Introduction
Alzheimer’s disease (AD) is an age-related,
progressive, irreversible neurodegenerative illness characterized
by memory deficits, neuronal loss, neurofibrillary tangles (NFTs)
and β-amyloid (Aβ) deposits in amyloid plaques. The etiology of AD
is largely unknown; however, there is growing evidence that chronic
stress may increase the risk of developing AD (1,2).
Stress initiates a sequence of events in the brain and peripheral
systems that enable organisms to deal with and adapt to new and
challenging situations (3).
However, when stress is maintained for extended periods of time,
most physiological systems are negatively affected by stress
(3). It has been reported that
chronic stress activates hippocampal glucocorticoid receptor,
increases neuronal metabolism and decreases cell survival and
neurogenesis. In addition, chronic stress promotes dendritic
atrophy and causes long-term potentiation and cognitive deficits
(4–6). In our previous study, we
demonstrated that chronic restraint stress (CRS) and stress-level
dexamethasone (5 mg/kg) exposure induced learning and memory
impairment and hippocampal neuronal damage in mice (7,8).
Recent studies have indicated that the endoplasmic
reticulum (ER) plays an important role in maintaining neurons in
neuropathological situations (9).
The ER is responsible for protein folding and the transport of
newly synthesized proteins. Moreover, the ER is a target for two
types of intracellular stresses: ER stress and oxidative stress
(10,11). ER stress response involves three
different signaling pathways: unfolded protein response (UPR),
ER-associated protein degradation (ERAD) and ER overload response
(EOR) (9,12). Under ER stress, unfolded proteins
are accumulated within the ER lumen and trigger an adaptive
response which acts to restore normal ER function. However, if ER
stress persists for extended periods of time and cellular
homeostasis cannot be restored, the ER stress response can lead to
cell apoptosis (11,13,14). ER stress has been reported in
different pathophysiological conditions, including
neurodegenerative diseases, such as AD (15,16). Previous studies have demonstrated
that chronic stress increases the accumulation of reactive oxygen
species (ROS) and induces oxidative damage in the cerebral cortex
and hippocampus (8,17). However, the effects of CRS on ER
stress in the frontal cortex and hippocampus have not yet been
established.
The model of CRS is most popular in the study of the
mechanisms of cognitive deficits induced by chronic stress
(18). We hypothesized that ER
stress plays an important role in the neuronal damage induced by
CRS. In the present study, male mice were repeatedly exposed to CRS
for 8 weeks. Cognitive function and histological damage in the
frontal cortex and hippocampus were assessed using the Morris water
maze and H&E staining. Furthermore, we measured the expression
levels of protein kinase C (PKC)α, 78 kDa glucose-regulated protein
(GRP78), mesencephalic astrocyte-derived neurotrophic factor (MANF)
and C/EBP-homologous protein (CHOP) in the frontal cortex and
hippocampus. The data presented in this study may contribute to a
more complete understanding of the mechanisms through which chronic
stress affects the development and progression of AD.
Materials and methods
Animals and treatment
The animal experiments were approved by the
Committee on the Care and Use of Laboratory Animals of Anhui
Medical University, Anhui, China. KM strain male mice (20–25 g)
were used in this study. The animals were obtained from the Anhui
Laboratory Animal Center. The mice were housed in standard cages (6
animals per cage), maintained under a 12-h dark/light cycle and
provided with food and water ad libitum.
The mice were randomly divided into the control
group and CRS group. The mice in the CRS group were placed in a
50-ml conical centrifuge tube with multiple punctures to allow
ventilation in their home cages for 2 h per day between 14:00 to
16:00 for 8 weeks (6 days each week) as previously reported
(19,20). The mice in the control group were
not subjected to stress and were allowed unrestricted access to
food and water.
Morris water maze test
The Morris water maze test was carried out as
previously described (21). The
Morris water maze consisted of a black circular pool (diameter, 120
cm; height, 60 cm) filled with water and the pool was divided into
4 quadrants. The animals (10 mice in each group) were placed into
the pool (facing the wall of the pool) and were allowed to
circumnavigate the pool in search of the escape platform (in the
center of a quadrant submerged 2 cm below the water surface) for 4
trials (60 sec per trial) per day from day 52 to 55 during exposure
to CRS. The escape latency (sec) was recorded to indicate the
learning results. After the final trial, the platform was removed
from the tank and each mouse was subjected to a 60-sec swim probe
test. The number of platform crossings (NPCs) and the swimming time
in the quadrant of the platform (STP) were recorded to indicate the
memory results.
Histological examination
Twenty-four hours after the final exposure to
restraint stress, the animals were sacrificed by cervical
dislocation. The brains were removed (5 animals in each group) and
fixed in 4% paraformaldehyde and embedded in paraffin. The brain
was cut into 5 μm-thick sections using a section cutter
(Leica Biosystems, Wetzlar, Germany). The sections were stained
with hematoxylin and eosin (H&E) and examined under a light
microscope (Olympus IX71; Olympus, Tokyo, Japan).
Immunohistochemistry
The brain paraffin sections were passed through a
gradient of xylene and ethyl alcohol to hydrate the tissue (5
sections in each group). The sections were treated with 0.3%
hydrogen peroxide to block endogenous peroxidase activity, then
washed with PBS. The sections were treated with primary antibody
overnight at 4°C. The primary antibodies to PKCα (BS646; 1:200),
GRP78 (BS6479; 1:200), CHOP (BS6814; 1:200) were from Bioworld
Technology Co. The sections were incubated with a biotinylated
anti-rabbit secondary antibody and subjected to diaminobenzidine
oxidation using the ABC kit (Zhongshan Golden Bridge Biotechnology
Co., Ltd., Beijing, China). The sections were re-stained with
hematoxylin and examined under a microscope (Olympus IX71). The
positive cells were stained brown. Three non-overlapping fields
(×400) in each area of the hippocampus CA1 region and the frontal
cortex of each section were analyzed in a blinded manner. The
integral optical density of immunopositive neurons in each section
was measured using the JD801 Image analysis system (Jiangsu Jieda
Technology, Jiangsu, China) to indicate the expression of PKCα,
GRP78 and CHOP.
