Introduction
Neuronal cytotoxicity occurs when there is an
imbalance in the systems that generate and scavenge reactive oxygen
species (ROS) in a number of pathological processes, including
neurodegenerative diseases (1,2).
Although glutamate is an important neurotransmitter in mammals,
high concentrations of this amino acid can contribute to neuronal
cell death. Moreover, glutamate toxicity induces the progressive
death of neuronal cells and this has been implicated in a variety
of neurological disorders, such as Parkinson’s disease, Alzheimer’s
disease, seizures, ischemia and trauma (3,4).
Glutamate has been shown to induce neuronal cell death through two
mechanisms: glutamate receptor-induced cytotoxicity and
ROS-mediated oxidative stress. HT22 mouse hippocampal neuronal
cells are a useful model for studying the mechanisms of
glutamate-induced oxidative stress which leads to cell death. Since
this cell line lacks functional ionotropic glutamate receptors, the
excessive accumulation of glutamate causes oxidative injury due to
the suppression of the cellular uptake of cysteine through the
cysteine/glutamate transport system; this, in turn, causes a
progressive depletion of glutathione (GSH), an intracellular
antioxidant molecule (5,6).
In addition, the excessive production of ROS induces
the activation of mitogen-activated protein kinase (MAPK) signaling
pathways, which, in turn, induces the transcription of numerous
apoptosis-related genes. MAPKs are serine/threonine protein kinases
involved in cell proliferation and differentiation, as well as in
inflammation, cancer and apoptosis. MAPKs can be subdivided into
three classes based on sequence homology: the extracellular
signal-regulated kinase (ERK), Jun N-terminal kinases (JNKs) and
the p38 kinases. MAPKs can be phosphorylated and then activated in
response to a variety of extracellular stimuli, leading to the
expression or activation of key molecules. Recent studies have
shown that ERK is also responsible for oxidative stress in
neurodegenerative disorders, ischemia and stroke (7–9).
There may be a close association between excessive ROS production
and MAPK activation in glutamate-induced neuronal cell death.
Additionally, glutamate toxicity is caused by increased
intracellular calcium influx due to GSH depletion, but not through
the glutamate receptor. Previous studies have demonstrated that
glutamate promotes an increase in calcium influx in HT22 cells, and
acts as a key regulator in response to oxidative stress in neuronal
cells (10,11).
Furthermore, glutamate-induced oxidative stress, ROS
production and calcium influx are crucial contributors to
mitochondrial dysfunction (12,13). Apoptosis-inducing factor (AIF) is
released from the mitochondria and translocates to the nucleus in
its truncated form (tAIF), promoting the transcription of
apoptosis-related genes. Therefore, the inhibition of the apoptotic
pathway activated by glutamate-induced oxidative stress may be an
excellent strategy for the prevention or treatment of
neurodegenerative disorders (14).
To counteract glutamate-induced oxidative stress
insults, neuronal cells have evolved defense mechanisms, such as
antioxidant enzymes. With the induction of antioxidant enzymes,
such as heme oxygenase-1 (HO-1) and NAD(P)H dehydrogenase [quinone]
1 (NQO1), neuronal cells are more resistant to the subsequent
provocation of glutamate-induced stress. The importance of HO-1 and
NQO1 expression in mediating neuroprotective properties has been
well characterized in neuro-degenerative disorders (15,16). HO-1 and NQO1 expression is
regulated through transcription factors, such as nuclear factor
(erythroid-derived 2)-like 2 (Nrf2, also known as NFE2L2),
cAMP-responsive element binding protein (CREB) and activating
protein-1 (AP-1). The promoter regions of the HO-1 and NQO1 genes
have antioxidant responsive elements (AREs) to which Nrf2 can
directly bind. The activation of Nrf2 by various stimulators is
followed by its translocation to the nucleus, where it modulates
the expression of ARE-driven genes (such as HO-1 and NQO1). Protein
kinase A (PKA)-mediated CREB activation also induces the expression
of these ARE-driven genes (HO-1 and NQO1). CREB activation mediates
the transcription of genes containing a cAMP-responsive element.
Previous studies have indicated that the PKA/CREB/Nrf2 signaling
pathway regulates the expression of the ARE-driven genes (HO-1 and
NQO1) and has subsequent neuroprotective effects (17–20).
We previously isolated a novel natural compound,
α-iso-cubebene, from Schisandra chinensis, an herb used for
food, tea and wine production, as well as in Traditional Chinese
Medicine (23). Recently, we
published a study on the beneficial bioactivities of
α-iso-cubebene, such as its anti-inflammatory, antiseptic and
immunomodulatory activities (24). Moreover, recent studies have
suggested that α-iso-cubebene inhibits amyloid β-induced
inflammation in microglia (21–23). However, the mechanisms underlying
its potential protective effects on glutamate-induced neuronal cell
death have not yet been fully investigated. In this study, we
investigated the neuroprotective effects of α-iso-cubebene against
glutamate-induced neuronal cell death using mouse
hippocampus-derived neuronal cells (HT22 cells). Our results
demonstrated that α-iso-cubebene attenuated glutamate-induced cell
death through the conservation of mitochondrial function in HT22
cells. The data from our study suggest that the expression of HO-1
and NQO1 may be at least partially responsible for the
neuroprotective effects of α-iso-cubebene, which involves the
activation of the Nrf2 and PKA/CREB signaling pathways.
Materials and methods
Reagents
Dulbecco’s modified Eagle’s medium (DMEM) and fetal
bovine serum (FBS) were purchased from Gibco-BRL (Grand Island, NY,
USA). 3-[4,5-Dimethythiazol-2-yl]-2,5-diphenyltetrazolium bromide
(MTT) and other reagents were purchased from Sigma-Aldrich (St.
Louis, MO, USA). Small interfering RNA (siRNA) against CREB and
Nrf2, and antibodies against α-tubulin (sc-23948), TATA-binding
protein (TBP; sc-204), ERK (sc-94), JNK (sc-571), p38 (sc-7149),
Nrf2 (sc-722), HO-1 (sc-10789) and NQO1 (sc-16464) were purchased
from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Antibodies against AIF (4642), COX IV (4850), phosphorylated
(p-)ERK (4370), p-JNK (4671), p-p38 (9215), p-PKA (4781), PKA
(4782), p-CREB (9198) and CREB (9197) were purchased from Cell
Signaling Technology (Beverly, MA, USA). The FuGENE HD transfection
reagent and the X-tremeGENE siRNA Transfection reagent were
obtained from Roche Diagnostics (Indianapolis, IN, USA). The
cytotoxicity detection kit [lactate dehydrogenase (LDH) assay] was
purchased from Roche Applied Science (Rotkreuz, Switzerland). The
APO-BrdU™ TUNEL Assay kit was purchased from Invitrogen (Carlsbad,
CA, USA). The nuclear extraction kit was purchased from Active
Motif (San Diego, CA, USA). The mitochondria Isolation kit was
purchased from Thermo Scientific (Rockford, IL, USA).
