Introduction
Obesity results in a low-grade and chronic
inflammatory state and β cells, adipocytes and other metabolic
cells respond to the excess inflow of nutrients and energy by
stress signals that trigger inflammation (1,2).
Adipocytes are not only storage units for triglycerides, but also
influence systemic lipid homeostasis through the production and
release of adipocyte-specific and adipocyte-enriched hormonal
factors, inflammatory mediators, such as the pro-inflammatory
cytokines, tumor necrosis factor-α (TNF-α), interleukin (IL)-6 and
monocyte chemoattractant protein-1 (MCP-1), and anti-inflammatory
adiponectin (3). The
dysregulation of lipolysis by the increased expression of adipose
inflammatory cytokines is an important factor contributing to
systemic insulin resistance (4).
The pathophysiology of adipocyte dysfunction under stress is
complex, therefore suggesting that the adaptive mechanisms that
operate in stressed adipocytes may have important implications for
understanding the mechanisms of obesity and other metabolic
disorders.
Autophagy is an evolutionarily conserved
lysosome-dependent system in found eukaryotes that regulates the
turnover of cellular proteins and organelles. During autophagy,
target proteins or organelles are delivered into double-membrane
autophagosomes for lysosomal degradation (5). This self-eating system has been
shown to control a variety of functions, including the control of
innate and adaptive immune responses by regulating cytokine
production (6) and to combat
persistent endoplasmic reticulum (ER) stress (7). Deficient autophagy by the
suppression of autophagy-related gene 7 (ATG7) renders hepatocytes
vulnerable to ER stress and insulin resistance, and conversely, the
restoration of hepatic autophagy by means of ATG7 overexpression
improves insulin sensitivity (8).
Macrophages derived from ATG16L1-deficient mice have been shown to
produce higher levels of IL-1β (9), and mice with a conditional deletion
of ATG7 in the intestinal epithelium show an enhanced IL-1β
expression (10). Changes in
adipose autophagy in obesity and metabolic disorders are now
rapidly being characterized; however, the precise role of autophagy
remains unclear and may depend on the disease state and
experimental model under investigation. For example, Ost et
al (11) demonstrated that
the autophagosome content was increased in isolated adipocytes
derived from obese and diabetic patients. By contrast, in rodents,
autophagy has been shown to be reduced both in vitro and in
the adipose tissue of animals (12).
ER stress and pro-inflammatory cytokines both play a
relevant role in adipose tissue inflammation (10,13). However, our understanding of the
molecular mechanisms of autophagy during inflammation and stress
remains incomplete. In this study, we used palmitate (PA) to
generate artificially hypertrophied mature adipocytes and examined
whether there was a resulting change in autophagy and inflammation.
Importantly, we used methodological approaches to examine the
changes in autophagy. We also examined the crosstalk between
autophagy and ER stress or inflammation. We aimed to determine
whether autophagy plays a protective or detrimental role in
stressed adipocyte inflammation in obesity.
Materials and methods
Reagents and antibodies
Fetal bovine serum (FBS), Dulbecco’s modified
Eagle’s medium (DMEM), 1% (v/v) streptomycin/penicillin and TRIzol
reagent were from Gibco Invitrogen, (Grand Island, NY, USA); the
Superscript III First-strand Synthesis System was from Promega
(Madison, WI, USA). Rapamycin (RAP) was purchased from Merck
Bioscience (Darmstadt, Germany). PA (P0500), bovine serum albumin
(BSA) (albumin, endotoxin), chloroquine (CQ), 4-phenyl butyrate
(4-PBA), 3-methyladenine (3-MA), SP600125 and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
were from Sigma-Aldrich (St. Louis, MO, USA). The PageRuler™
Prestained Protein Ladder was from Thermo Scientific (Kalamazoo,
MI, USA). Complete protease inhibitor mixture and immunoblot
polyvinylidene difluoride (PVDF membranes were from Roche
Diagnostics (Barcelona, Spain). RIPA lysis buffer and the BCA
protein assay kit were from Beijing ComWin Biotech Co., Ltd.
(Beijing, China). Western Chemiluminescent HRP substrate was
purchased from Millipore (Billerica, MA, USA). Rabbit
anti-microtubule-associated protein 1 light chain 3 (LC3) 1:1,000
(no. 2775), rabbit anti-activating transcription factor 4 (ATF4)
1:1,000 (no. 11815), rabbit anti-C/EBP homologous protein (CHOP)
1:1,000 (no. 5554), rabbit anti-p-eukaryotic translation initiation
factor 2α (eIF2α) 1:1,000 (no. 3398), rabbit anti-p-c-Jun
N-terminal kinase (JNK) and total JNK 1:1,000 (no. 9912) were from
Cell Signaling Technology (Beverly, MA, USA). Goat polyclonal
anti-BiP 1:200 (sc-1050) and goat polyclonal anti-glyceraldehyde
3-phosphate dehydrogenase (GAPDH) 1:200 (sc-48166) were from Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
Cell culture and PA treatment
The 3T3-L1 cells were obtained from the American
Type Culture Collection (ATCC; Rockville, MD, USA). The cells were
seeded and fed every 2 days in DMEM containing 25 mM glucose
supplemented with 50 U/ml penicillin, 50 μg/ml streptomycin,
100 mM MEM sodium pyruvate and 10% FBS. The cells were grown under
5% CO2 at 37°C. At confluence, differentiation was
induced by the addition of medium containing 0.5 mM
isobutylmethylxanthine, 1 μM dexamethasone (Sigma-Aldrich)
and 10 μg/ml insulin. After 48 h, this mixture was replaced
with fresh medium, and this was changed every 2 days. On days 6–8
after the induction of adipocyte differentiation, the cells were
used for the experiments. PA/BSA conjugates were prepared as
previously described (14). PA
was dissolved at 70°C in 0.1 M NaOH to obtain a 100 mM stock
solution. A 5% (w/v) solution of free fatty acid (FFA)-free BSA was
prepared in double distilled water. Subsequently, a 5 or 10 mM
PA/BSA mixture was prepared by suitable combination of the 2
above-mentioned solutions. Finally, the mixture was further diluted
in serum-free medium to obtain the required final concentrations of
0.5 mM PA/0.5% BSA or 1.0 mM PA/0.5% BSA.