Immunoblot analysis
Protein was extracted from the frozen tissue of the
frontal cortex and hippocampus (4 animals in each group). The
protein concentration was determined using the BCA Protein Assay
kit (Shanghai Sangon Biotechnology, Shanghai, China) and equal
amounts of protein were separated by SDS-PAGE and transferred onto
PVDF membranes. The membranes were blocked at room temperature for
1 h with 5% dry skim milk in Tris-buffered saline containing 1%
Tween-20 (TBS-T). The membranes were treated with antibodies to
GRP78, CHOP and MANF (provided by Professor Yuxian Shen, Anhui
Medical University) and β-actin (1:1,000) overnight at 4°C. The
membranes were then incubated with anti-rabbit IgG antibody
conjugated to HRP (1:10,000) for 1 h. After extensive washes, the
protein bands were detected using chemiluminescence reagents (ECL
kit; Amersham Biosciences, Little Chalfont, UK). The Tanon4500
Imaging System (Shanghai Tanon Technology, Shanghai, China) was
used to visualize the protein bands, and densitometry was performed
using ImageJ software. The relative density of the immunoreactive
bands was normalized to the density of the corresponding bands of
β-actin.
Data analysis
The data were analyzed by using SPSS 16.0
statistical analysis software. Statistical differences between the
two treatment groups were determined using an unpaired two-tailed
Student’s t-test, and a value of P<0.05 was considered to
indicate a statistically significant difference. Data are presented
as the means ± SD.
Results
CRS accelerates behavioral impairment in
male mice
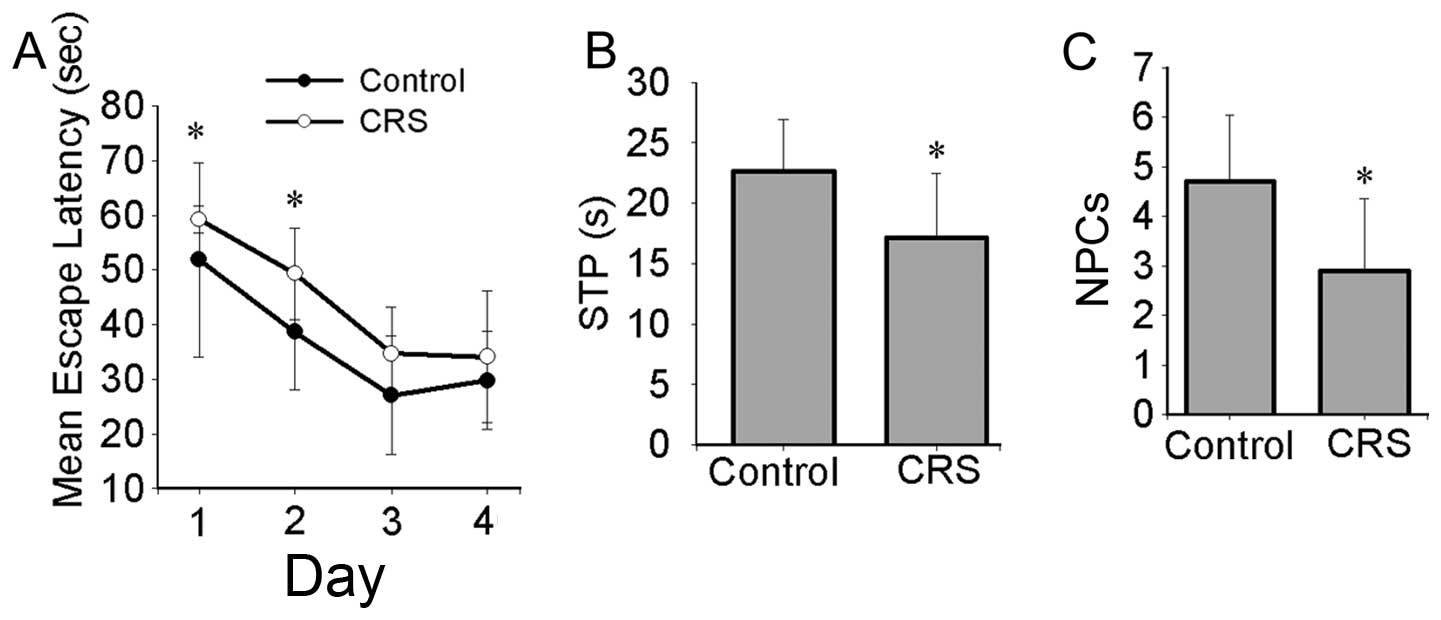
In the memory training experiment, the mean escape
latencies (day 1 and 2) differed significantly between the 2 groups
(day 1, 51.85.±17.74 vs. 59.19±2.44 sec; day 2, 38.65±10.70 vs.
49.35±8.36 sec for mice in the control group and mice exposed to
CRS, respectively) (Fig. 1A). In
the probe trial, the average number of platform crossings (NPCs)
and the swimming time in the quadrant of the platform (STP) also
differed significantly between the 2 groups (NPCs, 4.70±1.34 vs.
2.90±1.45; STP, 22.62±4.36 vs. 17.19±5.32 STP for mice in the
control group and mice exposed to CRS, respectively) (Fig. 1B and C).