Cell culture
The HT22 hippocampal cell line was obtained from
Professor Youn-Chul Kim, Wonkwang University (Iksan, Korea). The
cells were grown as monolayers in DMEM supplemented with 5%
heat-inactivated FBS. The cells were incubated at 37°C in a
humidified atmosphere containing 5% CO2. To avoid
changes in cell characteristics caused by extended periods of cell
culture, all experiments were conducted with cells between passages
15 and 25. Each cell suspension was subcultured by treatment with
trypsin/EDTA every 2 days in order to maintain exponential
growth.
Cell viability assay
The cells were incubated in wells of a 24-well plate
at a density of 4x104 cells/well. MTT solution (50
μg/ml) was added to each well. The plates were then
incubated for an additional 4 h at 37°C in a 5% CO2
atmosphere, after which the supernatant was removed. Formazan
crystals that had formed in viable cells were solubilized using
dimethyl sulfoxide (DMSO). The absorbance of each well was measured
at 570 nm by using a microplate reader (Wallace, Boston, MA,
USA).
LDH release assay
Extracellular LDH activity was
spectro-photometrically measured using a cytotoxicity detection kit
according to the manufacturer’s instructions. Briefly, the cells
were seeded in 96-well plates and then subjected to the indicated
treatments. For analysis, the supernatant was extracted from each
well, and catalyst solutions were added followed by incubation for
30 min at room temperature. The absorbance of each well was
measured at 490 nm using a microplate reader (Wallace).
Measurement of intracellular ROS and
calcium levels
To evaluate the levels of intracellular ROS and
calcium, the cells were treated with CM-H2DCFDA (an
indicator of ROS production) or Fluo-4-AM (an indicator of general
oxidative stress; both from Invitrogen), for 1 h at 37°C under 5%
CO2. The cells were then harvested and washed 3 times
with phosphate-buffered saline (PBS). The fluorescence intensity
was then measured by flow cytometry (using the flow cytometer at
the Bio-IT Fusion Technology Research Institute, Pusan National
University, Busan, Korea) at an excitation wavelength of 488 nm and
an emission wavelength of 525 nm. Data analyses were performed
using CXP software 2.0 (Beckman Coulter, Brea, CA, USA).
Measurement of sub-G1 cell
population
The cells were collected by centrifugation at 800
rpm for 3 min after treatment, washed twice in PBS, and fixed with
75% ethanol overnight. Prior to flow cytometric analysis, the fixed
cells were washed with PBS and incubated with a final concentration
of 50 μg/ml propidium iodide for 10 min in the dark. The
percentage of apoptotic cells was calculated as the percentage of
the sub-G1 peak as determined using CXP software 2.0 (Beckman
Coulter).
Terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling (TUNEL) assay
The DNA ladder pattern in the cells was evaluated by
TUNEL assay using an APO-BrdU™ TUNEL Assay kit (Invitrogen)
according to the manufacturer’s instructions. The results were
analyzed by flow cytometry (fluorescein isothiocyanate; excitation
at 488 nm and emission at 520 nm). Data analyses were performed
using CXP 2.0 (Beckman Coulter).
Assay for mitochondrial membrane
potential (MMP, Δψm)
MMP (Δψm) was determined by flow
cytometry using the J-aggregate forming lipophilic cationic probe,
5,5′,6,6′-tetra-chloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyanine
iodide (JC-1). The cells were stained with JC-1 and analyzed by
flow cytometry with CXP 2.0 analysis software (Beckman Coulter).
JC-1 red fluorescence, indicating an intact Δψm, was excited at 488
nm, and emission was detected using a 613±20 nm bandpass filter.
For each sample, 10,000 cells were acquired and analyzed by flow
cytometry. Data were analyzed using the fluorescence intensity of
the analyzed cell population.
Protein extracts and western blot
analysis
The cytosol, nuclear and mitochondrial extracts were
isolated with a nuclear extraction kit according to the
manufacturer’s instructions. The protein content of the cell
lysates was then determined using the Bradford protein assay
(Bio-Rad, Hercules, CA, USA). The protein in each sample was
resolved by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis, transferred onto a polyvinylidene difluoride
membrane, and exposed to the appropriate antibody. The proteins
were visualized by an enhanced chemiluminescence detection system
(Amersham Biosciences, Piscataway, NJ, USA) using horseradish
peroxidase-conjugated anti-rabbit or anti-mouse secondary antibody.
Images were acquired using an ImageQuant 350 analyzer (Amersham
Biosciences).
Transient transfection with siRNA
Transfection of the cells with siRNA was performed
using the X-tremeGENE siRNA Transfection Reagent (Roche Applied
Science) according to manufacturer’s instructions. Commercially
available mouse CREB- and Nrf2-specific siRNAs and negative control
siRNAs (both from Santa Cruz, Heidelberg, Germany) were used for
transfection. Briefly, X-tremeGENE siRNA transfection reagent (10
μl) was added to 100 μl of serum-free medium
containing 2 μg of each siRNA followed by incubation for 20
min at room temperature. Gene silencing was measured after 48 h by
western blot analysis.
Transient transfection and dual
luciferase assay
The cells were transfected with the CRE and
ARE-reporter plasmid, or an HO-1 promoter reporter plasmid using
FuGENE-HD reagent according to manufacturer’s instructions. A
Renilla luciferase control plasmid, pRL-CMV, was
co-transfected as an internal control for transfection efficiency.
Luciferase activity was assayed using a dual-luciferase assay kit
according to manufacturer’s instructions. Luminescence was measured
using a microplate luminometer (Wallac 1420).
Statistical analysis
Data are expressed as the means ± standard error
(SE). Statistical analysis was performed using the Statistical
Package for the Social Sciences (SPSS) software (version 18.0) to
identify significant differences based on either one- or two-way
analysis of variance (ANOVA) followed by Dunn’s post-hoc tests.
P-values <0.05 were considered to indicate statistically
significant differences. Each experiment was repeated at least 3
times.