Transmission electron microscopy
(TEM)
After the indicated treatments, mature 3T3-L1
adipocytes were fixed in phosphate buffer (pH 7.4) containing 2.5%
glutaraldehyde and 2% paraformaldehyde at room temperature for 60
min. The cells were post-fixed in 1% OsO4 at room
temperature for 60 min, dehydrated through graded ethanol
solutions, and embedded in Quetol 812 (Nisshin EM Co., Tokyo,
Japan). Areas containing cells were block-mounted and cut into 70
nm sections that were stained with uranyl acetate (saturated
aqueous solution) and lead citrate and examined under a
transmission electron microscope (H-7100; Hitachi, Ibaraki,
Japan).
Immunofluorescence
The cells were grown on round glass coverslips in 35
mm cell culture dishes. Following a 20-min fixation with
pre-chilled methanol, the coverslips were washed with
phosphate-buffered saline (PBS) and permeabilized with 0.2% Triton
X-100-PBS for 30 min. The coverlips were then incubated with rabbit
polycolonal LC3 primary antibodies (1:200) at 37°C for 90 min in
the dark, followed by 3 washes (10 min each) in PBS. Subsequently,
the coverlips were incubated with goat-anti rabbit IgG-FITC
secondary antibodies (1:1,000) at 37°C for 1 h in dark and washed 3
times (10 min each time) in PBS. Digital images were obtained at an
original magnification of x20 using a Nikon C1 confocal microscope,
with NIS-Element software (Nikon, Melville, NY, USA).
Western blot analysis
The cells were washed with PBS and lysed in lysis
buffer (0.5% Triton X-100, 10 mM HEPES pH 7.9, 50 mM NaCl, 100 mM
EDTA, 0.5 M sucrose) containing 0.1% protease inhibitor cocktail
(Roche Diagnostics). The lysates were then incubated on ice for 30
min and centrifuged at 8,000 × g for 10 min. Equal amounts of
protein were subjected to sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE; 10–15%), transferred onto PVDF
membranes. Molecular weights were estimated by comparison with a
pre-stained protein ladder. Non-specific binding was blocked using
5% skim milk. The membranes were then incubated with specific
primary antibodies overnight at 4°C. The membranes were washed with
PBS-Tween-20 and incubated with peroxidase-conjugated secondary
antibodies [anti-rabbit IgG, HRP-linked sntibody 1:5,000 (no.
7074); Cell Signaling Technology; and mouse anti-goat IgG,
HRP-linked antibody 1:5,000 (no. sc-2354); Santa Cruz
Biotechnology, Inc.] Protein bands were detected using the Western
Chemiluminescent HRP substrate (Millipore). Immunoblots were
quantified by densitometric analysis using ImageTool 3.0 software.
The quantification of protein phosphorylation was normalized to the
corresponding total protein expression, and the relative expression
level of a certain protein was normalized to GAPDH.
Reverse transcription-quantitative
(real-time) polymerase chain reaction (RT-qPCR)
Total RNA was extracted using TRIzol reagent
(Invitrogen); samples of 1 μg total RNA were reverse
transcribed into cDNA using a cDNA synthesis kit (Promega).
Quantitative PCR (qPCR) was performed using an ABI 7900 sequencer
(Life Technologies, Carlsbad, CA, USA) with the SYBR-Green PCR
Master Mix (Promega) and d(N)6 random hexamer with
primers obtained from Invitrogen. The thermocycling parameters were
as follows: 95°C for 10 min, 40 cycles of 95°C for 15 sec, 60°C for
1 min. Each sample was run in triplicate and normalized against
36B4 RNA. The fold changes were determined using the ΔΔCt
method. The primers (mouse) used were as follows: IL-6 forward,
5′-CTGGGAAA TCGTGGAAATG-3′ and reverse, 5′-CCAGAGGAAATTTT
CAATAGGC-3′; MCP-1 forward, 5′-AGCCAACTCTCACTG AAGCCA-3′ and
reverse, 5′-AGTAGCAGCAGGTGAGT GGG-3′; and 36B4 forward,
5′-CGACCTGGAAGTCCAA CTAC-3′ and reverse,
5′-ATCTGCTGCATCTGCTTG-3′.
Cell viability
Cell viability was assessed by MTT assay. Briefly,
mature adipocytes were seeded in a 96-well plate and incubated
under different conditions. The culture medium was then removed,
and 200 μl/well were diluted in MTT solution (0.5 mg/ml) in
PBS for 1 h at 37°C. During the incubation time, MTT was converted
to an insoluble formazan. At the end of the incubation time, the
medium containing MTT was aspirated, and each well was gently
washed with PBS. Dimethyl sulfoxide (DMSO) was then used to
solubilize the precipitated formazan, and the concentration was
determined using a microplate reader at OD 570 nm.
Statistical analysis
The results are expressed as the means ± SEM.
Comparisons of a single variable in >2 groups were analyzed by
one-way ANOVA followed by Tukey’s multiple comparison tests
(GraphPad Prism). Statistical analysis was performed using the
paired and unpaired t-test between 2 groups, using SPSS 12.0
software (SPSS, Inc., Chicago, IL, USA). Values of P<0.05 were
considered to indicate statistically significant differences.