CRS enhances neuronal degeneration in the
frontal cortex and hippocampus CA1 region
In order to investigate the effects of chronic
stress on cortical and hippocampal neurons, we examined the
neuronal histomorphological changes in the frontal cortex and
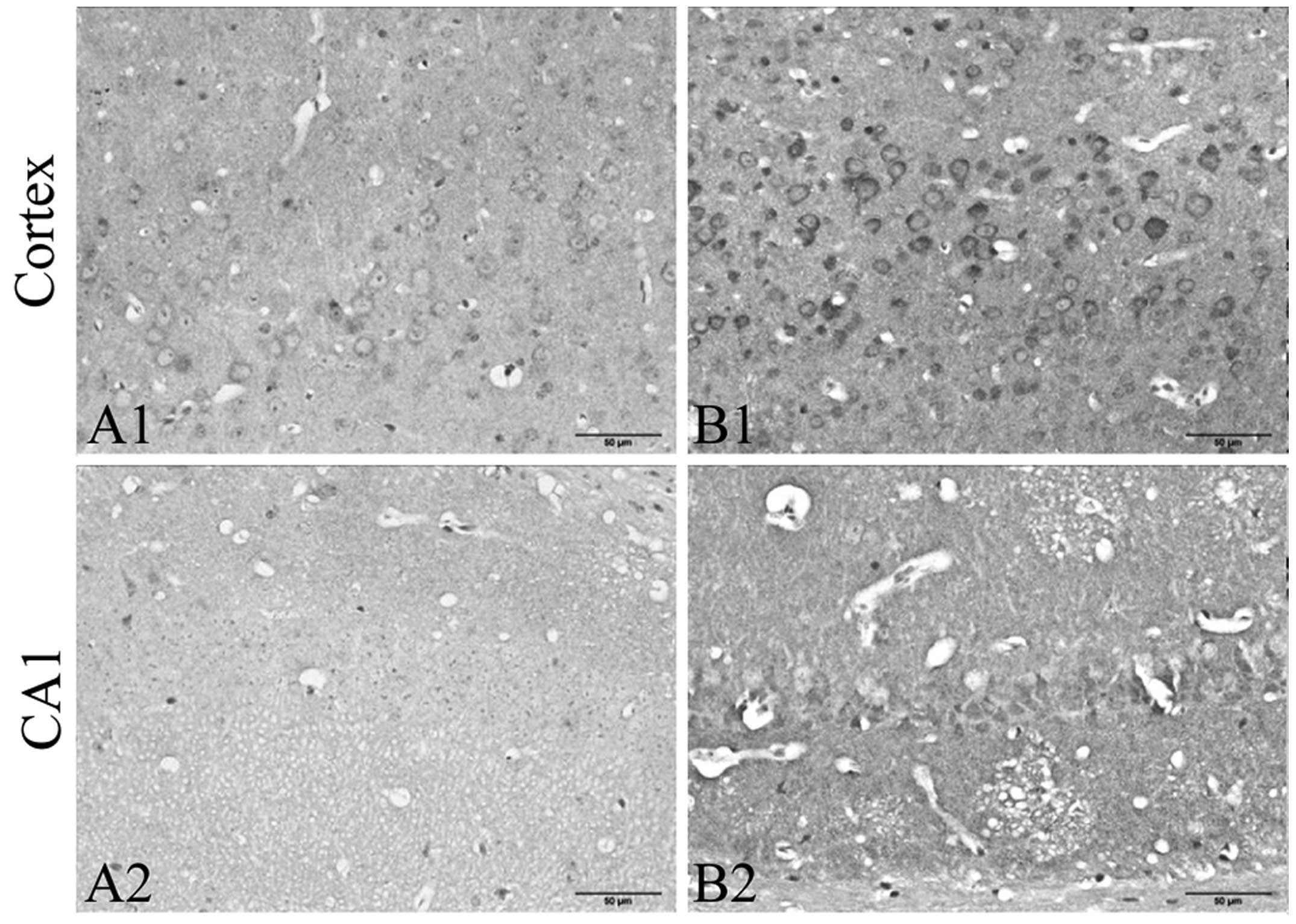
hippocampus (Fig. 2). In the
control group mice, no obvious neuronal abnormalities were observed
in the frontal cortex and hippocampus CA1 region (Fig. 2A, A1 and A2). However, in the CRS
group, more degenerative neurons were observed in the frontal
cortex and hippocampus CA1 region (Fig. 2B, B1 and B2). Acidophilic
degeneration and nuclear pyknosis was observed.
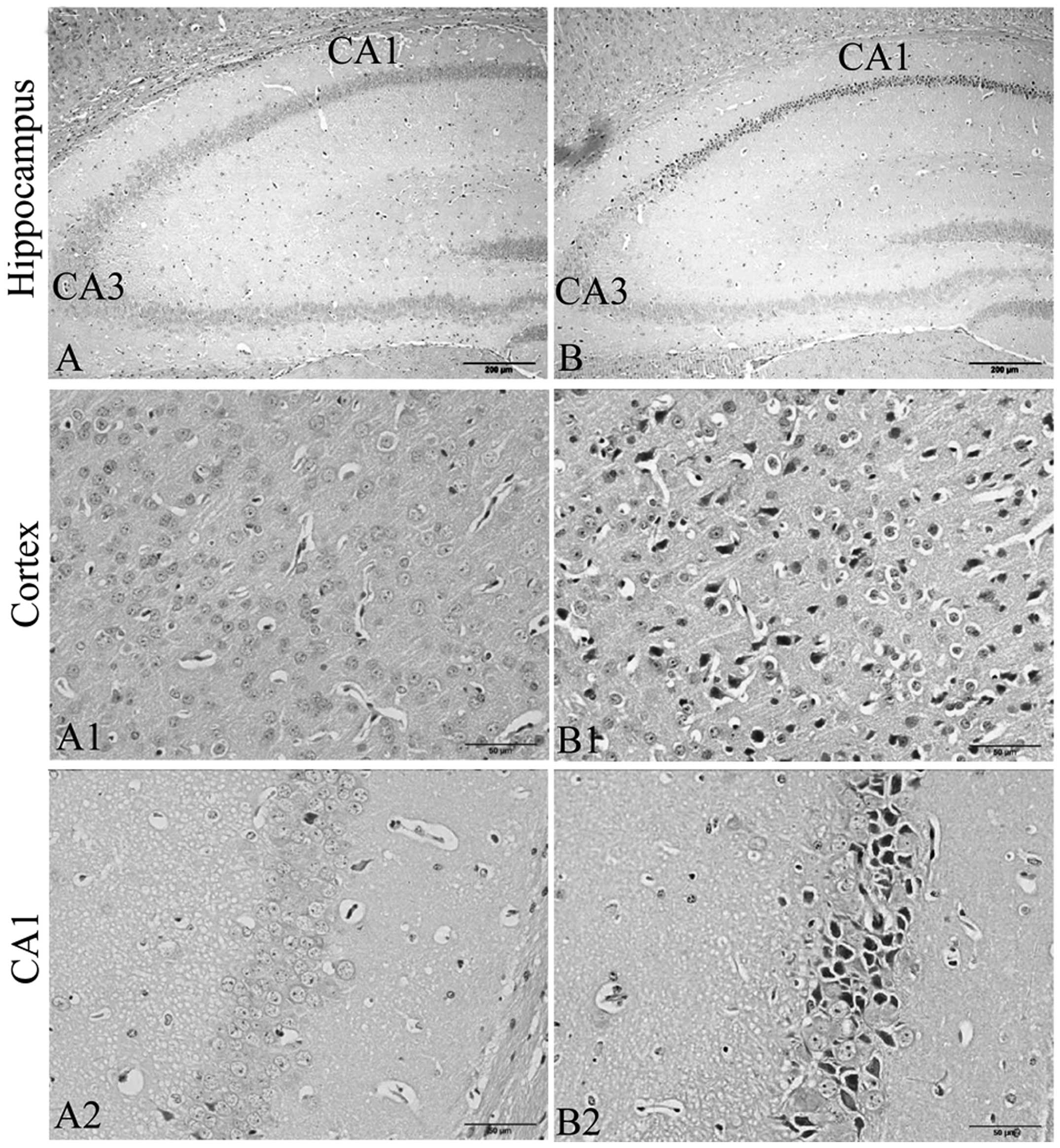 | Figure 2Exposure to chronic restraint stress
(CRS) enhances neuronal degenerative changes in the frontal cortex
and hippocampus CA1 region (H&E staining, hippocampus,
magnification, ×100; cortex and CA1, ×400; n=5). In the
degenerating cells, acidophilic degeneration and nuclear pyknosis
was observed. In the CRS group, more degenerative neurons were
observed in the frontal cortex and hippocampus CA1 areas. (A)
Hippocampus, (A1) frontal cortex, and (A2) hippocampus CA1 region
in the control group. (B) Hippocampus, (B1) frontal cortex, abd
(B2) hippocampus CA1 region in the group exposed to CRS. |
Effects of CRS on the expression of PKCα,
GRP78, CHOP and MANF in the frontal cortex and hippocampus
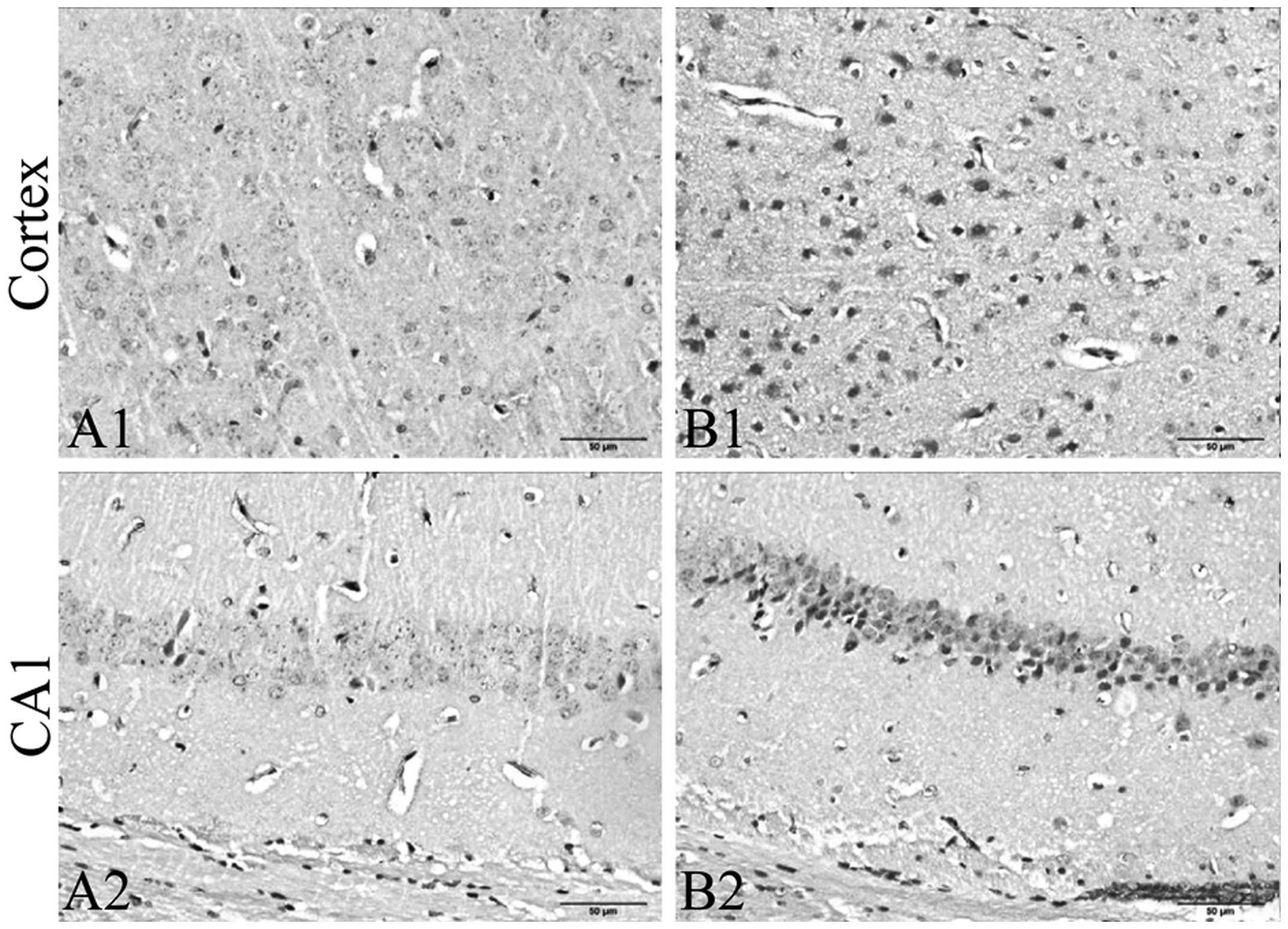
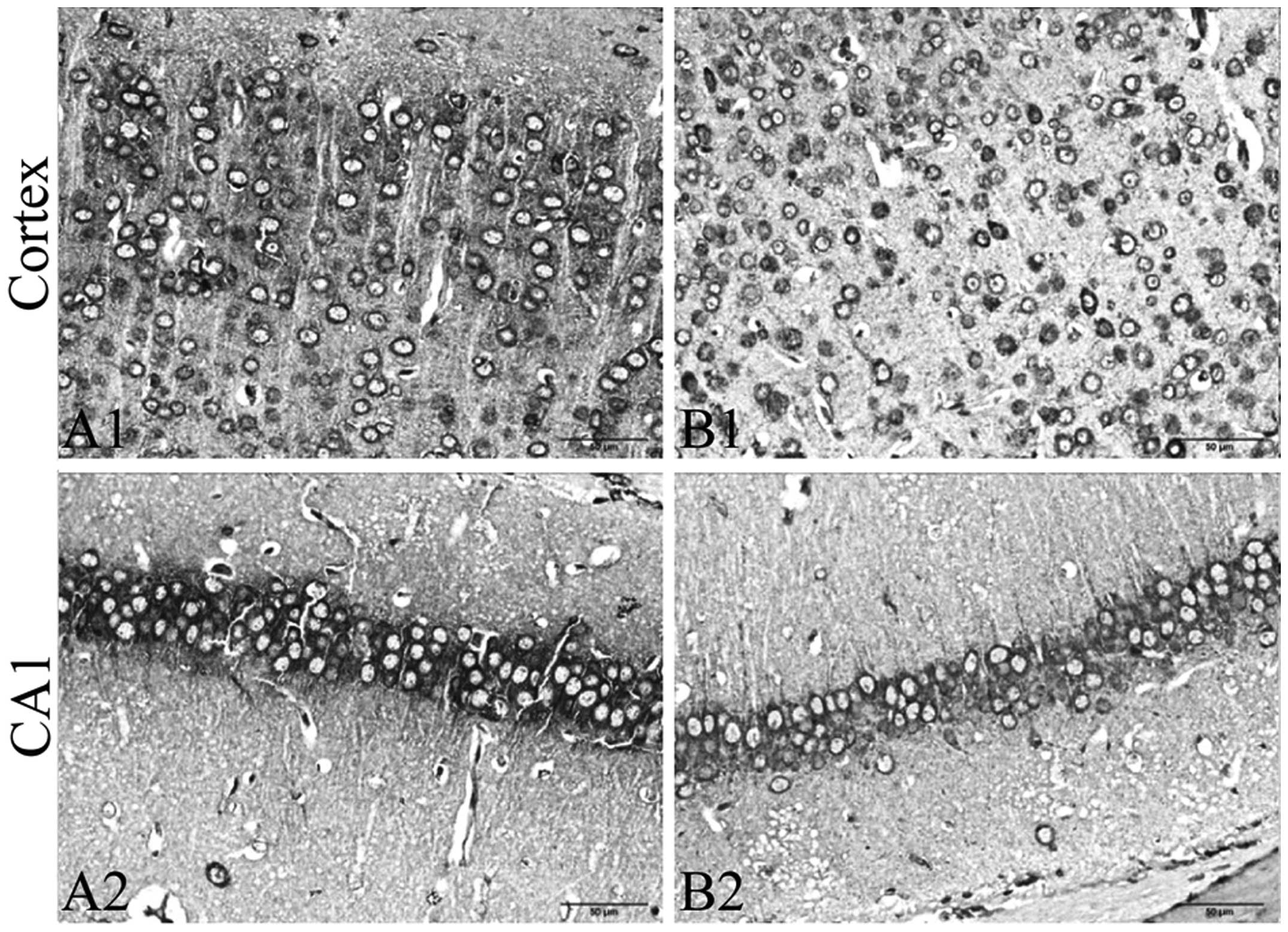
To evaluate the effects of CRS on ER stress, we
detected the expression of PKCα, GRP78 and CHOP in the frontal
cortex and hippocampus by immunohistochemistry. We further detected
the expression of MANF, GRP78 and CHOP in the tissue of the frontal
