Introduction
Nuclear factor of activated T-cells (NFAT) is a
family of transcription factors consisting of four closely related
proteins, i.e., nuclear factor of activated T-cells, cytoplasmic 1
(NFATc1) (NFATc, NFAT2), NFATc2 (NFATp, NFAT1), NFATc3 (NFATx,
NFAT4) and NFATc4 (NFAT3), as well as a more distant relative,
NFAT5 [tonicity response element-binding protein (TonEBP)]. In the
basal state, NFAT is hyperphosphorylated in the cytoplasm.
Subsequent to cell stimulation and calcium release, NFAT is
dephosphorylated by the phosphatase calcineurin and translocates to
the nucleus, where it cooperates with other factors and
co-activators to promote de novo gene transcription
(1). A number of recent findings
have pointed to the important roles of NFAT in modulating
phenotypes associated with malignancy and tumor progression
(1,2). NFAT isoforms are overexpressed in
human solid tumors and hematological malignancies (3,4)
and appear to play a role in cancer cell autonomous functions, such
as invasive migration, differentiation and the survival of cancer
cells. The understanding of the roles of NFAT in tumor progression
may provide aid the development of effective therapeutics targeting
the NFAT pathway in cancer progression and metastasis.
Cyclooxygenase-2 (COX-2) converts arachidonic acid
into bioactive lipids, including prostaglandin E2 (PGE2), which has
been found to be elevated in various types of tumors (5,6).
Studies have documented that the localized expression of COX-2 and
its catalyzed product, PGE2, are sufficient in initiating the
development of and promoting the progression of tumors in
situ. COX-2 is normally expressed at very low or undetectable
levels and its expression is rapidly induced at sites of
inflammation and proliferation in response to stimuli, such as
growth factors and tumor promoters (7). COX-2 expression and PGE2 levels are
elevated in a variety of human cancers and are associated with
increased angiogenesis, tumor invasion and resistance to apoptosis
(8–12). It is well known that COX-2 is
frequently overexpressed in colorectal cancer, and PGE2 has been
identified as the principal prostanoid promoting cell growth and
survival in colorectal tumors. PGE2 is able to exert pleiotropic
effects in colorectal tumors, promoting cell proliferation,
survival, angiogenesis, migration and invasion (13). However, the mechanisms responsible
for the malicious activity of COX-2 remain unclear in glioblastoma
multiforme (GBM).
A link between NFAT activity and COX-2 is evident
from previous studies. NFAT has been reported to regulate COX-2
expression in human T lymphocytes (14). Putative NFAT recognition sequences
are present in the human COX-2 proximal promoter, and deletion
analysis has shown that they are important for its transcriptional
activation (14). A previous
study also demonstrated that these sites are essential for the
induction of COX-2 expression by NFAT in colon carcinoma cells
(15). However, it is not known
whether NFAT can modulate COX-2 expression in GBM.
NFATc1 is a transcription factor of the NFAT family
that is regulated by calcium and the phosphatase calcineurin
(16). Evidence suggests that
NFAT proteins, and NFATc1 in particular, regulate important
cellular processes, such as proliferation and apoptosis in
different cell types (17,18).
However, little information is available on the expression and
activation of the NFAT transcription pathway and its function in
cancer cells. In the present study, we identified NFATc1 as
ectopically expressed and highly activated in U251 cells both in
vivo and in vitro. The inhibition of NFATc1 signaling in
cultured U251 cells by treatment with small interfering RNA (siRNA)
for the knock down of NFATc1 expression led to a marked decrease in
cell invasion. We also demonstrated that the invasive effect of
NFATc1 is mediated by the direct binding of activated NFATc1 to the
COX-2 promoter, resulting in the upregulation of COX-2
transcription and increased GBM cell invasion. These results
provide important novel insight into the mechanisms of oncogenic
NFATc1 activation in GBM.
Materials and methods
Cell culture
The human U251 cell line was obtained from the
American Type Culture Collection (ATCC, Manassas, VA, USA). The
cells were cultured as monolayers in DMEM and fetal bovine serum
(FBS) (both from Gibco, Carlsbad, CA, USA) supplemented with 10%
heat-inactivated FBS, 100 mg/l streptomycin and 100 U/ml penicillin
at 37°C in a humidified atmosphere of 95% air and 5%
CO2.