Results
Autophagy is activated in response to
PA
As the lipidation of LC3 and its association with
autophagsome membranes has been established as a useful marker of
autophagy (15), we detected LC3
expression by western blot analysis and fluorescence microscopy. A
reliable marker of autophagy is the conversion of the ATG protein,
LC3, from a soluble form (LC3-I) to a lipidized form (LC3-II),
which stably associates with the membranes of autophagosomes
(16). This conversion can be
detected by measuring the accumulation of the LC3-II form. Our
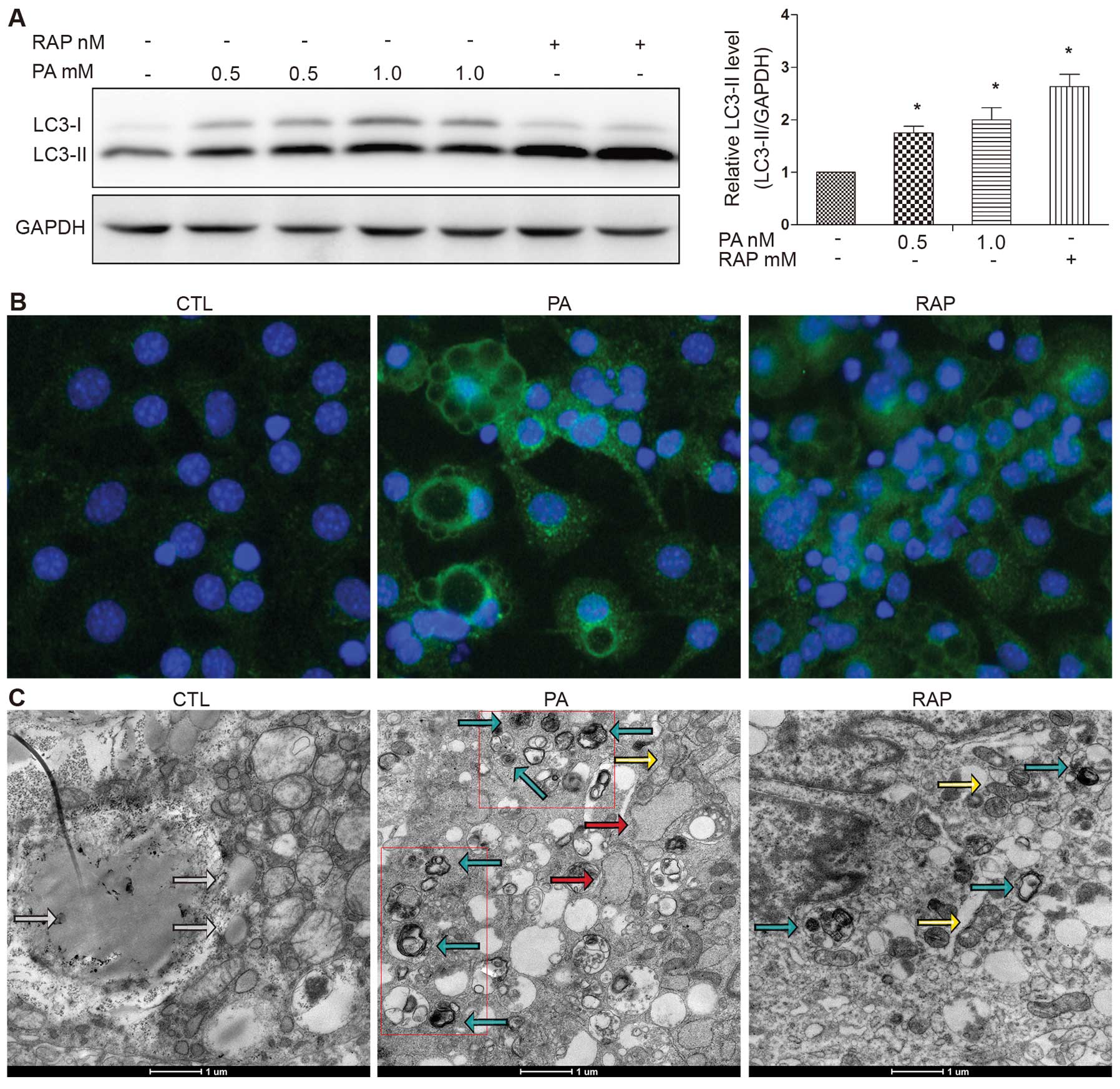
results demonstrated that treatment with PA increased LC3-II
formation in response to treatment with PA (0.5 or 1.0 mM) for 12 h
(Fig. 1A). RAP, a known mammalian
target of rapamycin (mTOR) pathway inhibitor, is known to induce
autophagy (17). RAP was used as
a positive control. We observed significant LC3-II accumulation
following treatment with RAP (100 nM) for 12 h (Fig. 1A). The number of LC3-II puncta was
increased and the LC3-II puncta were clearly visible in the PA (0.5
mM)-treated group compared with the control group (cells treated
with 0.5% BSA), shown as green fluorescent granules in the
cytoplasm, mainly around the nucleus. The number of LC3-II puncta
was also increased in the cells treated with RAP (Fig. 1B).
To gain insight into the morphological changes
induced by PA, electron microscopy was performed using the mature
3T3-L1 adipocytes. Treatment with PA (0.5 mM) for 12 h induced the
formation of autophagosomes, which were recognized at the
ultrastructural level as double-membrane vacuolar structures
containing visible cytoplasmic contents. Autolysosomes were
recognized as single-membrane vacuolar structures containing
high-density materials and some contained multivesicular body-like
vesicles (Fig. 1C). These
characteristics were also observed in the cells treated with RAP
(100 nM) for 12 h. Abundant lipid droplets, but rare evidence of
autophagosomes were observed in the control cells (Fig. 1C).
PA induces ER stress and subsequent
autophagy
ER stress is a potential molecular mechanism of
lipotoxicity, and we thus examined whether PA induces ER stress in
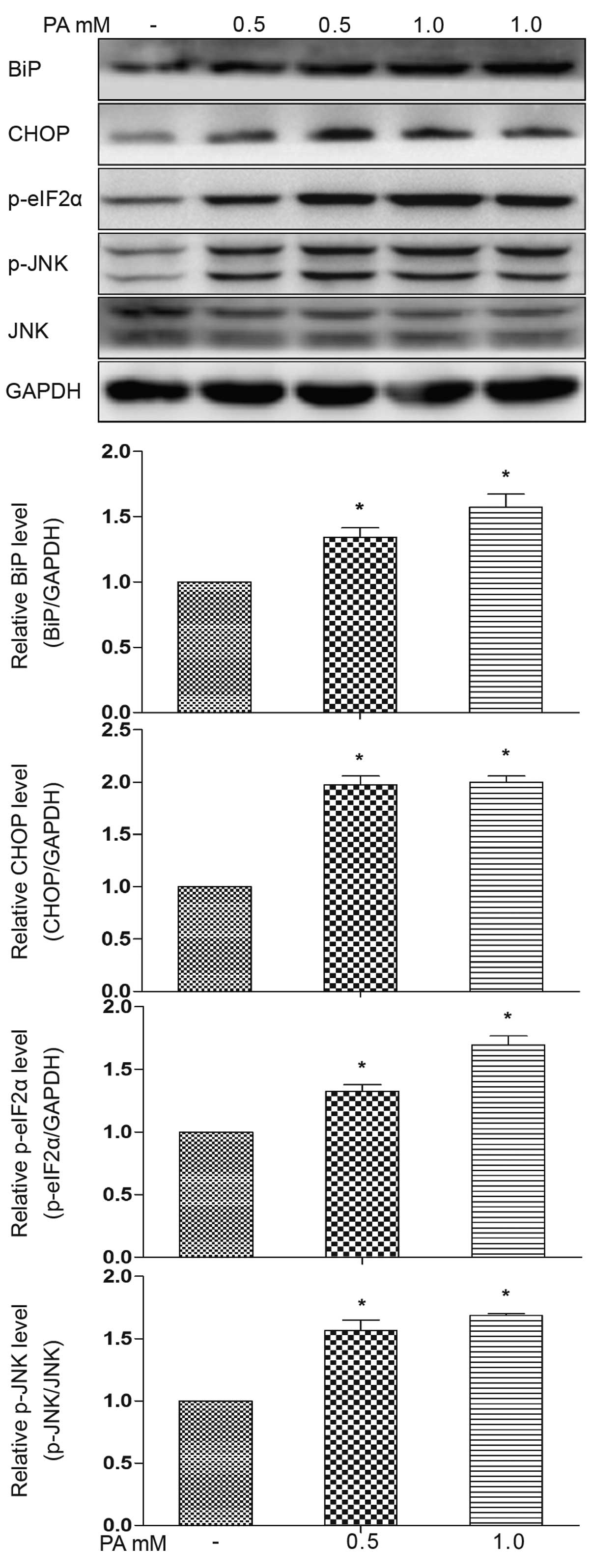
mature adipocytes. We observed an increase in ER stress protein
markers, evidenced by an increase in the expression of BiP, CHOP
and eIF2α phosphoralytion, as well as in JNK phosphoralytion
following treatment with PA (0.5 or 1.0 mM) for 12 h (Fig. 2). These results suggest the
activation of the PKR-like endoplasmic reticulum kinase
(PERK)-associated unfolded protein response (UPR) pathways, as well
as inositol-requiring enzyme 1 (IRE1) pathways. Increased CHOP
expression occurs downstream of the main pathways activated
following ER stress, namely PERK, ATF6 and IRE1 (18). In addition, electron microscopy
was performed on the PA-treated adipocytes (for 12 h) and revealed
a dilated ER (Fig. 1C, red
arrow).