cortex and hippocampus by immunoblot analysis. The results from
immunohistochemistry revealed that exposure to CRS significantly
increased the expression of PKCα (Fig. 3) and CHOP (Fig. 4A–C), and decreased the expression
of GRP78 (Fig. 5A–C) in the
frontal cortex and hippocampus CA1 region. The results of
immunoblot analysis revealed that exposure to CRS significantly
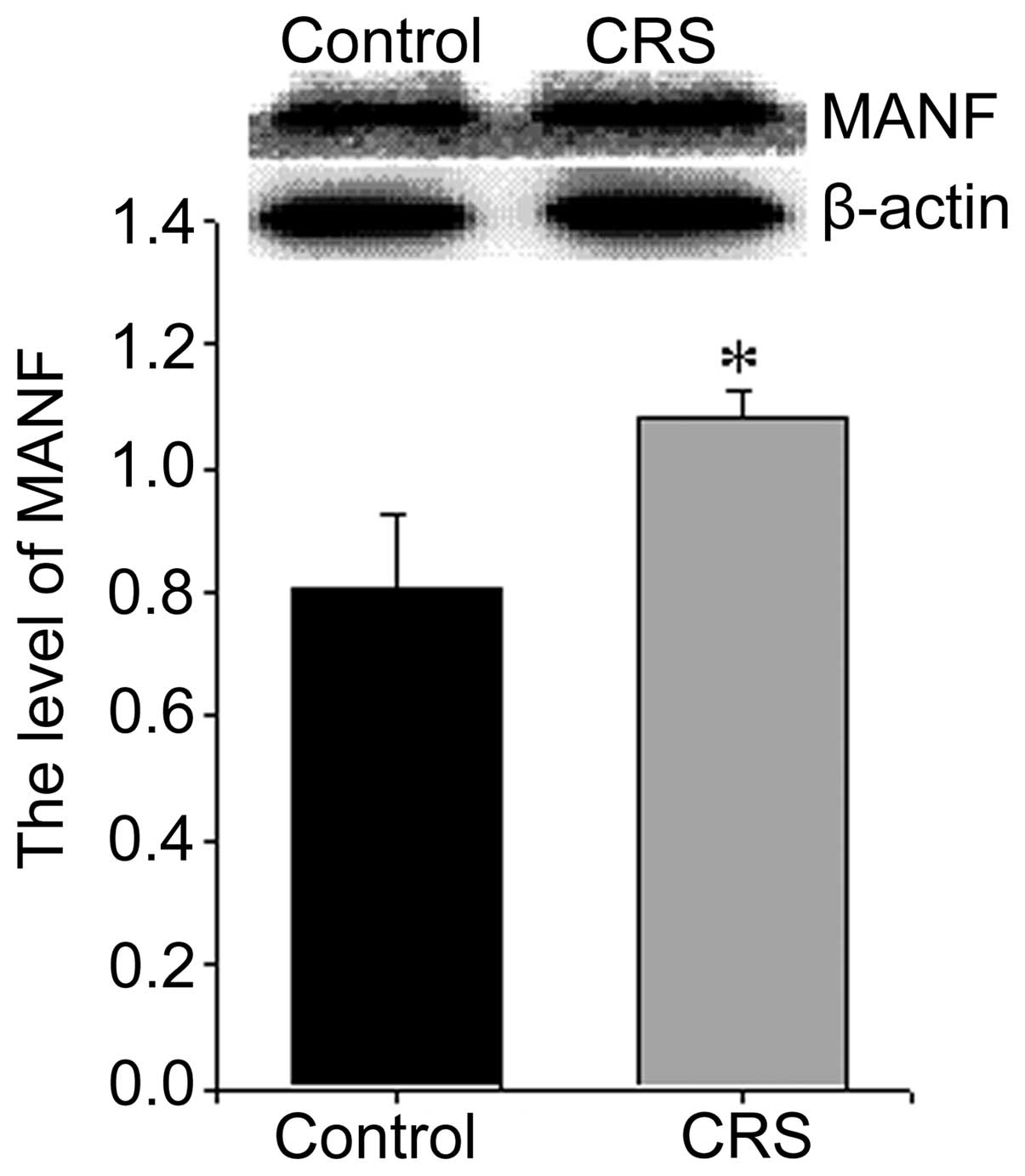
increased the expression of MANF (Fig. 6) and CHOP (Fig. 4D) and decreased the expression of
GRP78 (Fig. 5D).
Discussion
The results of the present study demonstrated that
exposure to CRS for 8 weeks caused a significant impairment in
cognitive function and neuronal damage in the frontal cortex and
hippocampus CA1 region of male mice. Moreover, exposure to CRS (for
8 weeks) significantly increased the expression of PKCα, CHOP and
MANF, decreased the expression of GRP78. To the best of our
knowledge, this is the first study to describe the effects of
chronic stress on ER stress in the frontal cortex and
hippocampus.
Chronic stress has been reported to be associated
with many neurodegenerative diseases, such as depression, AD and
Parkinson’s disease (22–24). The chronic stress-induced
neurodegenerative diseases are an outcome of different mechanisms,
such as central neurotransmitters, neurohormonal factors,
particularly those linked with the hypothalamic-pituitary-adrenal
(HPA) axis and free radical generation (25,26). It has been reported that exposure
to acute restraint stress (150 min of immobilization) significantly
increases neuronal damage in the frontal cortex and striatum both
at 1 and 24 h following exposure to restraint stress (27). The present study demonstrated that