Results
Inhibitory effects of α-iso-cubebene on
the glutamate-induced death of HT22 cells
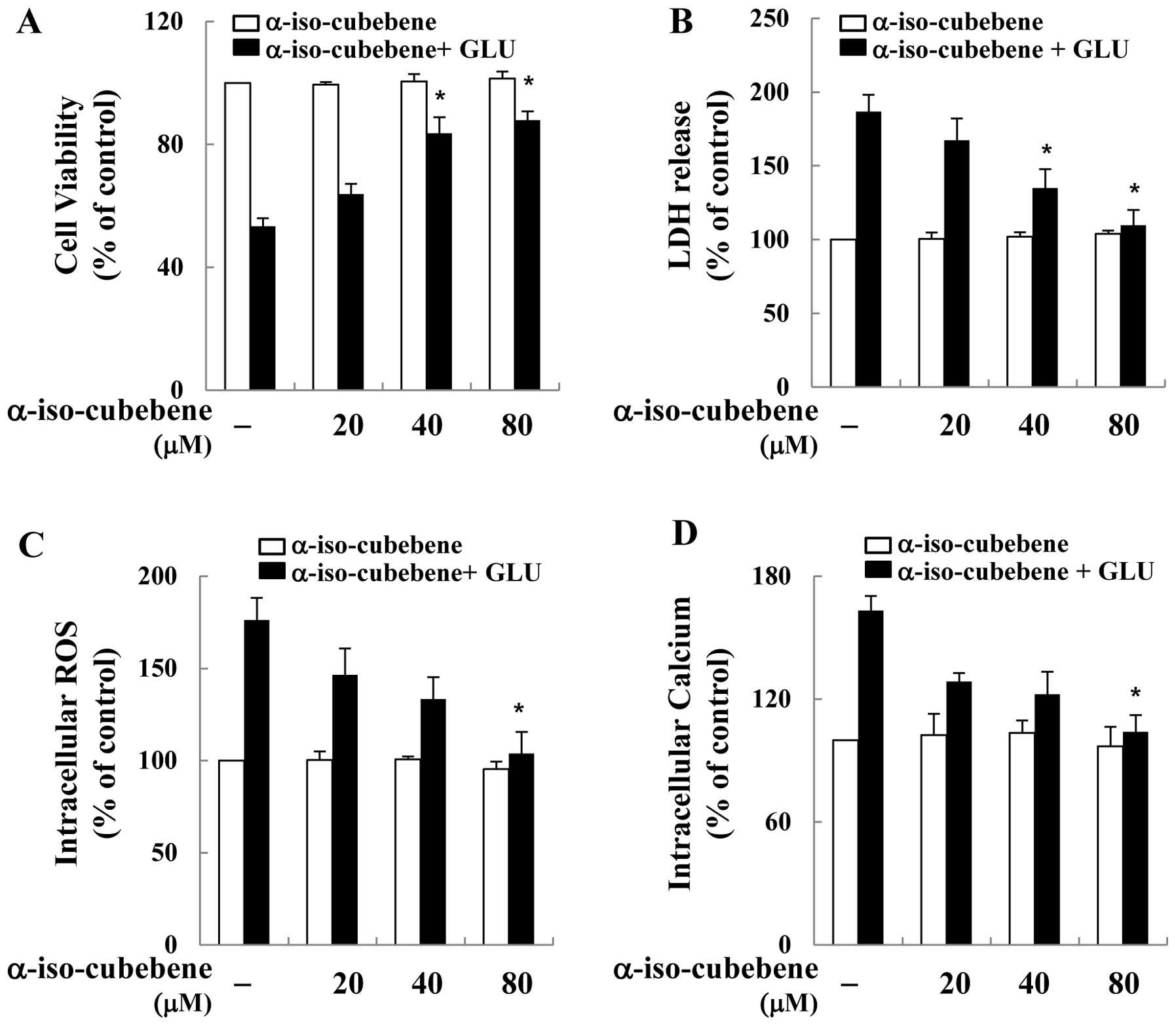
To examine the ability of α-iso-cubebene to exert
neuronal protective effects against glutamate-induced cell death,
its effects on glutamate-induced cell death on the murine
hippocampal neuronal cell line, HT22, were investigated. To
accomplish this, the HT22 cells were first pre-treated with
non-cytotoxic concentrations of α-iso-cubebene [isolated from
Schisandra chinensis (20–80 μM)] for 12 h, and then
treated with glutamate for 12 h. Cell viability was then measured
by MTT assay. Cell viability was recovered by α-iso-cubebene in a
dose-dependent manner (Fig. 1A).
The neuroprotective effects of α-iso-cubebene were also verified by
LDH release assay. The results revealed that glutamate
significantly increased LDH release from the HT22 cells. However,
the cells treated with α-iso-cubebene prior to glutamate incubation
displayed a dose-dependent attenuation of LDH release (Fig. 1B).
It is fairly well established that glutamate
toxicity is mediated by ROS production and oxidative stress-induced
cell death (25). Hence, in the
current study, we investigated whether the neuro-protective effects
of α-iso-cubebene involved the blockage of glutamate-induced
oxidative stress in hippocampal neuronal cells. Cellular oxidative
stress was determined by H2DCF-DA, based on the
ROS-mediated conversion of H2DCF-DA to fluorescent DCF.
Glutamate increased intracellular ROS production in the HT22 cells.
By contrast, at the tested concentrations, α-iso-cubebene
profoundly reduced the glutamate-induced production of ROS
(Fig. 1C). In addition,
glutamate-mediated cytotoxicity results in GSH depletion through
the suppression of cysteine uptake and increased intracellular
calcium concentration. Abnormal levels of GSH and calcium in cells
consequently induce ROS overproduction (13). Therefore, in this study, in order
to determine the association between the protective effects of
α-iso-cubebene on neuronal cells and glutamate-induced cell death,
its inhibitory activity against glutamate-induced calcium influx