We further addressed the cause-effect relationship
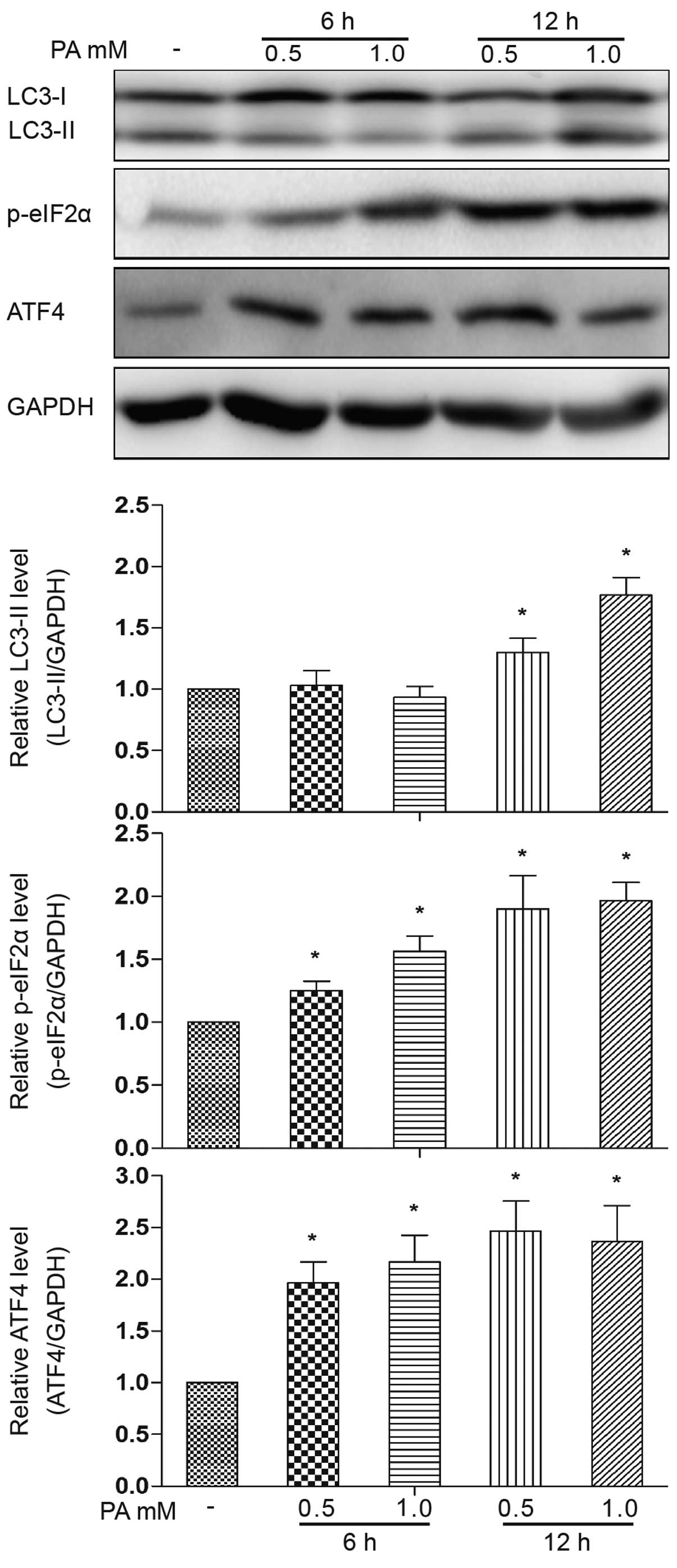
among ER stress and autophagy. The phosphorylation of eIF2α and the
ATF4 protein expression level were increased following treatment
with PA (0.5 and 1.0 mM) for 6 h (Fig. 3). No obvious accumulation of
LC3-II was observed following treatment with PA for 6 h, but only
at 12 h of treatment. Based on this observation, we then
investigated the association between ER stress and autophagy by
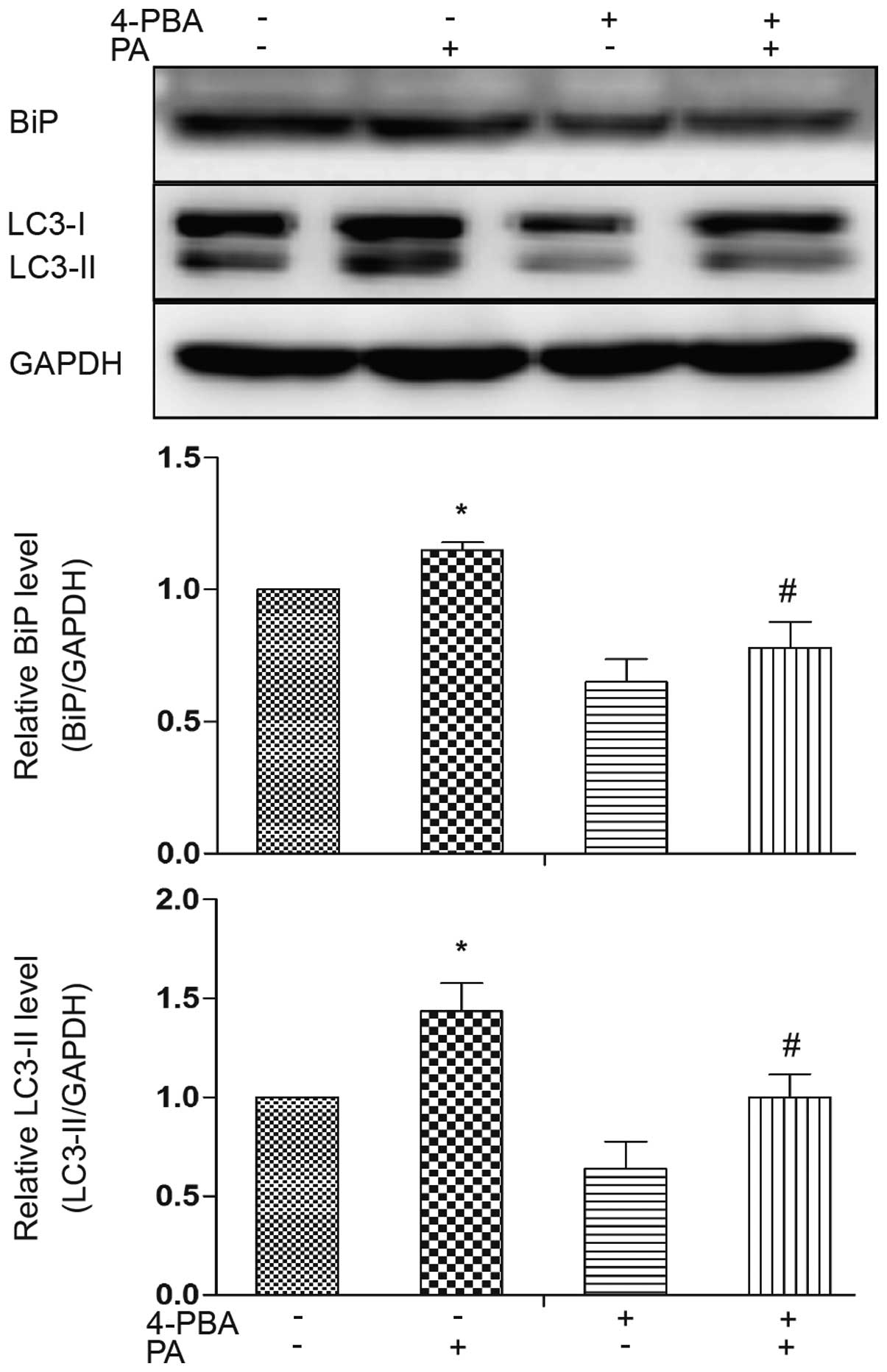
treating the adipocytes with 4-PBA, an ER stress inhibitor. ER
stress was ameliorated by treatment with 4-PBA, as evidenced by a
decrease in the expression level of the ER stress marker, BiP
(Fig. 4). Western blot analysis