exposure to CRS for 8 weeks caused a significant impairment in
cognitive function and neuronal damage in the frontal cortex and
hippocampus CA1 region in male mice. However, it remains unclear as
to whether ER stress is involved in chronic stress-induced learning
and memory impairment.
ER stress is originally an adaptive response that
can be induced by various stimuli during normal ER function, such
as the accumulation of unfolded or misfolded proteins and changes
in ER Ca2+ homeostasis. ER stress triggers the
activation of several signaling pathways to cope with the abnormal
load in the ER lumen (28). The
UPR pathway induces the upregulation of ER chaperones, such as
GRP78 and the EOR pathway induces the activation of nuclear
factor-κB (NF-κB), leading to the production of cytokines (29). However, ER stress may also
contribute to cell suicide when abnormalities become extensive
(30). Furthermore, studies have
demonstrated that ER stress may play an important role in the
development of neurodegenerative diseases (13) and ER stress may occur prior to
neuronal cell death (31–33). Previous studies have also reported
that oxidative stress contributes to ER stress (33–35). In our previous study, we
demonstrated that exposure to CRS increased ROS accumulation and
induced oxidative damage in the cerebral cortex and hippocampus
(8). This suggests that ER stress
is involved in the oxidative damage induced by chronic stress in
the frontal cortex and hippocampus.
The activation of PKC isoforms has been shown to be
associated with ER stress (28,36). PKC is involved in the induction of
GRP78 (37), a major ER chaperone
and a crucial regulator of ER homeostasis. PKCα, a classic PKC
isoform, has been reported to mediate growth arrest in human
