar
|
|
4
|
Maroudas A, Bayliss MT, Uchitel-Kaushansky
N, Schneiderman R and Gilav E: Aggrecan turnover in human articular
cartilage: use of aspartic acid racemization as a marker of
molecular age. Arch Biochem Biophys. 350:61–71. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thomas B, Thirion S, Humbert L, et al:
Differentiation regulates interleukin-1beta-induced
cyclooxygenase-2 in human articular chondrocytes: role of p38
mitogen-activated protein kinase. Biochem J. 362:367–373. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Crofford LJ, Lipsky PE, Brooks P, Abramson
SB, Simon LS and van de Putte LB: Basic biology and clinical
application of specific cyclooxygenase-2 inhibitors. Arthritis
Rheum. 43:4–13. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Burdan F, Chalas A and Szumilo J:
Cyclooxygenase and prostanoids - biological implications. Postepy
Hig Med Dosw (Online). 60:129–141. 2006.(In Polish).
|
|
8
|
Droge W: Free radicals in the
physiological control of cell function. Physiol Rev. 82:47–95.
2002.PubMed/NCBI
|
|
9
|
Tormos KV, Anso E, Hamanaka RB, et al:
Mitochondrial complex III ROS regulate adipocyte differentiation.
Cell Metab. 14:537–544. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nathan C and Cunningham-Bussel A: Beyond
oxidative stress: an immunologist's guide to reactive oxygen
species. Nat Rev Immunol. 13:349–361. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cadet J and Wagner JR: DNA base damage by
reactive oxygen species, oxidizing agents, and UV radiation. Cold
Spring Harb Perspect Biol. 5:pii: a0125592013. View Article : Google Scholar
|
|
12
|
Sasaki K, Hattori T, Fujisawa T, Takahashi
K, Inoue H and Takigawa M: Nitric oxide mediates
interleukin-1-induced gene expression of matrix metalloproteinases
and basic fibroblast growth factor in cultured rabbit articular
chondrocytes. J Biochem. 123:431–439. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Woo CC, Kumar AP, Sethi G and Tan KH:
Thymoquinone: potential cure for inflammatory disorders and cancer.
Biochem Pharmacol. 83:443–451. 2012. View Article : Google Scholar
|
|
14
|
Kim KJ, Lee OH and Lee BY:
Low-molecular-weight fucoidan regulates myogenic differentiation
through the mitogen-activated protein kinase pathway in C2C12
cells. Br J Nutr. 106:1836–1844. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang H, Xi S, Xu Y, et al: Sodium arsenite
induces cyclooxygenase-2 expression in human uroepithelial cells
through MAPK pathway activation and reactive oxygen species
induction. Toxicol In Vitro. 27:1043–1048. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yoon YM, Kim SJ, Oh CD, et al: Maintenance
of differentiated phenotype of articular chondrocytes by protein
kinase C and extracellular signal-regulated protein kinase. J Biol
Chem. 277:8412–8420. 2002. View Article : Google Scholar
|
|
17
|
Asahina I, Sampath TK and Hauschka PV:
Human osteogenic protein-1 induces chondroblastic, osteoblastic,
and/or adipocytic differentiation of clonal murine target cells.
Exp Cell Res. 222:38–47. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qi Z, Yin F, Lu L, et al: Baicalein
reduces lipopolysaccharide-induced inflammation via suppressing
JAK/STATs activation and ROS production. Inflamm Res. 62:845–855.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pamenter ME, Ali SS, Tang Q, et al: An in
vitro ischemic penumbral mimic perfusate increases NADPH
oxidase-mediated superoxide production in cultured hippocampal
neurons. Brain Res. 1452:165–172. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dycus DL, Au AY, Grzanna MW, Wardlaw JL
and Frondoza CG: Modulation of inflammation and oxidative stress in
canine chondrocytes. Am J Vet Res. 74:983–989. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu SM and Kim SJ: Production of reactive
oxygen species by withaferin A causes loss of type collagen
expression and COX-2 expression through the PI3K/Akt, p38, and JNK
pathways in rabbit articular chondrocytes. Exp Cell Res.
319:2822–2834. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brouilette S, Singh RK, Thompson JR,
Goodall AH and Samani NJ: White cell telomere length and risk of
premature myocardial infarction. Arterioscler Thromb Vasc Biol.
23:842–846. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bubici C, Papa S, Dean K and Franzoso G:
Mutual cross-talk between reactive oxygen species and nuclear
factor-kappa B: molecular basis and biological significance.
Oncogene. 25:6731–6748. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tiku ML, Shah R and Allison GT: Evidence
linking chondrocyte lipid peroxidation to cartilage matrix protein
degradation. Possible role in cartilage aging and the pathogenesis
of osteoarthritis. J Biol Chem. 275:20069–20076. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sauter E, Buckwalter JA, McKinley TO and
Martin JA: Cytoskeletal dissolution blocks oxidant release and cell
death in injured cartilage. J Orthop Res. 30:593–598. 2012.
View Article : Google Scholar
|
|
26
|
Korbecki J, Baranowska-Bosiacka I,
Gutowska I and Chlubek D: The effect of reactive oxygen species on
the synthesis of prostanoids from arachidonic acid. J Physiol
Pharmacol. 64:409–421. 2013.PubMed/NCBI
|
|
27
|
Yu SM and Kim SJ: Thymoquinone-induced
reactive oxygen species causes apoptosis of chondrocytes via
PI3K/Akt and p38kinase pathway. Exp Biol Med (Maywood).
238:811–820. 2013. View Article : Google Scholar
|
|
28
|
Kim MJ, Nepal S, Lee ES, Jeong TC, Kim SH
and Park PH: Ethanol increases matrix metalloproteinase-12
expression via NADPH oxidase-dependent ROS production in
macrophages. Toxicol Appl Pharmacol. 273:77–89. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sawicki G: Synergistic effect of
inhibitors of MMPs and ROS-dependent modifications of contractile
proteins on protection hearts subjected to oxidative stress. Curr
Pharm Des. 20:1345–1348. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Torres M and Forman HJ: Redox signaling
and the MAP kinase pathways. Biofactors. 17:287–296. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang X, Liu JZ, Hu JX, et al:
ROS-activated p38 MAPK/ERK-Akt cascade plays a central role in
palmitic acid-stimulated hepatocyte proliferation. Free Radic Biol
Med. 51:539–551. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lin CF, Young KC, Bai CH, et al: Blockade
of reactive oxygen species and Akt activation is critical for
anti-inflammation and growth inhibition of metformin in phosphatase
and tensin homolog-deficient RAW264.7 cells. Immunopharmacol
Immunotoxicol. 35:669–677. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Eo SH, Cho HS and Kim SJ: Resveratrol
regulates type II collagen and COX-2 expression via the ERK, p38
and Akt signaling pathways in rabbit articular chondrocytes. Exp
Ther Med. 7:640–648. 2014.PubMed/NCBI
|
|
34
|
Lee WK, Chung KW, Kim GH and Kim SJ:
Gallotannin causes differentiation and inflammation via ERK1/2 and
p38 kinase pathways in rabbit articular chondrocytes. Mol Med Rep.
7:701–707. 2013.
|