revealed that LC3-II accumulation was decreased in the presence of
4-PBA compared to treatment with PA alone (Fig. 4). These results indicate a
potential causal involvement of ER stress in the induction of
autophagy induced by PA.
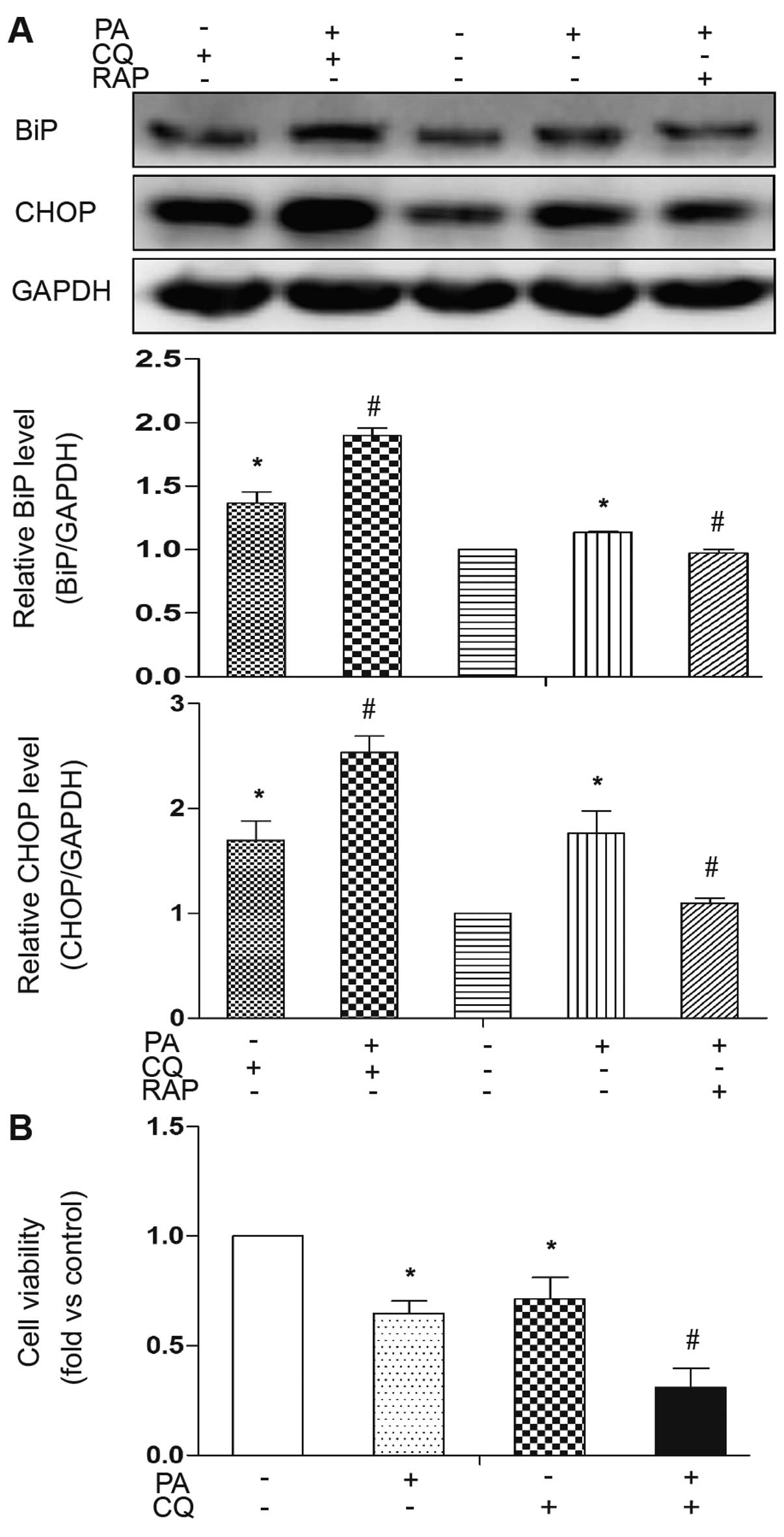
The inhibition of autophagy renders cells
susceptible to ER stress and apoptosis
To address the role of PA-induced autophagy in
mature adipocytes, the cells were co-treated with CQ, a lysosome
inhibitor, to block the autophagic flux, as previously described
(19). The pharmacological
inhibition of autophagy by CQ significantly increased the
expression levels of the PA-induced ER stress markers, BiP and
CHOP, indicating the extent of cellular stress when PA-induced
autophagy was inhibited (Fig.