was examined. Calcium influx in HT22 cells was significantly
reduced by α-iso-cubebene treatment, whereas that of the
glutamate-treated cells was increased compared to the controls
(untreated cells; Fig. 1D).
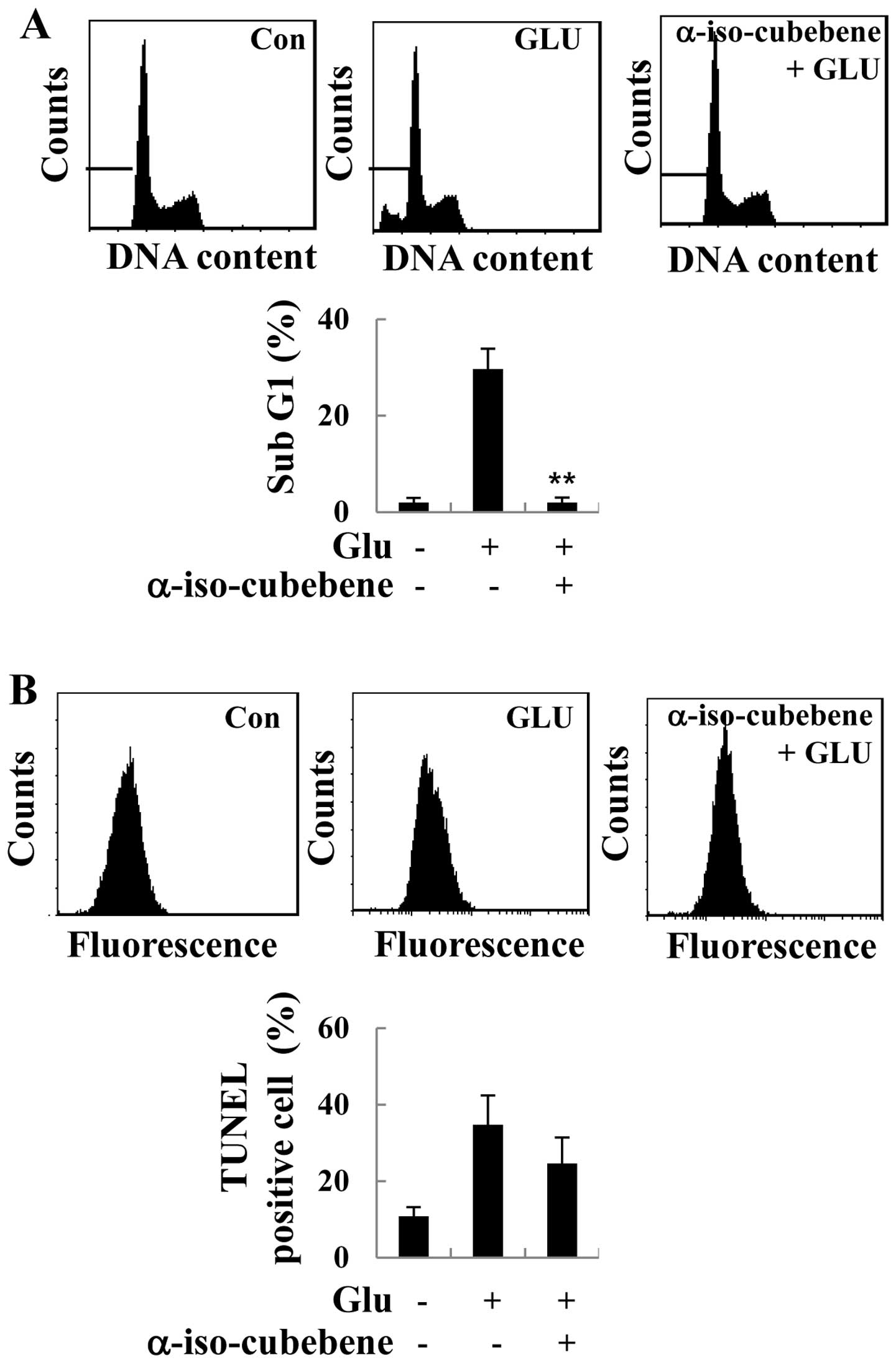
α-iso-cubebene attenuates
glutamate-induced apoptosis in HT22 cells
It has been suggested that excessive glutamate
levels results in apoptosis (12). Thus, in this study, in order to
investigate the role of α-iso-cubebene in glutamate-induced
apoptosis in HT22 cells, the cells were treated with glutamate for
the indicated periods of time, and the effects of α-iso-cubebene on
cell cycle distribution were analyzed. As demonstrated by our
results, the ratio of sub-G1 cells was increased following
treatment with glutamate, indicating an increase in
glutamate-induced apoptosis. However, glutamate-induced apoptosis
was decreased in the α-iso-cubebene-pre-treated cells compared to
the controls (untreated cells; Fig.
2A). Additionally, to verify the specific type of cell death
elicited by glutamate, TUNEL assay was performed to assess the
typical DNA ladder pattern of the glutamate-treated HT22 cells. The
HT22 cells that had undergone 24 h of glutamate treatment showed an
increase in apoptosis compared to the controls, whereas
pre-treatment with α-iso-cubebene resulted in a significant
decrease in cell apoptosis (Fig.
2B).
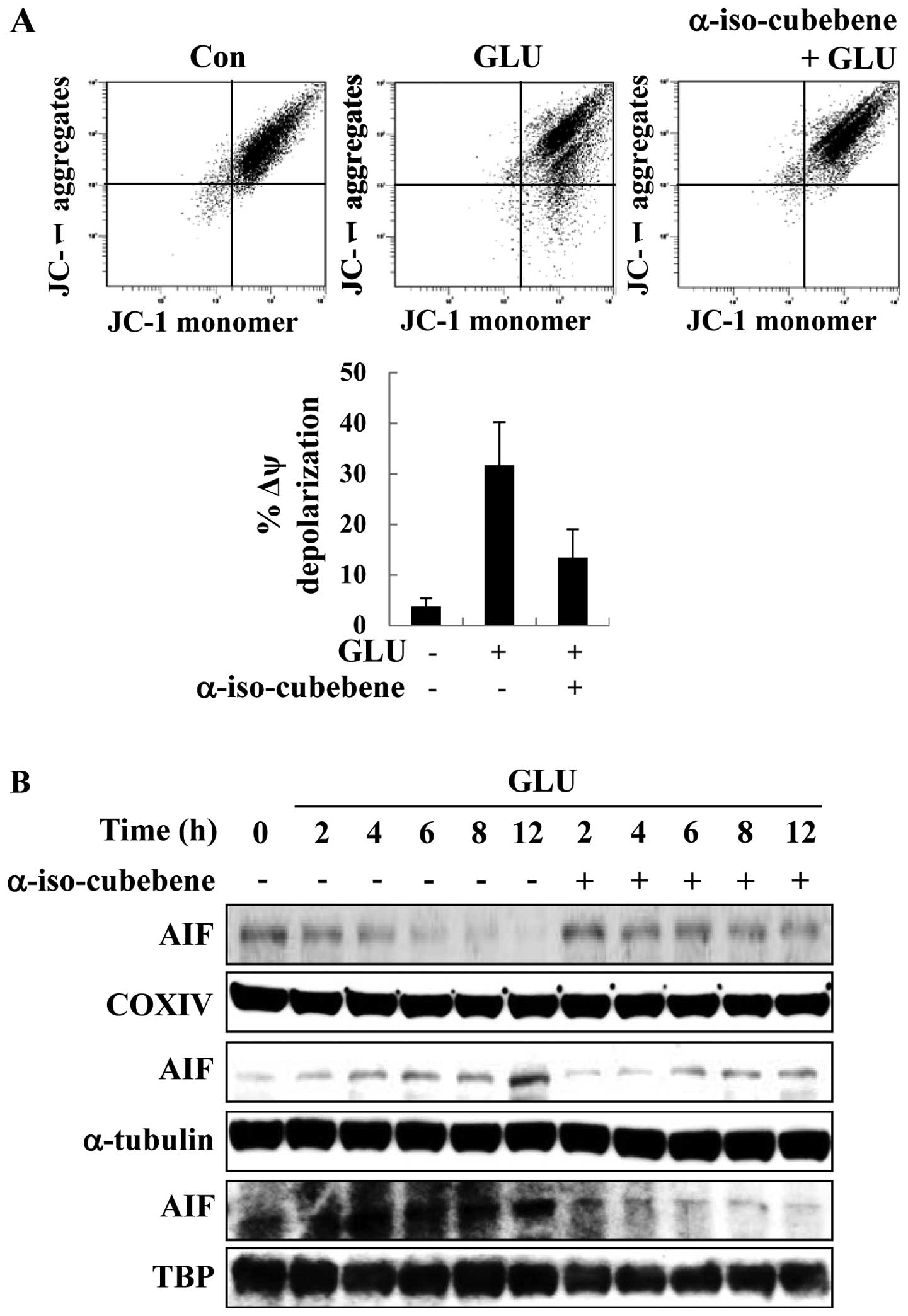
α-iso-cubebene modulates
glutamate-induced mitochondrial function and AIF localization
Mitochondrial membrane disruption is a primary
process in apoptosis; therefore, the JC-1 stain was used to detect
the glutamate-induced changes in MMP. With JC-1 staining, the HT22
cells treated with glutamate displayed a significantly increased
green fluorescence intensity compared to the controls (untreated
cells), indicating the glutamate-induced disruption of MMP
(Δψm). By contrast, pre-treatment with α-iso-cubebene
reversed the glutamate-induced increase in green fluorescence
(Fig. 3A). Thus, mitochondrial
function was effectively protected by treatment with
α-iso-cubebene.