Immunofluorescence staining
Immunofluorescence staining was carried out as
previously described (19). The
U251 cells were grown on Poly-D-lysine-coated, 8-chamber slides
with 5,000 cells/chamber. After 24 h, U251 cells were left
untreated or treated with CsA (1 μg/ml) for 60 min.
Thereafter, the cells were washed with phosphate-buffered saline
(PBS) and fixed for 10 min with 4% paraformaldehyde in PBS,
permeabilized with 0,25% Triton X-100 in PBS and blocked for 1 h in
PBS supplemented with 5% FBS. The cells were incubated overnight at
4°C with rabbit anti-p-NFATc1 (1:100 dilution; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) antibody, followed by
washing and incubating with Cy3-labeled anti-rabbit secondary
antibody (1:200 dilution; Sigma, St. Louis, MO, USA) for 2 h. In
the control samples, the U251 cells were treated with an equal
volume of DMEM. The nuclei were counterstained with Hoechst 33258.
The results were observed and photographed under a fluorescence
microscope (Leica Microsystems, Wetzlar, Germany).
Production of recombinant retroviruses
and infection of U251 cells
The CA-NFATc1 (constitutively active form of NFATc1)
mutant (pMSCV-caNFATc1) and the control retroviral plasmids were
provided by Dr Neil Clipstone (Northwestern University, Chicago,
IL, USA). Recombinant retroviruses were produced as previously
described (20) by
co-transfecting either the pMSCV-GFP or pMSCV-caNFATc1 proviral
vectors together with pVSV-G (Clontech Laboratories, Inc., Mountain
View, CA, USA), encoding the glycoprotein of the vesicular
stomatitis virus, into the GP293 pantropic packaging cell line
(Clontech Laboratories, Inc.) using Lipofectamine Plus (Invitrogen,
Carlsbad, CA, USA). The medium was replaced after 24 h, and viral
supernatants were harvested 2 days posttransfection and stored at
−80°C. For infection, 5×104 cells were plated per well
in a 6-well plate. The following day, the medium was replaced with
2 ml of viral supernatant containing 8 μg/ml polybrene
(Sigma), and the plates were centrifuged at 2,000 rpm for 1.5 h at
room temperature. After the removal of the viral supernatant, the
cells were expanded in growth medium for subsequent analysis and
typically used within 5–7 days of infection. To ensure
reproducibility, each experiment was repeated 3 times using cells
derived from independent viral infections and independently derived
retroviral stocks.
Transient transfection of NFATc1
siRNA
siRNA oligos for the knockdown of endogenous NFATc1
proteins were prepared using the ON-TARGETplus SMARTpool siRNA from
Dharmacon, Inc. (Lafayette, CO, USA). The cells were transfected
with NFATc1 (100 nM) using the DharmaFECT siRNA transfection
reagent (Dharmacon, Inc.) according to the manufacturer’s
instructions. ON-TARGETplus non-targeting siRNA (Dharmacon, Inc.)
was used as a negative control (control siRNA) and the selective
silencing of NFATc1 was confirmed by western blot analysis.
Cellular protein preparation and western
blot analysis
The U251 cells were treated with CA-NFATc1 mutant or
NFATc1 siRNA. Cellular protein was extracted using the Cell Protein
Extraction kit; (Millipore, Billerica, MA, USA). Protein
concentrations were determined using the Coomassie (Bradford)
Protein Assay kit (Thermo Fisher Scientific, Waltham, MA, USA).
Equal amounts of protein were separated by SDS-PAGE using 8%
separating gels followed by transfer to nitrocellulose membranes.
Following transfer, the membranes were blocked using 5% non-fat
dried milk in PBS; pH 7.2 and incubated overnight at 4°C with the
primary antibody (pAb), including anti-NFATc1 (sc-17834),
anti-COX-2 (sc-70879) and anti-β-actin (sc-130656) antibodies
(1:1,000 dilution; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA). The membranes were washed 3 times with PBS, 0,1% Tween-20
(PBST) and then incubated with secondary antibodies (horseradish
peroxidase-conjugated, goat antibodies to rabbit (sc-2025) and goat
antibodies to mouse (sc2027; dilution of 1:5,000; Santa Cruz
Biotechnology, Inc.) for 2 h at 24°C. Following washing 3 times
with PBS, 0.1% Tween-20, the immunoreactive bands were visualized
using enhanced chemiluminescence detection reagents. Autoradiograms
were scanned and the labeled bands were quantified using the
Sigma-Gel software (Sigma).