embryonal rhabdomyosarcoma cells by inducing the activation of
JNKs, p38 kinase and ERKs (38),
which are involved in the induction of ER stress (37,39). GRP78 is a molecular chaperone only
located in the ER. GRP78 facilitates protein folding, stabilizes
calcium homeostasis and protects cells against oxidative stress and
apoptotic death (40,41). With ER stress, the level of the ER
marker, GRP78, has been shown to increase in a time-dependent
manner, peaking at 24 h and decreasing at 48 h (42). CHOP, also known as growth arrest
and DNA damage-inducible gene 153 (GADD153), is an important
mediator of ER stress-induced cell death (43). CHOP is expressed at low levels
under physiological conditions. It is upregulated during severe and
prolonged ER stress and plays a crucial role in cell arrest and
apoptosis (44,45). In a previous study, the apoptotic
initiator, CHOP, showed the highest level at 48 h during ER stress
(42). MANF is also an ER stress
responsive protein with neuroprotective effects in animal models of
neurodegeneration. ER stress can cause the upregulation of MANF
(46). In the present study, we
found that exposure to CRS significantly increased the expression
of PKCα, CHOP and MANF, and decreased the expression of GRP78 in
the frontal cortex and hippocampus CA1 region. These data suggest
that CRS induces the ER overload response in the cortex and
hippocampus. Furthermore, ER-resident proteins, such as GRP78 are
susceptible to oxidative damage (47). Oxidation of the ER proteins may
dissociate GRP78 and cause further activation of the ER stress
signals (48). CRS can promote
ROS accumulation, which may dissociate GRP78 and induce the
decrease of GRP78 in the frontal cortex and hippocampus.