Apoptosis correlates with the levels of proteins
that regulate mitochondrial-dependent apoptosis and the apoptotic
pathway induced by AIF signaling (14). As previously demonstrated, the
disruption of MMP leads to the release of the pro-apoptotic
factors, cytochrome c, AIF and endonuclease G (Endo G). Upon
these changes, the normal form of AIF is cleaved and released from
the mitochondria (14). In this
study, to determine the effects of α-iso-cubebene on AIF
distribution, mitochondrial, cytosolic and nuclear fractions of AIF
were analyzed by western blot analysis. COX IV is specific to the
inner mitochondrial membrane protein, α-tubulin is specific to the
cytoplasmic protein and TBP is specific to the nuclear fraction.
COX IV, α-tubulin and TBP are commonly used for determining the
mitochondrial, cytosolic and nuclear fractions in western blot
analysis. Following treatment with glutatame, the level of AIF in
the mitochondrial fractions was decreased at the indicated time
points (Fig. 3). By contrast,
treatment with glutamate increased the cytosolic and nuclear
fractions of AIF after 12 h, and treatment with α-iso-cubebene
reversed the glutamate-induced translocation of AIF at this time
point (Fig. 3B). Thus, these data
suggest that α-iso-cubebene effectively suppresses the
glutamate-induced disruption of MMP and the translocation of
AIF.
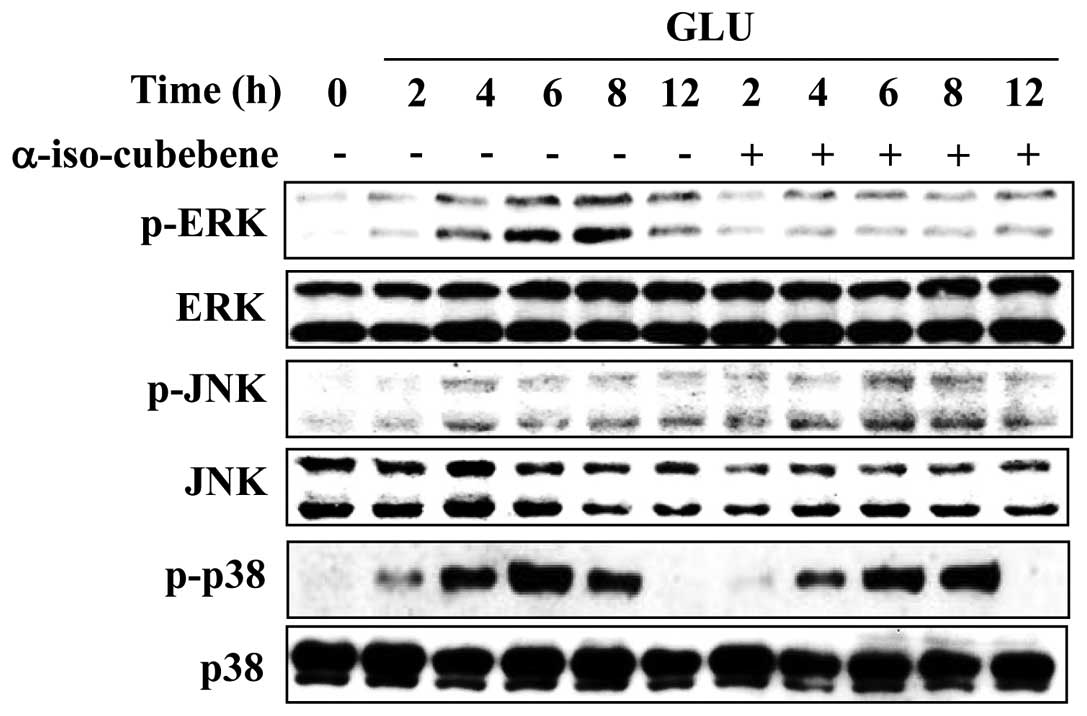
α-iso-cubebene suppresses the activation
of ERK induced by glutamate
Emerging evidence indicates that MAPK activation and
oxidative stress are involved in the pathophysiology of
neurodegenerative diseases. MAPK activation and oxidative stress
also induce apoptosis in neuronal cells (26). Therefore, the effects of
α-iso-cubebene on the glutamate-mediated activation of ERK, JNK and
p38 MAPKs were examined. The levels of p-ERK, p-p38 and p-JNK were
increased after 4–8 h of glutamate treatment (Fig. 4). However, the levels of p-ERK
were significantly reduced following treatment with
α-iso-cubebene.
α-iso-cubebene-induced activation of
PKA/CREB/Nrf2 and the expression of HO-1 and NQO1
Previous data have indicated a critical role of
ARE-driven genes in the host defense against a variety of
physiological insults, such as oxidative stress, hypoxia and
pro-inflammatory cytokines, as well as in the regulation of normal
immune function (15). In this
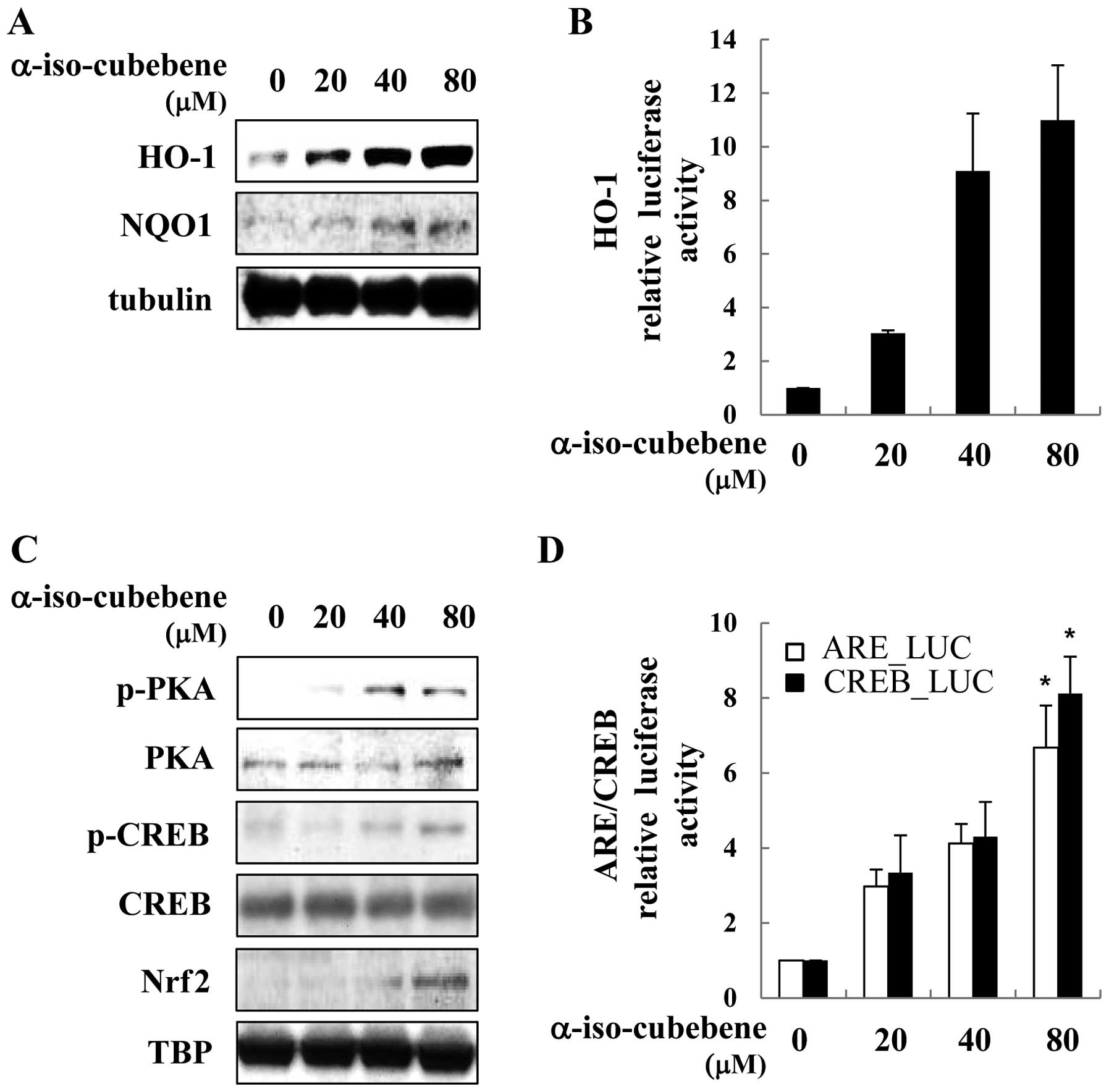
study, α-iso-cubebene increased ARE-driven gene (HO-1 and NQO1)
expression in a time and dose-dependent manner. The HT22 cells
treated with α-iso-cubebene had increased expression levels of HO-1
and NQO1 (Fig. 5A). To determine
the effects of α-iso-cubebene on HO-1 expression, the HT22 cells
were transiently transfected with HO-1 promoter reporter plasmid.