RNA isolation and quantitative polymerase
chain reaction (qPCR)
Total RNA was extracted from the cells and cDNA
synthesis and amplification were performed as described in a
previous study (4). Primers were
designed as: COX-2 forward, 5′-CAAAAGCTGGGAAGCCTTCTCTAACC-3′ and
reverse, 5′-GCCCAGCCCGTTGGTGAAAG-3′; matrix metalloproteinase
(MMP)-2 forward, 5′-ATGGATCCTGGCTTTCCC-3′ and reverse,
5′-GCTTCCAAACTTCACGCTC-3′; MMP-9 forward, 5′-TGACAGCGACAAGAAGTG-3′
and reverse, 5′-CAGTGAAGCGGTACATAGG-3′; GAPDH forward,
5′-GCACCGTCAAGGCTGAGAAC-3′ and reverse, 5′-TGGTGAAGACGCCAGTGGA-3′.
Comparative qPCR was performed in triplicate, including no template
controls. Relative expression was calculated using the comparative
Ct method.
Reporter gene assay
Reporter gene assay was carried out as previously
described (19). A 2,004-bp-long
COX-2 promoter region spanning −2,069 to −66 bp upstream of the
translational start site was cloned by PCR and subcloned into the
pGL3 vector (pGL3-COX-2). The sequence was confirmed by DNA
sequencing. The U251 cells were seeded in 12-well plates
(2×106 cells/plate) overnight and transiently
co-transfected with pGL3-COX-2 or the internal control, pRL-TK
(internal control (Promega Corp., Madison, WI, USA), along with
indicated plasmids using Lipofectamine 2000 transfection reagent
(Invitrogen), following the manufacturer’s instructions. Firefly
and Renilla luciferase activities in the cell extracts were
measured using a dual-luciferase reporter assay system (Promega
Corp.). The relative luciferase activity was then calculated by
normalizing COX-2 promoter luciferase activity to control Renilla
luciferase activity. The results were expressed as the percentage
of relative luciferase activity of the control group, which was set
to 1.
In vitro Matrigel invasion assay
Polycarbonate filters (9 mm, 8.0-μm pore
size) were pre-coated with 50 μl of Matrigel
(Becton-Dickinson, San Jose, CA, USA), diluted to 1 mg/ml in
serum-free medium (DMEM), and applied for 1 h at 37°C until gelled,
after which cell suspension was applied. The cells were grown under
standard conditions (see above) and weaned from 10% fetal calf
serum to serum-free medium in 2 steps to 5 and 0%, over a 24 h
period. Following trypsinization, the cells were diluted so that
1×103 cells were added per well. The lower chamber was
filled with 500 μl 20% fetal calf serum medium. The cells
subjected to the various treatments were incubated for 24 h and
then washed once (5 min) with PBS (pH 7.4). Non-invading cells were
removed from the upper surface of the membrane by scrubbing, and
cells on the lower surface of the filter were fixed for 30 min in
methanol and glacial acetic acid (3:1), air-dried briefly and
stained with Giemsa. A total of 5 random fields were counted at
×200 magnification. Data represent the average cells of 5 fields
compared between the experimental groups and the control group.
These separate wells were used per test condition, and these
experiments were performed 3 times.
Tissue samples and patients
Tumor specimens were obtained from patients admitted
for diagnosis and treatment at the Fourth Affiliated Hospital of
Harbin Medical University (Harbin, China). The diagnosis was made
according to World Health Organization criteria. The present study
was approved by the Ethics Committee of the Fourth Affiliated
Hospital of Harbin Medical University and was based on the criteria
of the Helsinki convention. Approval for the use of tissue samples
was obtained from our institutional review board. Fresh surgical
samples from glioma patients and non-neoplastic brain tissues
(temporal lobectomy; temporal lobe epilepsy surgery) were
immediately snap-frozen in liquid nitrogen upon surgical removal.
The formalin-fixed, paraffin-embedded archival tissue blocks were
retrieved and the matching H&E-stained slides were screened for
representative tumor regions by a neuropathologist. The tissue
samples included 5 diffuse astrocytomas (grade II), 8 anaplastic
astrocytomas (grade III) and 50 GBMs (grade IV astrocytoma). In
addition, 10 non-neoplastic brain tissues from epilepsy surgical
resections were also included.
Immunohistochemistry (IHC)
The perfused brains were cryoprotected in a solution
of 20% sucrose in 0.1 M of potassium phosphate buffer overnight.