5A). Conversely, treatment with RAP to induce autophagy
reversed these effects in response to PA (Fig. 5A). In addition, the expression
levels of the ER stress markers were also increased by treatment
with CQ without PA treatment, indicating that basal autophagy plays
a role against cellular stress (Fig.
5A). The detrimental effects of the inhibition of PA-induced
autophagy were confirmed using a cell viability assay. Cell
viability significantly decreased by co-treatment with PA and CQ
(Fig. 5B). Treatment with CQ
alone also decreased cell viability, indicating that basal
autophagy plays a role against cell death. These results
demonstrate that the activation of autophagy is an adaptive
response to PA and functions as a mechanism for cellular
self-protection against ER stress and cell death.
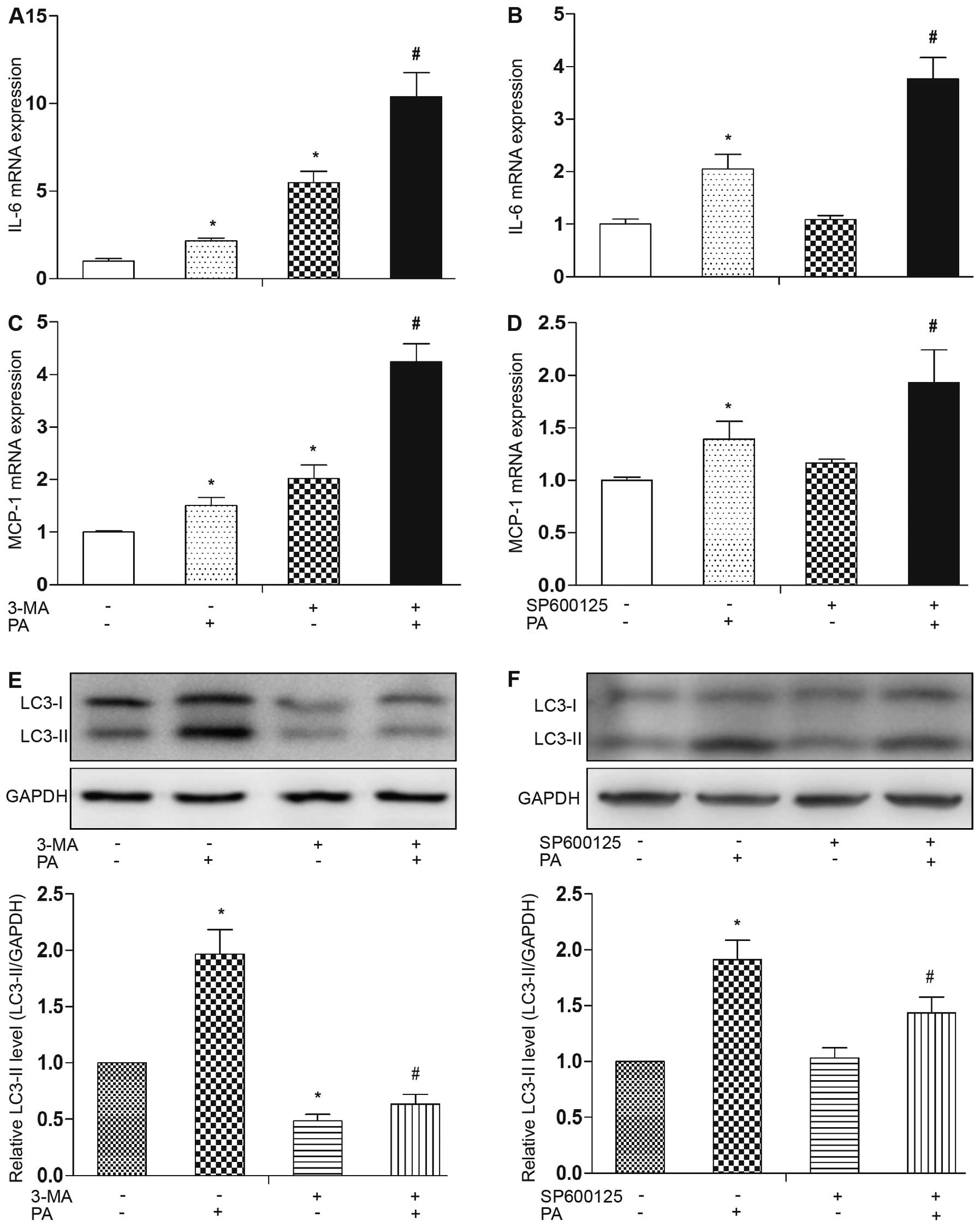
ER stress-induced autophagy is partially
JNK-dependent and mediates the limitation of PA-induced
inflammatory cytokine expression
Pre-loading of the cells with 0.5 mM/l of the
saturated fatty acid, PA, for 12 h resulted in an increase in the
mRNA expression levels of the inflammatory cytokines, IL-6 and
MCP-1, in the mature adipocytes (Fig.
6A and C). In order to determine the role of autophagy in
PA-induced inflammatory cytokine expression, we pre-treated the
adipocytes with the autophagy inhibitor, 3-MA, for 1 h followed by
treatment with PA for 12 h. 3-MA effectively inhibited the
induction of autophagy by PA, as evidenced by the decrease in
LC3-II accumulation (Fig. 6E).