In conclusion, to the best of our knowledge, the
present study demonstrates for the first time that ER stress may be
involved in the impairment of cognitive function and neuronal
damage in the frontal cortex and hippocampus induced by CRS. It
should be noted that the present study only examined the effects of
CRS on cognitive function, neuronal damage and the expression of
PKCα, GRP78, CHOP and MANF in the frontal cortex and hippocampus.
The close interaction between chronic stress and ER warrants
further investigation. Despite its preliminary character, this
study clearly indicates that ER stress is implicated in the
learning and memory impairment induced by CRS. Manipulation of the
ER stress pathway signals may provide novel therapeutic
interventions for chronic stress-related neurodegenerative
diseases, such as AD.
Acknowledgments
The authors are grateful for the support provided by
the National Natural Science Foundation of China (81173624) and the
Doctor Foundation of Anhui Medical University (XJ201011). They also
would like to thank Dake Huang, Shan Huang and Li Gui (Synthetic
Laboratory of Basic Medicine College, Anhui Medical University) for
their excellent technical assistance.
References
|
1
|
Wilson RS, Evans DA, Bienias JL, Mendes de
Leon CF, Schneider JA and Bennett DA: Proneness to psychological
distress is associated with risk of Alzheimer’s disease. Neurology.
61:1479–1485. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Csernansky JG, Dong H, Fagan AM, et al:
Plasma cortisol and progression of dementia in subjects with
Alzheimer-type dementia. Am J Psychiatry. 163:2164–2169. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sandi C: Stress, cognitive impairment and
cell adhesion molecules. Nat Rev Neurosci. 5:917–930. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
McEwen BS and Magarinos AM: Stress and
hippocampal plasticity: implications for the pathophysiology of
affective disorders. Hum Psychopharmacol. 16:S7–S19. 2001.
View Article : Google Scholar
|
|
5
|
Kim JJ and Diamond DM: The stressed
hippocampus, synaptic plasticity and lost memories. Nat Rev
Neurosci. 3:453–462. 2002.PubMed/NCBI
|
|
6
|
Rothman SM and Mattson MP: Adverse stress,
hippocampal networks, and Alzheimer’s disease. Neuromolecular Med.
12:56–70. 2010. View Article : Google Scholar :
|
|
7
|
Li WZ, Li WP, Yao YY, et al:
Glucocorticoids increase impairments in learning and memory due to
elevated amyloid precursor protein expression and neuronal
apoptosis in 12-month old mice. Eur J Pharmacol. 628:108–115. 2010.
View Article : Google Scholar
|
|
8
|
Wang Y, Kan H, Yin Y, et al: Protective
effects of ginsenoside Rg1 on chronic restraint stress induced
learning and memory impairments in male mice. Pharmacol Biochem
Behav. 120:73–81. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ansari N and Khodagholi F: Molecular
mechanism aspect of ER stress in Alzheimer’s disease: current
approaches and future strategies. Curr Drug Targets. 14:114–122.
2013. View Article : Google Scholar
|
|
10
|
Schroder M and Kaufman RJ: The mammalian
unfolded protein response. Annu Rev Biochem. 74:739–789. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lin JH, Li H, Yasumura D, et al: IRE1
signaling affects cell fate during the unfolded protein response.
Science. 318:944–949. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Minamino T and Kitakaze M: ER stress in
cardiovascular disease. J Mol Cell Cardiol. 48:1105–1110. 2010.
View Article : Google Scholar
|
|
13
|
Nakagawa T, Zhu H, Morishima N, et al:
Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and
cytotoxicity by amyloid-beta. Nature. 403:98–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Marciniak SJ, Yun CY, Oyadomari S, et al:
CHOP induces death by promoting protein synthesis and oxidation in
the stressed endoplasmic reticulum. Genes Dev. 18:3066–3077. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Katayama T, Imaizumi K, Manabe T, Hitomi
J, Kudo T and Tohyama M: Induction of neuronal death by ER stress
in Alzheimer’s disease. J Chem Neuroanat. 28:67–78. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Costa RO, Ferreiro E, Martins I, et al:
Amyloid beta-induced ER stress is enhanced under mitochondrial
dysfunction conditions. Neurobiol Aging. 33:824.e5–824.e16. 2012.
View Article : Google Scholar
|
|
17
|
Moceri VM, Kukull WA, Emanuel I, van Belle
G and Larson EB: Early-life risk factors and the development of
Alzheimer’s disease. Neurology. 54:415–420. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu Y, Zhuang X, Gou L, et al: Protective
effects of nizofenone administration on the cognitive impairments
induced by chronic restraint stress in mice. Pharmacol Biochem
Behav. 103:474–480. 2013. View Article : Google Scholar
|
|
19
|
Yin D, Tuthill D, Mufson RA and Shi Y:
Chronic restraint stress promotes lymphocyte apoptosis by
modulating CD95 expression. J Exp Med. 191:1423–1428. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Derin N, Yargicoglu P, Aslan M, Elmas O,
Agar A and Aiciguzel Y: The effect of sulfite and chronic restraint
stress on brain lipid peroxidation and anti-oxidant enzyme
activities. Toxicol Ind Health. 22:233–240. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yin Y, Ren Y, Wu W, et al: Protective
effects of bilobalide on Aβ(25–35) induced learning and memory
impairments in male rats. Pharmacol Biochem Behav. 106:77–84. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vanitallie TB: Stress: a risk factor for
serious illness. Metabolism. 51:40–45. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kaufman D, Banerji MA, Shorman I, et al:
Early-life stress and the development of obesity and insulin
resistance in juvenile bonnet macaques. Diabetes. 56:1382–1386.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pittenger C and Duman RS: Stress,
depression, and neuroplasticity: a convergence of mechanisms.