The brain sections were cut on a freezing microtome (Leica SM2000R)
and mounted on gelatinized slides. The sections were dried at
40–50°C for 2 h and were maintained at −20°C until analysis.
Briefly, IHC was performed as follows: the sections were incubated
overnight at room temperature with the primary antibody (NFATc1,
1:100) diluted in PBS with Tween-20 (PBST). The negative controls
received only PBST. The slides were washed with PBST and incubated
with the secondary antibodies (1:1,000 in PBST) for 90 min. The
slides were washed again with PBST and incubated with
streptavidin-horseradish peroxidase (1:200 in PBST) for 60 min. The
reactions were developed with 0.04% 3,3′-diaminobenzidine (DAB) +
0.03% H2O2. The DAB reactions were
intensified with an OsO4 solution (0.04%) for 30 min.
The slides were counterstained with hematoxylin, dehydrated and
mounted with Permount. The samples were visualized and images were
captured using a light microscope (Olympus BX51; Olympus Optical,
Tokyo, Japan). The degree of immunostaining of the sections was
viewed and scored separately by 2 independent investigators, and
the scores were determined by combining the proportion of
positively stained tumor cells and the intensity of staining.
Scores from the 2 investigators were averaged for further
comparative evaluation of NFATc1 expression. The proportion of
positively stained tumor cells was graded as previously described
(21).
Statistical analysis
All the data are presented as the means ± standard
error. All analyses were performed with one-way analysis of
variance using SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA). A
value of P<0.05 was considered to indicate a statistically
significant difference.
Results
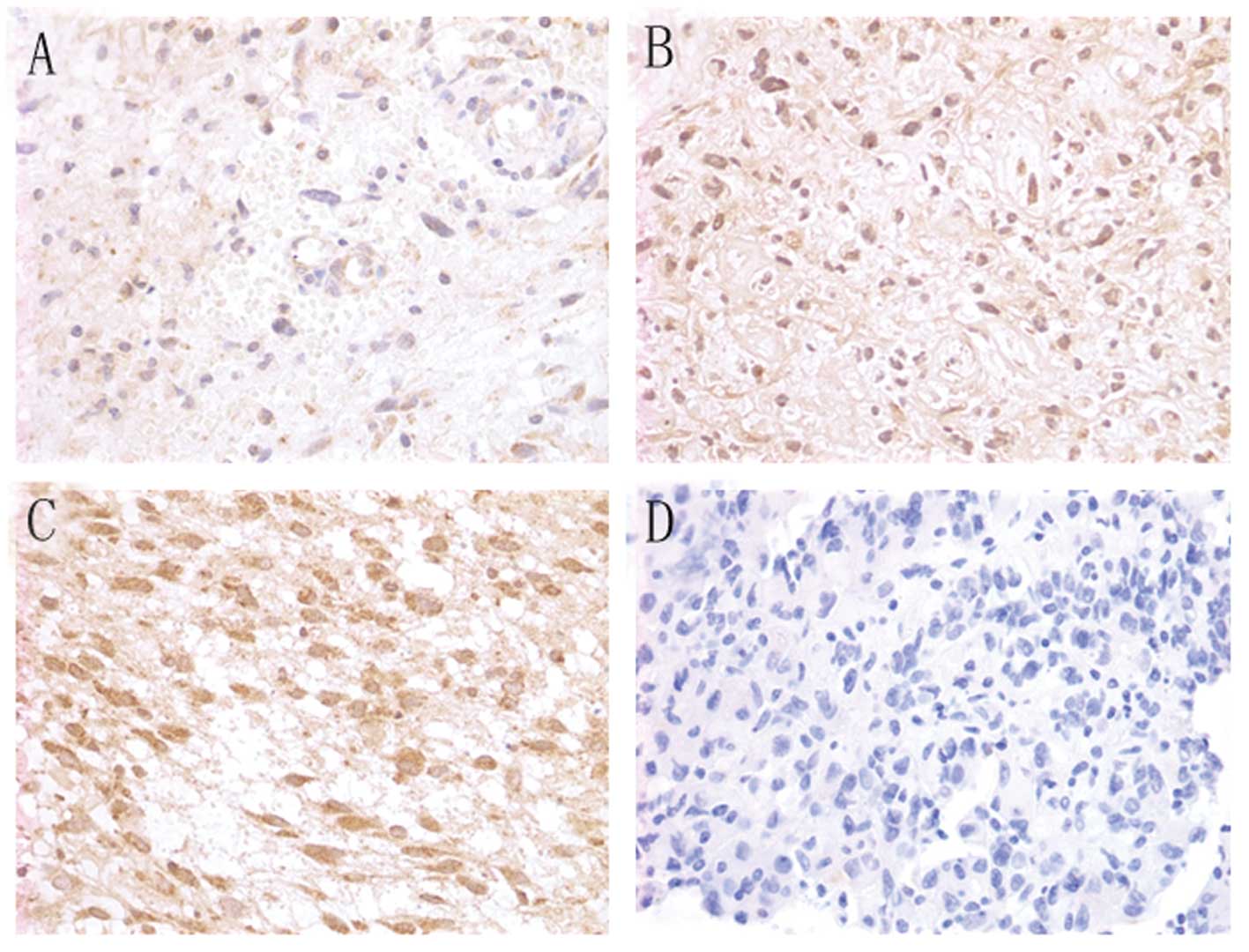
NFATc1 is constitutively activated in
glioma tissues