RT-qPCR revealed that treatment with 3-MA further increased the
mRNA expression levels of MCP-1 and IL-6 compared to treatment with
PA alone (Fig. 6A and C). Since
the induction of autophagy by ER stress is in part mediated by JNK
activation (20), we then
examined the role of JNK in PA-induced autophagy in mature
adipocytes using the specific JNK inhibitor, SP600125. As
anticipated, the enhanced accumulation of LC3-II was decreased by
pre-treatment with SP600125 compared to treatment with PA alone
(Fig. 6F). Importantly, the
expression of IL-6 and MCP-1 was further increased by SP600125 in
the PA-treated adipocytes (Fig. 6B
and D). These results demonstrate that PA-induced autophagy is
partially JNK-dependent and that the activation of JNK-dependent
autophagy plays a role in limiting inflammatory cytokine
expression.
Discussion
It has become well accepted that changes in
inflammation in adipocytes and the infiltration of adipose tissue
by immune cells are key characteristics of obesity in animal models
and humans (21). Increased
exposure to fatty acids, whether due to the increased fat content
of modern diets or aberrant lipolysis in adipocytes has been
suggested as one of the key activators of both altered metabolic
and immune signaling in obesity. Indeed, the mechanisms through
which fatty acids contribute to the emergence of inflammation and
the adaptive response remain an important avenue for further
research. In this study, we used PA treatment as an in vitro
method to generate artificially hypertrophied adipocytes and to
examine the effects on the cells, including autophagy, ER stress
and inflammation, as well as the crosstalk between these cellular
events. We demonstrate that autophagy plays a role in limiting
cellular stress and inflammation as a positive response to PA in
adipocytes.
Autophagy is known to play a vital role in adipose
tissue to maintain the cellular concentration of ATP in diabetic
adipocytes (11). Accordingly, it
has been demonstrated that autophagy is essential for the
completion of adipogenesis, functioning as a potential survival
mechanism for cells during the differentiation process (22). On the other hand, the ER
stress-induced downregulation of adiponectin appears to be mediated
by an autophagy-dependent mechanism (23). Indeed, multiple characteristics of
autophagy, such as an increase the number of autophagic vacuoles
(AVs), has been observed in human obesity and type 2 diabetes
(22). Our data demonstrated that
autophagy was activated in mature adipocytes in response to PA. We
used temporal analysis of LC3-II formation to show a rapid and
transient effect of PA on autophagosome formation in adipocytes.
Our data from TEM and LC3 staining clearly indicated that PA
induced an autophagic flux in these cells. Autophagy can be
activated under various circumstances, as occurs with the Toll-like
receptor (TLR) (24). Both
adipocytes and adipose tissue inflammatory cells express TRL4
(25), and FFA has been suggested
to be a trigger of metabolism-associated inflammation through
TLR2/4 (26). We hypothesized
that the effects of faty acids on autophagy may differ
substantially depending on the fatty acid used, the cell type
examined and the duration of treatment. For example, as previously
demonstrated, 8 h treatment of PA was sufficient to activate
autophagosome formation in human hepatic cells, whereas prolonged
treatment beyond 24 h blocked the autophagic flux and increased ER
stress (27). In this study, we
demonstrate that PA induces ER stress in mature adipocytes and an
increase in the autophagic flux, which occurs at least in part in
response to ER stress and is JNK-dependent. This conclusion was
based on the use of the molecular chaperone, 4-PBA, which abolishes
UPR induction, as well as on the use of a JNK inhibitor (SP600125).
Indeed, it has been demonstrated that in other cell types in
response to various stimuli, ER stress initiates the UPR which
leads to a myriad of compensatory cellular effects, one of which is
the induction of autophagy (28).
Thus, our data confirm that in mature adipocytes, PA elicits this
ER stress-JNK-autophagy axis, and based on previously reported data
(29), it was important to
determine the functional consequences of the PA-induced increase in
the autophagic flux.
It has been postulated that ER stress-induced
autophagy may have evolved as a mechanism used by cells to dispose
of misfolded proteins that cannot be degraded by endoplamic
reticulum-related degradation, consequently, assisting ER
homeostasis (30). Thus, it can
be hypothesized that deficient autophagy may also serve to augment
ER stress and promote cell dysfunction, even cell death. In this
study, when we used CQ, a lysosome inhibitor, to block the
autophagic flux, this rendered cells susceptible to cell death, as
evidenced by the increase in CHOP expression, which contributed to
ER stress-induced apoptosis and decreased cell viability.
Conversely, this effect was reversed by the activation of autopahgy
using RAP. Therefore, our data clearly indicate that autophagy is
induced in adipocytes subsequent to the activation of the ER
stress-JNK pathway and plays a protective role against PA-induced
cellular stress and death.
In addition to cell death, inflammation was also
evident following treatment with PA. This was detected by measuring
the expression levels of pro-inflammatory cytokine. To determine
the role of autophagy in inflammation, we used the autophagy
inhibitor, 3-MA. Notably, a further increase in the expression
levels of MCP-1 and IL-6 was observed following pre-treatment with
3-MA compared to treatment with PA alone, which was also observed
in response to treatment with the JNK inhibitor, SP600125. Our
results strongly suggest that the activation of ER
stress-JNK-autophagy plays a role in limiting PA-induced
inflammation. The inhibition of autophagy in humans and mouse
adipose tissue explants has been shown to lead to a significant
increase in IL-1β, IL-8 mRNA expression and protein secretion
(31). Previous studies about
autophagy on inflammation is still unclear. Autophagy was
demonstrated attenuate vascular endothelial inflammation through
cAMP signaling pathway (32). The
pro-inflammatory cytokine, IL-1β, is degraded in autophagosomes,
which limits its availability for inflammasome-dependent activation
(33). The suppression of
autophagy may further increase inflammatory cytokine expression
induced by ER stress, which interacts with a number of inflammatory
signaling pathways (12). Further
studies on the mechanisms of autophagy during inflammation are
warranted.
In conclusion, in this study, we demonstrated that
treatment with PA induced ER stress and, consequently, cell death
and inflammation. The activation of autophagy occurred as an
effective protective cellular response against PA-induced cellular
stress in a JNK-dependent manner. Importantly, we present evidence
of the role of autophagy in limiting PA-induced inflammation. This
further validates the potential value of UPR-JNK targeted therapies
as a promising approach for the treatment of obesity induced
inflammation and insulin resistance.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant nos. 81370904 and 81400823). We
thank Professor Huaidong Song (Ruijin Hospital, Shanghai Jiao Tong
University School of Medicine) for kindly providing us with the
technological support and instructions for carrying out our
study.