Neuropsychopharmacology. 33:88–109. 2008. View Article : Google Scholar
|
|
25
|
Zafir A and Banu N: Modulation of in vivo
oxidative status by exogenous corticosterone and restraint stress
in rats. Stress. 12:167–177. 2009. View Article : Google Scholar
|
|
26
|
Jankord R and Herman JP: Limbic regulation
of hypothalamo-pituitary-adrenocortical function during acute and
chronic stress. Ann NY Acad Sci. 1148:64–73. 2008. View Article : Google Scholar
|
|
27
|
Ahmad A, Rasheed N, Ashraf GM, et al:
Brain region specific monoamine and oxidative changes during
restraint stress. Can J Neurol Sci. 39:311–318. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kuo TC, Huang WJ and Guh JH: WJ9708012
exerts anticancer activity through PKC-α related crosstalk of
mitochondrial and endoplasmic reticulum stresses in human
hormone-refractory prostate cancer cells. Acta Pharmacol Sin.
32:89–98. 2011. View Article : Google Scholar
|
|
29
|
Wang XZ, Lawson B, Brewer JW, et al:
Signals from the stressed endoplasmic reticulum induce
C/EBP-homologous protein (CHOP/GADD153). Mol Cell Biol.
16:4273–4280. 1996.PubMed/NCBI
|
|
30
|
Groenendyk J and Michalak M: Endoplasmic
reticulum quality control and apoptosis. Acta Biochim Pol.
52:381–395. 2005.PubMed/NCBI
|
|
31
|
Watts LT, Rathinam ML, Schenker S and
Henderson GI: Astrocytes protect neurons from ethanol-induced
oxidative stress and apoptotic death. J Neurosci Res. 80:655–666.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Loh KP, Huang SH, De Silva R, Tan BK and
Zhu YZ: Oxidative stress: apoptosis in neuronal injury. Curr
Alzheimer Res. 3:327–337. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu M, Yang S, Elliott MH, et al: Oxidative
and endoplasmic reticulum stresses mediate apoptosis induced by
modified LDL in human retinal Müller cells. Invest Ophthalmol Vis
Sci. 53:4595–4604. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xu C, Bailly-Maitre B and Reed JC:
Endoplasmic reticulum stress: cell life and death decisions. J Clin
Invest. 115:2656–2664. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Park GB, Kim YS, Lee HK, et al:
Endoplasmic reticulum stress-mediated apoptosis of EBV-transformed
B cells by cross-linking of CD70 is dependent upon generation of
reactive oxygen species and activation of p38 MAPK and JNK pathway.
J Immunol. 185:7274–7284. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pino SC, O'Sullivan-Murphy B, Lidstone EA,
et al: Protein kinase C signaling during T cell activation induces
the endoplasmic reticulum stress response. Cell Stress Chaperones.
13:421–434. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lien YC, Kung HN, Lu KS, Jeng CJ and Chau
YP: Involvement of endoplasmic reticulum stress and activation of
MAP kinases in beta-lapachone-induced human prostate cancer cell
apoptosis. Histol Histopathol. 23:1299–1308. 2008.PubMed/NCBI
|
|
38
|
Mauro A, Ciccarelli C, De Cesaris P, et
al: PKCα-mediated ERK, JNK and p38 activation regulates the
myogenic program in human rhabdomyosarcoma cells. J Cell Sci.
115:3587–3599. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Song MS, Park YK, Lee JH and Park K:
Induction of glucose-regulated protein 78 by chronic hypoxia in
human gastric tumor cells through a protein kinase
C-epsilon/ERK/AP-1 signaling cascade. Cancer Res. 61:8322–8330.
2001.PubMed/NCBI
|
|
40
|
Liu H, Bowes RC III, van de Water B,
Sillence C, Nagelkerke JF and Stevens JL: Endoplasmic reticulum
chaperones GRP78 and calreticulin prevent oxidative stress,
Ca2+ disturbances, and cell death in renal epithelial
cells. J Biol Chem. 272:21751–21759. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yu Z, Luo H, Fu W and Mattson MP: The
endoplasmic reticulum stress-responsive protein GRP78 protects
neurons against excitotoxicity and apoptosis: suppression of
oxidative stress and stabilization of calcium homeostasis. Exp
Neurol. 155:302–314. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yin QQ, Dong CF, Dong SQ, et al: AGEs
induce cell death via oxidative and endoplasmic reticulum stresses
in both human SH-SY5Y neuroblastoma cells and rat cortical neurons.
Cell Mol Neurobiol. 32:1299–1309. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar
|
|
45
|
Tajiri S, Oyadomari S, Yano S, et al:
Ischemia-induced neuronal cell death is mediated by the endoplasmic
reticulum stress pathway involving CHOP. Cell Death Differ.
11:403–415. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Parkash V, Lindholm P, Peranen J, et al:
The structure of the conserved neurotrophic factors MANF and CDNF
explains why they are bifunctional. Protein Eng Des Sel.
22:233–241. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hayashi T, Saito A, Okuno S, Ferrand-Drake
M, Dodd RL and Chan PH: Oxidative injury to the endoplasmic
reticulum in mouse brains after transient focal ischemia. Neurobiol
Dis. 15:229–239. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Rabek JP, Boylston WH III and
Papaconstantinou J: Carbonylation of ER chaperone proteins in aged
mouse liver. Biochem Biophys Res Commun. 305:566–572. 2003.
View Article : Google Scholar : PubMed/NCBI
|