To determine whether NFATc1 is constitutively
activated in glioma, the levels of NFATc1 expression were assessed
by IHC using anti-NFATc1 antibody on glioma tissues. Among the
glioma samples, 92.1% (58/63) demonstrated positive nuclear
staining, whereas normal tissue did not stain. NFATc1 staining was
predominantly nuclear in vivo. There was no significant
difference in the constitutive activation frequency between the
low- and high-grade gliomas. However, the expression levels of
NFATc1 were significantly higher in the high-grade gliomas (grades
III and IV) compared with the low-grade gliomas (grade II)
(Fig. 1), which supports the
hypothesis that NFATc1 activation is associated with the
progression of glioma.
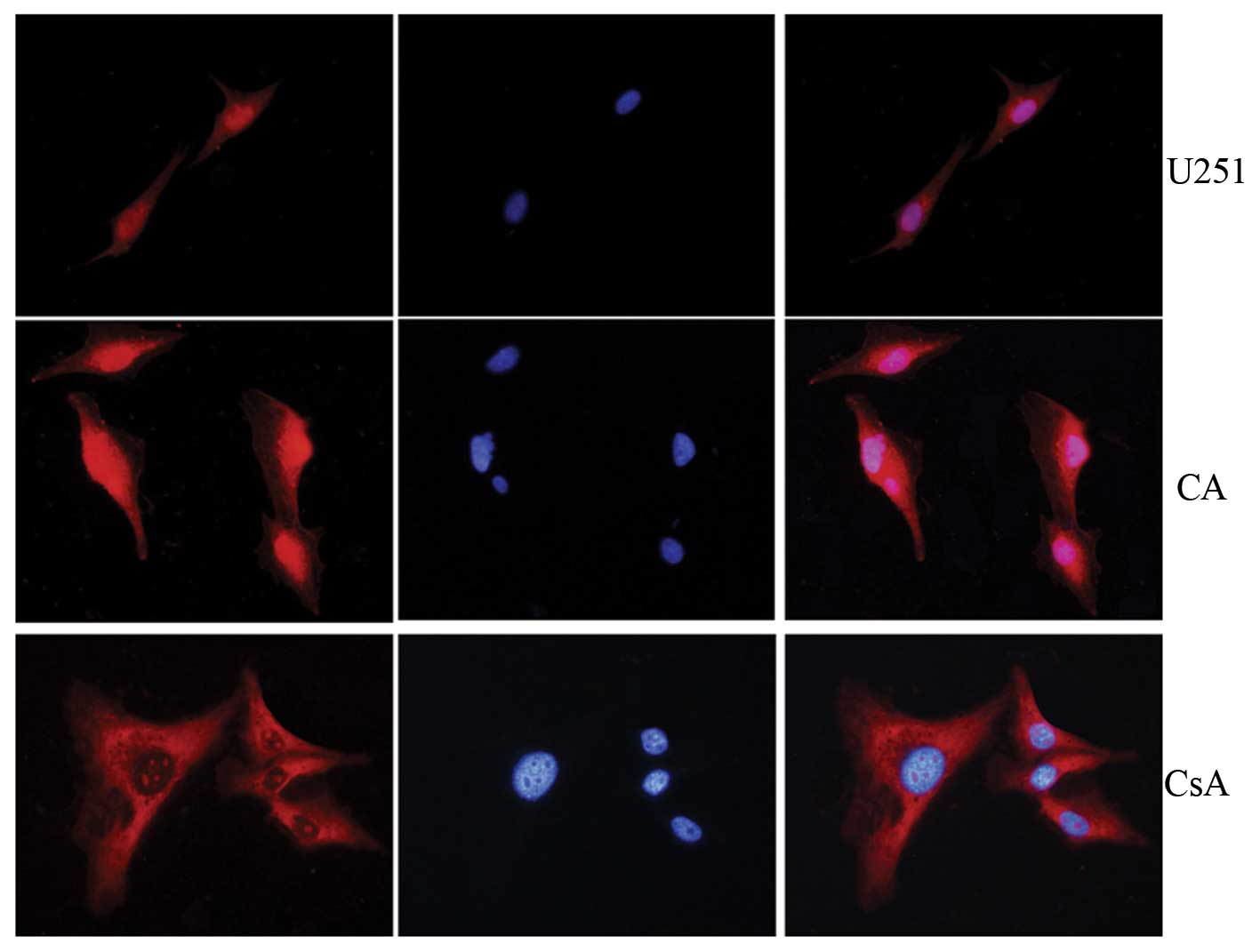
NFATc1 is activated in U251 cells and is
repressible by cyclosporin A (CsA); CA-NFATc1 promotes NFATc1
activation
Immunofluorescence staining of the U251 cells