Treatment with α-iso-cubebene increased HO-1 promoter activity in a
dose-dependent manner (Fig. 5B).
Transcription factors that ARE-driven genes are associated with
CREB and Nrf2 (27). Therefore,
we investigated whether α-iso-cubebene enhances CREB and Nrf2
activation. The evaluation of the phosphorylation status of PKA and
CREB revealed that the maximum levels of their activation occurred
4 h following treatment with α-iso-cubebene (data not shown).
α-iso-cubebene increased PKA and CREB phosphorylation, thereby
affecting the total levels of PKA and CREB. In addition to CREB and
Nrf2 activation, we examined the downstream effects of
α-iso-cubebene on Nrf2 activation. To accomplish this, we
investigated the α-iso-cubebene-induced Nrf2 nuclear translocation
in HT22 cells (Fig. 5C). To
elucidate the mechanisms underlying these effects, we used a
luciferase reporter gene driven by Nrf2-binding to ARE and
CRE-binding to CRE to analyze Nrf2 and CREB transactivity. The
results revealed that α-iso-cubebene significantly increased ARE
and CRE transactivity in the HT22 cells (Fig. 5D).
Role of the CREB/Nrf2 pathway in the
neuroprotective effects of α-iso-cubebene
CREB and Nrf2 have been identified as key mediators
in the regulation of the crosstalk between the neuro-protective
signals induced by antioxidant gene cell response (28). In this study, we confirmed that
α-iso-cubebene markedly inhibited glutamate-induced cell death and
that this effect correlated with the activation of the CREB and
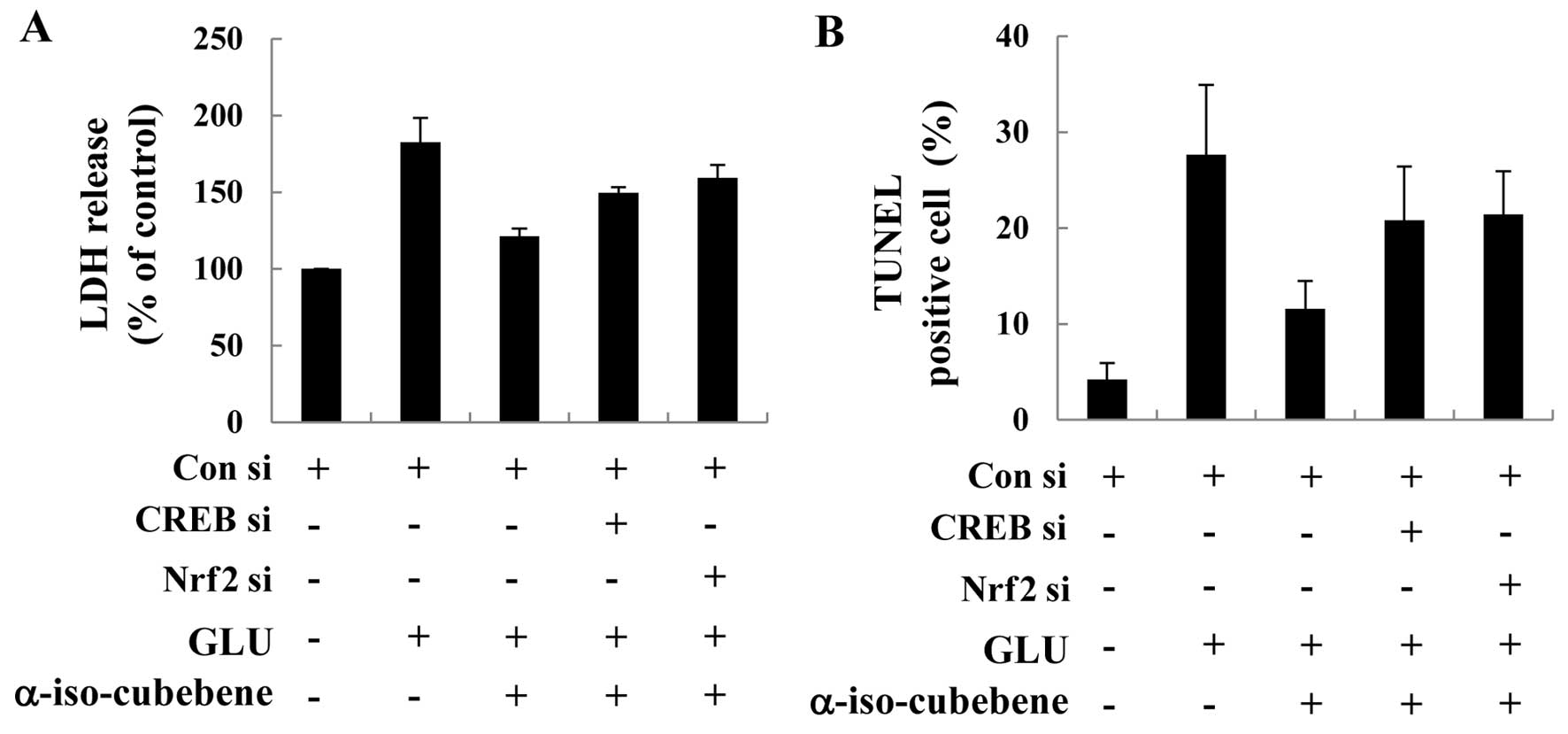
Nrf2 pathways. For these experiments, we used CREB and Nrf2 siRNA
systems to silence the CREB and Nrf2 proteins, and then determined
the effects on LDH release and performed TUNEL assay in HT22 cells.