Abbreviations:
|
ATF4
|
activating transcription factor 4
|
|
ATG
|
autophagy-related gene
|
|
AV
|
autophagic vacuole
|
|
CHOP
|
C/EBP homologous protein
|
|
CQ
|
chloroquine
|
|
ER
|
endoplasmic reticulum
|
|
IL-6
|
interleukin-6
|
|
IRE1
|
inositol-requiring enzyme 1
|
|
JNK
|
c-Jun N-terminal kinase
|
|
LC3
|
microtubule-associated protein 1 light
chain 3
|
|
3-MA
|
3-methyladenine
|
|
MCP-1
|
monocyte chemoattractant protein-1
|
|
mTOR
|
mammalian target of rapamycin
|
|
4-PBA
|
4-phenyl butyrate
|
|
PERK
|
PKR-like endoplasmic reticulum
kinase
|
|
eIF2α
|
eukaryotic translation initiation
factor 2α
|
|
PA
|
palmitate
|
|
RAP
|
rapamycin
|
|
TLR
|
toll-like receptor
|
|
UPR
|
unfolded protein response
|
|
TNF-α
|
tumor necrosis factor-α
|
References
|
1
|
Harvey AE, Lashinger LM and Hursting SD:
The growing challenge of obesity and cancer: an inflammatory issue.
Ann NY Acad Sci. 1229:45–52. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
González-Chávez A, Elizondo-Argueta S,
Gutiérrez-Reyes G and Leon-Pedroza JI: Pathophysiological
implications between chronic inflammation and the development of
diabetes and obesity. Cir Cir. 79:209–216. 2011.PubMed/NCBI
|
|
3
|
Wang Z and Nakayama T: Inflammation, a
link between obesity and cardiovascular disease. Mediators Inflamm.
2010:5359182010.PubMed/NCBI
|
|
4
|
Takahashi K, Yamaguchi S, Shimoyama T, et
al: JNK- and IkappaB-dependent pathways regulate MCP-1 but not
adiponectin release from artificially hypertrophied 3T3-L1
adipocytes preloaded with palmitate in vitro. Am J Physiol
Endocrinol Metab. 294:E898–E909. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kuballa P, Nolte WM, Castoreno AB and
Xavier RJ: Autophagy and the immune system. Annu Rev Immunol.
30:611–646. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yin JJ, Li YB, Wang Y, et al: The role of
autophagy in endoplasmic reticulum stress-induced pancreatic beta
cell death. Autophagy. 8:158–164. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang L, Li P, Fu S, Calay ES and
Hotamisligil GS: Defective hepatic autophagy in obesity promotes ER
stress and causes insulin resistance. Cell Metab. 11:467–478. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Saitoh T, Fujita N, Jang MH, et al: Loss
of the autophagy protein Atg16L1 enhances endotoxin-induced
IL-1beta production. Nature. 456:264–268. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hotamisligil GS: Endoplasmic reticulum
stress and the inflammatory basis of metabolic disease. Cell.
140:900–917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ost A, Svensson K, Ruishalme I, et al:
Attenuated mTOR signaling and enhanced autophagy in adipocytes from
obese patients with type 2 diabetes. Mol Med. 16:235–246. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yoshizaki T, Kusunoki C, Kondo M, et al:
Autophagy regulates inflammation in adipocytes. Biochem Biophys Res
Commun. 417:352–357. 2012. View Article : Google Scholar
|
|
13
|
Hummasti S and Hotamisligil GS:
Endoplasmic reticulum stress and inflammation in obesity and
diabetes. Circ Res. 107:579–591. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Listenberger LL, Ory DS and Schaffer JE:
Palmitate-induced apoptosis can occur through a
ceramide-independent pathway. J Biol Chem. 276:14890–14895. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–26. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rubinsztein DC, Gestwicki JE, Murphy LO
and Klionsky DJ: Potential therapeutic applications of autophagy.
Nat Rev Drug Discov. 6:304–312. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ravikumar B, Vacher C, Berger Z, et al:
Inhibition of mTOR induces autophagy and reduces toxicity of
polyglutamine expansions in fly and mouse models of Huntington
disease. Nat Genet. 36:585–595. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Oyadomari S and Mori M: Roles of
CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ.
11:381–389. 2004. View Article : Google Scholar
|
|
19
|
Klionsky DJ, Abdalla FC, Abeliovich H, et
al: Guidelines for the use and interpretation of assays for
monitoring autophagy. Autophagy. 8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ogata M, Hino S, Saito A, et al: Autophagy
is activated for cell survival after endoplasmic reticulum stress.
Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Olefsky JM and Glass CK: Macrophages,
inflammation, and insulin resistance. Annu Rev Physiol. 72:219–246.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li H, Zhou B, Xu L, et al: The reciprocal
interaction between autophagic dysfunction and ER stress in adipose
insulin resistance. Cell Cycle. 13:565–579. 2014. View Article : Google Scholar
|
|
23
|
Zhou L and Liu F: Autophagy: roles in
obesity-induced ER stress and adiponectin downregulation in
adipocytes. Autophagy. 6:1196–1197. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Martinez J, Verbist K, Wang R and Green
DR: The relationship between metabolism and the autophagy machinery
during the innate immune response. Cell Metab. 17:895–900. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schäffler A and Schölmerich J: Innate
immunity and adipose tissue biology. Trends Immunol. 31:228–235.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yin J, Peng Y, Wu J, Wang Y and Yao L:
Toll-like receptor 2/4 links to free fatty acid-induced
inflammation and β-cell dysfunction. J Leukoc Biol. 95:47–52. 2014.
View Article : Google Scholar
|
|
27
|
González-Rodríguez A, Mayoral R, Agra N,
et al: Impaired autophagic flux is associated with increased
endoplasmic reticulum stress during the development of NAFLD. Cell
Death Dis. 5:e11792014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Levine B and Klionsky DJ: Development by
self-digestion: molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ishida Y and Nagata K: Autophagy
eliminates a specific species of misfolded procollagen and plays a
protective role in cell survival against ER stress. Autophagy.
5:1217–1219. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qin L, Wang Z, Tao L and Wang Y: ER stress
negatively regulates AKT/TSC/mTOR pathway to enhance autophagy.
Autophagy. 6:239–247. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jansen HJ, van Essen P, Koenen T, et al:
Autophagy activity is up-regulated in adipose tissue of obese
individuals and modulates proinflammatory cytokine expression.
Endocrinology. 153:5866–5874. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen ML, Yi L, Jin X, et al: Resveratrol
attenuates vascular endothelial inflammation by inducing autophagy
through the cAMP signaling pathway. Autophagy. 9:2033–2045. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Harris J, Hartman M, Roche C, et al:
Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for
degradation. J Biol Chem. 286:9587–9597. 2011. View Article : Google Scholar : PubMed/NCBI
|