indicated that NFATc1 was accumulated in the nucleus. Treatment of
the U251 cells with the calcineurin inhibitor, CsA, caused the
rapid inhibition and nuclear export of NFATc1. CA-NFATc1 treatment
further induced the accumulation of NFATc1 in the cell nuclei and
significantly increased NFATc1 staining (Fig. 2).
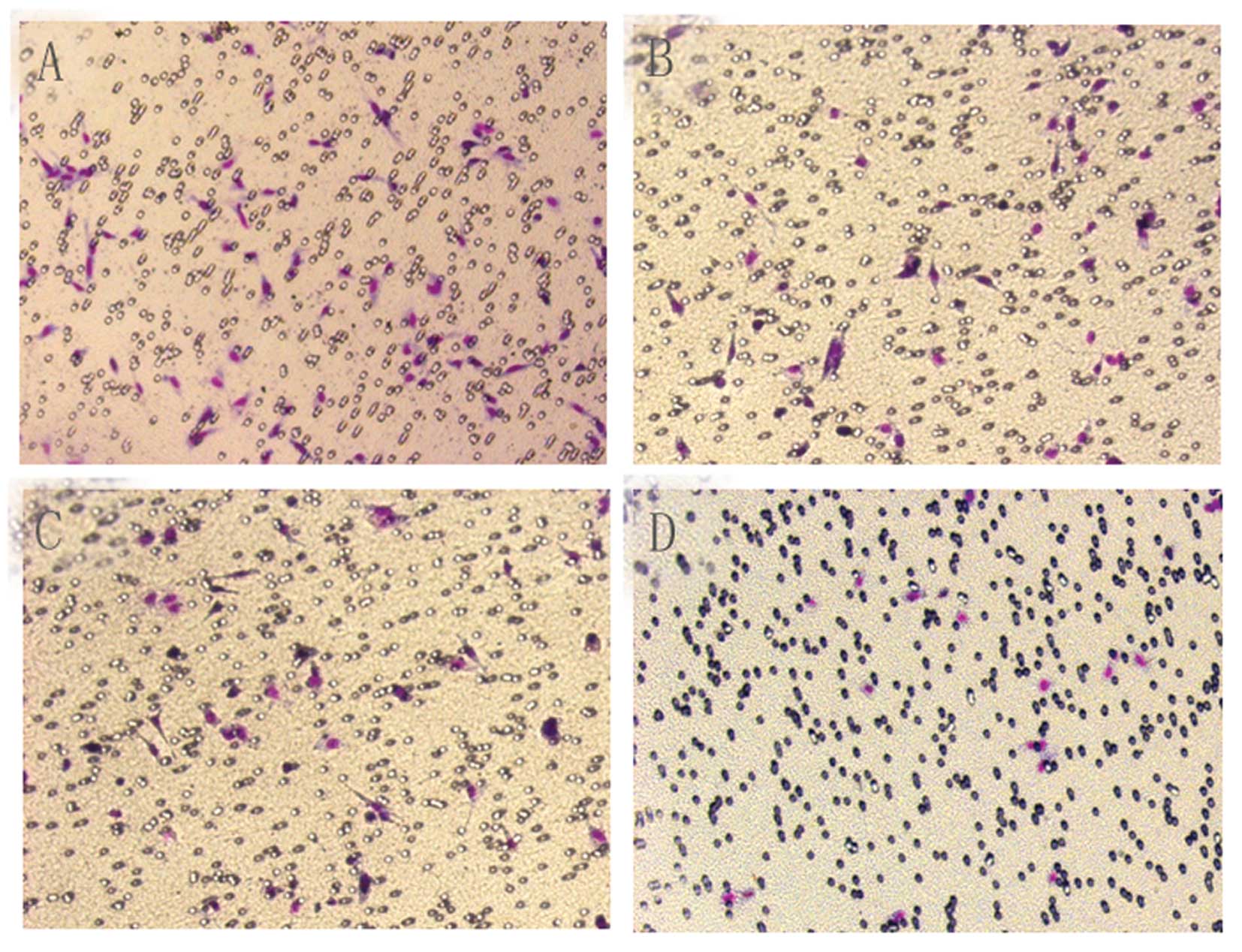
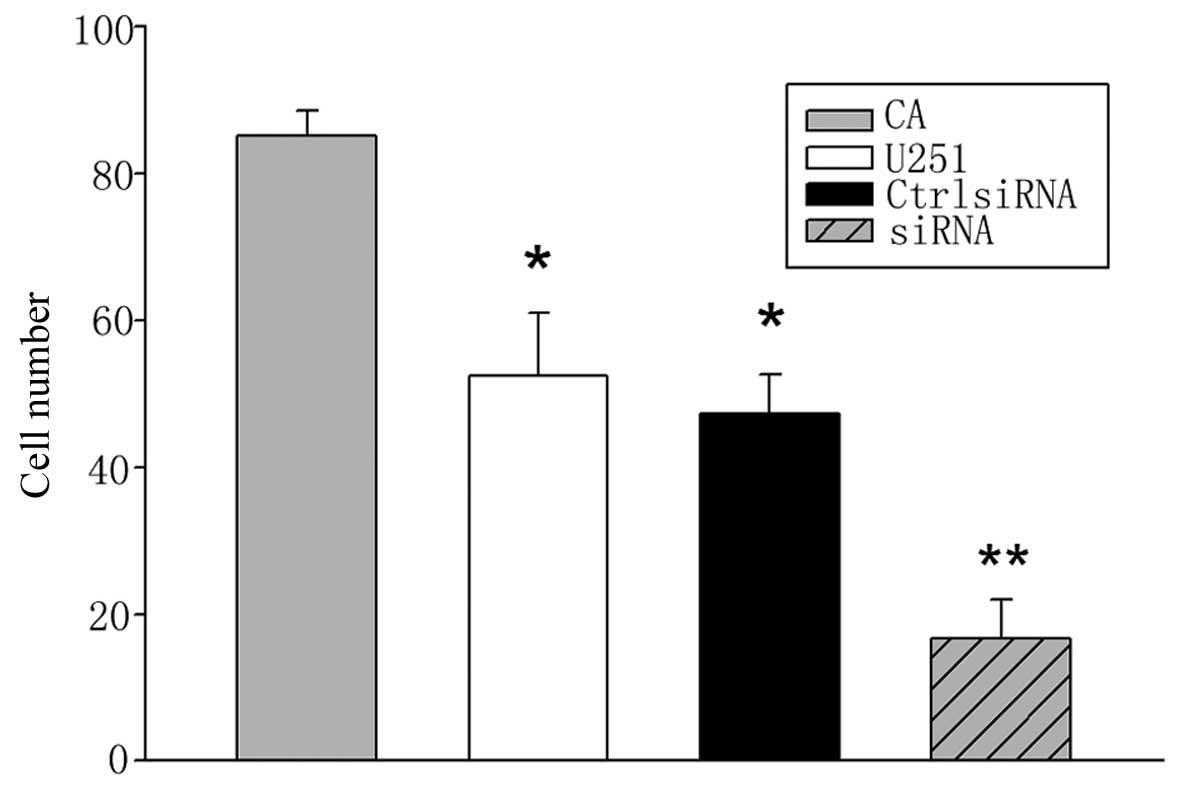
NFATc1 plays an important role in the
invasion of U251 cells
When the transfectants were allowed to invade the
Matrigel in an in vitro invasion assay, the
CA-NFATc1-transfected U251 cells were significantly more invasive
compared with the untreated controls (Figs. 3 and 4). Conversely, transfection of the U251
cells with NFATc1 siRNA resulted in a reduction in the invasion
ability compared with the controls (Figs. 3 and 4). The viability of the U251 cells was
significantly affected by NFATc1 siRNA treatment (Figs. 3 and 4).