The HT22 cells were transfected with control siRNA or CREB siRNA or
Nrf2 siRNA for 48 h. Western blot analysis of the treated cells
revealed that CREB and Nrf2 siRNA caused significantly knocked down
CREB and Nrf2 by 48 h compared to the control siRNA-transfected
cells (data not shown). The HT22 cells were transfected with CREB
and Nrf2 siRNA, followed by treatment with α-iso-cubebene and
glutamate. The inhibitory effects of α-iso-cubebene on the
glutamate-induced release of LDH and the number of TUNEL-positive
cells were abolished by CREB and Nrf2 siRNA, suggesting the
involvement of the PKA/CREB/Nrf2 pathway in the
α-iso-cubebene-induced neuroprotection in HT22 cells (Fig. 6).
Discussion
The results of this study demosntrated that
α-iso-cubebene significantly protected HT22 neuronal cells from
glutamate-induced oxidative death by inhibiting the production of
intracellular ROS and calcium. These findings support a
neuro-protective role of α-iso-cubebene through the PKA/CREB/Nrf2
signaling pathway and its downstream antioxidant enzymes. HT22, an
immortalized mouse hippocampal neuronal cell line, has been used as
an in vitro model of Alzheimer’s disease, a disease
characterized by neuronal cell death, particularly in the
hippocampus. Moreover, the loss of neuronal cells due to oxidative
stress appears to be a common cause contributing to
neurodegenerative diseases (29,30). Glutamate is a major contributor to
neuronal cell death through oxidative toxicity in HT22 cells. The
mechanisms of glutamate-induced toxicity in HT22 cells involve
excessive glutamate levels, leading to the depletion of GSH, an
intracellular antioxidant material, by disrupting the
glutamate/cysteine antiporter system. Following GSH depletion, a
series of biological processes occur, including ROS accumulation,
calcium influx and lipid peroxidation (30). In this study, we assessed
glutamate-induced HT22 cell death and neurotoxicity by MTT and LDH
release assays and confirmed that α-iso-cubebene markedly
attenuated the glutamate-induced decrease in cell viability and the
increase in LDH release. We also examined glutamate-induced
apoptotic cell death in the HT22 cell line, which is typically
characterized by hypodiploid DNA content and DNA fragmentation.
These apoptotic features were detected by cell cycle analysis and
TUNEL assays. Of note, treatment with α-iso-cubebene markedly
attenuated these apoptotic features, indicating that α-iso-cubebene
may be qualified for inhibiting glutamate-induced apoptotic cell
death. We also examined the effects of α-iso-cubebene on
glutamate-induced cell death in HT22 cells by evaluating the
inhibition of intracellular ROS and calcium accumulation. Since
these events are known to play a major role in glutamate-induced
neuronal cell death (3,4), the inhibition of intracellular ROS
and calcium influx in its early stages may be a central point for
the α-iso-cubebene-induced neuroprotective effects.
The disruption of MMP induced by oxidative stress is
regulated by several anti- and pro-apoptotic proteins, such as
Bcl-2 and Bax. During the apoptotic process, Bcl-2 and Bax change
their location, allowing Bax to enter the outer mitochondrial
membrane, permeabilizing it, and causing the release of cytochrome
c and the translocation of AIF. Mitochondrial apoptosis is
divided into two types: caspase-dependent and caspase-independent.
Caspase-independent apoptosis is mainly dependent on promoting the
AIF translocation and disrupting the function and structure of the
nucleus (14). Consistent with
previous data (12), the present
results suggest that glutamate-induced HT22 cell death occurs
through mitochondrial dysfunction and the release of AIF from the
mitochondria. However, α-iso-cubebene prevented both
glutamate-induced mitochondrial dysfunction and the abnormal
distribution of AIF.
Protein phosphatases play an important role in
controlling the magnitude and duration of MAPK activation (8). Thus, this study focused on the
possible role of MAPK family members, which are known to play
crucial roles during oxidative stress in many cell types (7,8).
Previous studies have suggested that the inhibition of ERK
activation protects cells against glutamate-induced death (7,8).
In agreement with these studies, a previous study also found that
glutamate led to the persistent activation of ERK, which was
associated with the death of HT22 cells (8). Our results indicated that
glutamate-induced oxidative toxicity led to the phosphorylation of
MAPKs, including JNK, ERK, and p38, resulting in persistent
neuronal cell death. By contrast, α-iso-cubebene was able to
significantly suppress the phosphorylation of ERK at the examined
time points, albeit with little effect on JNK and p38
phosphorylation.
HO-1 and NQO1 are binding sites for the Nrf2
transcription factor and, as such, key signaling antioxidant
enzymes for the regulation of neuroprotection through oxidative
responses. A recent study demonstrated an inverse correlation
between the antioxidant enzyme level and neuroprotection (31). In this study, we examined whether
α-iso-cubebene increases the expression of HO-1 and NQO1. Treatment
with α-iso-cubebene induced the expression of HO-1 and HO-1 in the
HT22 cells. Additionally, a number of neuroprotective drugs have
been shown to inhibit glutamate-induced neuronal cell death by
stimulating the PKA/CREB/Nrf2 pathway (20,32). In agreement with these studies,
the results of the present study demonstrated that α-iso-cubebene
induced the activation of the PKA/CREB/Nrf2 pathway, thus providing
a basis for the neuroprotective effects against glutamate-induced
toxicity in HT22 cells.
In conclusion, α-iso-cubebene was selected as a
candidate for this fundamental research for investigating the
neuroprotective drugs from natural products. α-iso-cubebene exerted
neuroprotective effects against glutamate-induced apoptosis in HT22
cells by suppressing the generation of ROS and calcium influx, as
well as mitochondrial disruption, events thought to be involved in
HT22 cell apoptosis. α-iso-cubebene inhibited the glutamate-induced
neuronal cell death by inducing HO-1 and NQO1 expression through
the PKA/CREB/Nrf2 pathway. These data suggest that α-iso-cubebene
may have therapeutic potential as a neuroprotective drug for the
treatment of neurodegenerative diseases.
Acknowledgments
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education, Science and Technology
(2012R1A1A3010601) and the Bio-industry Technology Development
Program (311054-03-3-HD120), Ministry for Food, Agriculture,
Forestry and Fisheries, Republic of Korea.
References
|
1
|
Coyle JT and Puttfarcken P: Oxidative
stress, glutamate, and neurodegenerative disorders. Science.
262:689–695. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tan S, Sagara Y, Liu Y, Maher P and
Schubert D: The regulation of reactive oxygen species production
during programmed cell death. J Cell Biol. 141:1423–1432. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Elphick LM, Hawat M, Toms NJ, Meinander A,
Mikhailov A, Eriksson JE and Kass GE: Opposing roles for caspase
and calpain death proteases in L-glutamate-induced oxidative
neurotoxicity. Toxicol Appl Pharmacol. 232:258–267. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Murphy TH, Miyamoto M, Sastre A, Schnaar
RL and Coyle JT: Glutamate toxicity in a neuronal cell line
involves inhibition of cystine transport leading to oxidative
stress. Neuron. 2:1547–1558. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Behl C, Lezoualc’h F, Trapp T, Widmann M,
Skutella T and Holsboer F: Glucocorticoids enhance oxidative
stress-induced cell death in hippocampal neurons in vitro.
Endocrinology. 138:101–106. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Maher P and Schubert D: Signaling by
reactive oxygen species in the nervous system. Cell Mol Life Sci.
57:1287–1305. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Stanciu M, Wang Y, Kentor R, et al:
Persistent activation of ERK contributes to glutamate-induced
oxidative toxicity in a neuronal cell line and primary cortical
neuron cultures. J Biol Chem. 275:12200–12206. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ravassard P, Pachoud B, Comte JC, et al:
Paradoxical (REM) sleep deprivation causes a large and rapidly
reversible decrease in long-term potentiation, synaptic
transmission, glutamate receptor protein levels, and ERK/MAPK
activation in the dorsal hippocampus. Sleep. 32:227–240.
2009.PubMed/NCBI
|
|
9
|
Kumamaru E, Numakawa T, Adachi N, et al:
Glucocorticoid prevents brain-derived neurotrophic factor-mediated
maturation of synaptic function in developing hippocampal neurons
through reduction in the activity of mitogen-activated protein
kinase. Mol Endocrinol. 22:546–558. 2008. View Article : Google Scholar
|
|
10
|
Fukui M, Song JH, Choi J, Choi HJ and Zhu
BT: Mechanism of glutamate-induced neurotoxicity in HT22 mouse
hippocampal cells. Eur J Pharmacol. 617:1–11. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Choi DW: Calcium-mediated neurotoxicity:
Relationship to specific channel types and role in ischemic damage.
Trends Neurosci. 11:465–469. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ankarcrona M, Dypbukt JM, Bonfoco E,
Zhivotovsky B, Orrenius S, Lipton SA and Nicotera P:
Glutamate-induced neuronal death: a succession of necrosis or
apoptosis depending on mitochondrial function. Neuron. 15:961–973.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gunter TE and Pfeiffer DR: Mechanisms by
which mitochondria transport calcium. Am J Physiol. 258:C755–C786.
1990.PubMed/NCBI
|
|
14
|
Zhang Y and Bhavnani BR: Glutamate-induced
apoptosis in neuronal cells is mediated via caspase-dependent and
independent mechanisms involving calpain and caspase-3 proteases as
well as apoptosis inducing factor (AIF) and this process is
inhibited by equine estrogens. BMC Neurosci. 7:492006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Calkins MJ, Johnson DA, Townsend JA, et
al: The Nrf2/ARE pathway as a potential therapeutic target in
neurodegenerative disease. Antioxid Redox Signal. 11:497–508. 2009.
View Article : Google Scholar
|
|
16
|
van Muiswinkel FL and Kuiperij HB: The
Nrf2-ARE signalling pathway: promising drug target to combat
oxidative stress in neurodegenerative disorders. Curr Drug Targets
CNS Neurol Disord. 4:267–281. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Satoh T, McKercher SR and Lipton SA:
Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs.
Free Radic Biol Med. 65:645–657. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee J, Kim CH, Simon DK, et al:
Mitochondrial cyclic AMP response element-binding protein (CREB)
mediates mitochondrial gene expression and neuronal survival. J
Biol Chem. 280:40398–40401. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Baxter PS, Martel MA, McMahon A, Kind PC
and Hardingham GE: Pituitary adenylate cyclase-activating peptide
induces long-lasting neuroprotection through the induction of
activity-dependent signaling via the cyclic AMP response
element-binding protein-regulated transcription co-activator 1. J
Neurochem. 118:365–378. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Park SY, Kim JH, Lee SJ and Kim Y:
Involvement of PKA and HO-1 signaling in anti-inflammatory effects
of surfactin in BV-2 microglial cells. Toxicol Appl Pharmacol.
268:68–78. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Choi YW, Kim HJ, Park SS, et al:
Inhibition of endothelial cell adhesion by the new
anti-inflammatory agent alpha-iso-cubebene. Vascul Pharmacol.
51:215–224. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee SK, Kim SD, Lee HY, et al:
α-Iso-cubebene, a natural compound isolated from Schisandra
chinensis fruit, has therapeutic benefit against polymicrobial
sepsis. Biochem Biophys Res Commun. 426:226–231. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee YJ, Shim JW, Lee YJ, et al:
Identification of a novel compound that stimulates intracellular
calcium increase and CXCL8 production in human neutrophils from
Schisandra chinensis. Biochem Biophys Res Commun. 379:928–932.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Park SY, Park SJ, Park NJ, Joo WH, Lee SJ
and Choi YW: α-Iso-cubebene exerts neuroprotective effects in
amyloid beta stimulated microglia activation. Neurosci Lett.
555:143–148. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Maher P and Davis JB: The role of
monoamine metabolism in oxidative glutamate toxicity. J Neurosci.
16:6394–6401. 1996.PubMed/NCBI
|
|
26
|
Ziady AG, Sokolow A, Shank S, Corey D, et
al: Interaction with CREB binding protein modulates the activities
of Nrf2 and NF-κB in cystic fibrosis airway epithelial cells. Am J
Physiol Lung Cell Mol Physiol. 302:L1221–L1231. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang T, Gu J, Wu PF, et al: Protection by
tetrahydroxystilbene glucoside against cerebral ischemia:
Involvement of JNK, SIRT1, and NF-kappaB pathways and inhibition of
intracellular ROS/RNS generation. Free Radic Biol Med. 47:229–240.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Katoh Y, Itoh K, Yoshida E, Miyagishi M,
Fukamizu A and Yamamoto M: Two domains of Nrf2 cooperatively bind
CBP, a CREB binding protein, and synergistically activate
transcription. Genes Cells. 6:857–868. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Arundine M and Tymianski M: Molecular
mechanisms of glutamate-dependent neurodegeneration in ischemia and
traumatic brain injury. Cell Mol Life Sci. 61:657–668. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tofighi R, Johansson C, Goldoni M, Ibrahim
WN, Gogvadze V, Mutti A and Ceccatelli S: Hippocampal neurons
exposed to the environmental contaminants methylmercury and
polychlorinated biphenyls undergo cell death via parallel
activation of calpains and lysosomal proteases. Neurotox Res.
19:183–194. 2011. View Article : Google Scholar
|
|
31
|
Zhang H, Liu H, Davies KJ, Sioutas C,
Finch CE, Morgan TE and Forman HJ: Nrf2-regulated phase II enzymes
are induced by chronic ambient nanoparticle exposure in young mice
with age-related impairments. Free Radic Biol Med. 52:2038–2046.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang H, Guo Z, Wu F, et al: PKA-mediated
protein phosphorylation protects ezrin from calpain I cleavage.
Biochem Biophys Res Commun. 333:496–501. 2005. View Article : Google Scholar : PubMed/NCBI
|