NFATc1 siRNA inhibits the induction of
COX-2 expression in U251 cells, whereas CA-NFATc1 increases COX-2
expression
We examined the expression of COX-2, a gene with
important implications in the progression of GBM, which has been
reported to be regulated by NFAT in several cell types (22,23). qPCR analysis of COX-2 expression
revealed high mRNA expression levels of COX-2 in the U251 cells.
Indeed, COX-2 has been identified as an inducible early gene in
response to several stimuli in different cell types (24,25). We found that the induction of
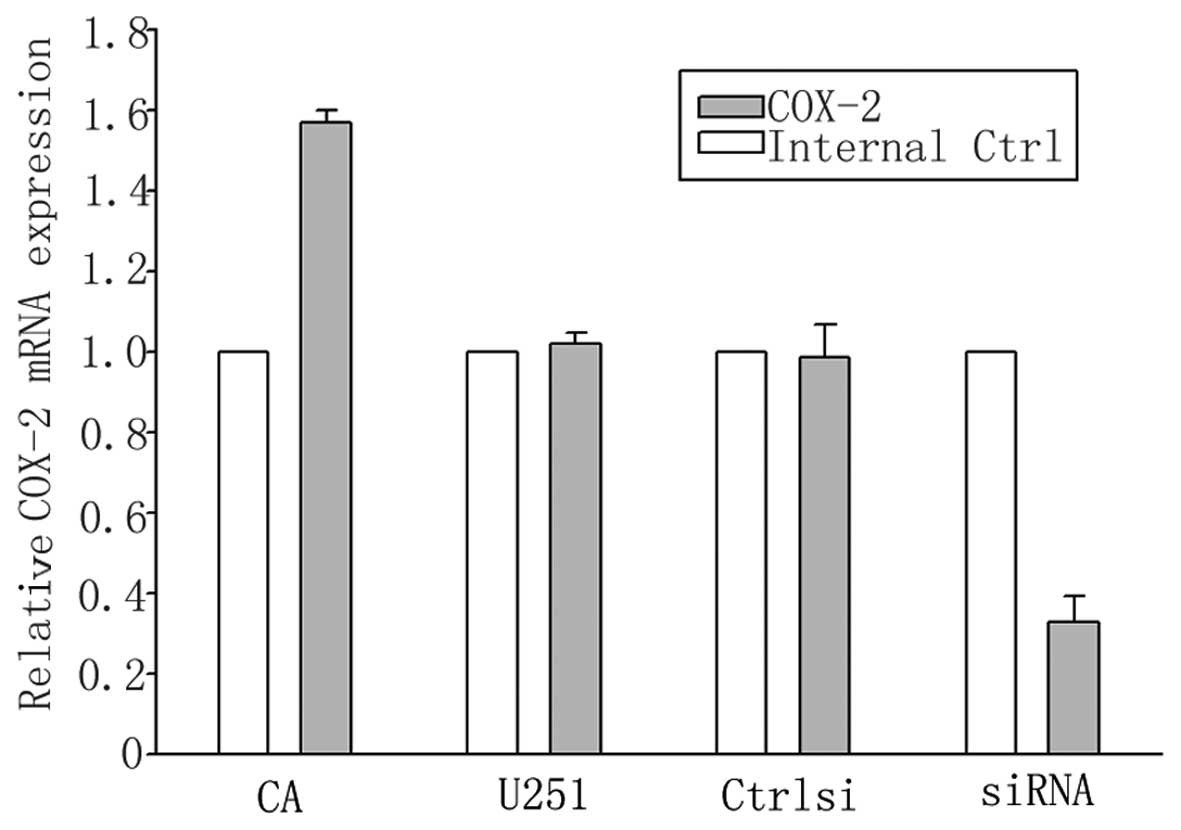
COX-2 mRNA expression was suppressed by NFATc1 siRNA (Fig. 5), indicating that the increase in
the mRNA levels mainly occurs at the transcriptional level,
requiring new RNA synthesis. On the other hand, CA-NFATc1 treatment
was found to increase COX-2 mRNA expression (Fig. 5). These results support the
hypothesis that COX-2 behaves as an inducible early gene, and that
NFATc1 regulates the expression of COX-2 in U251 cells.
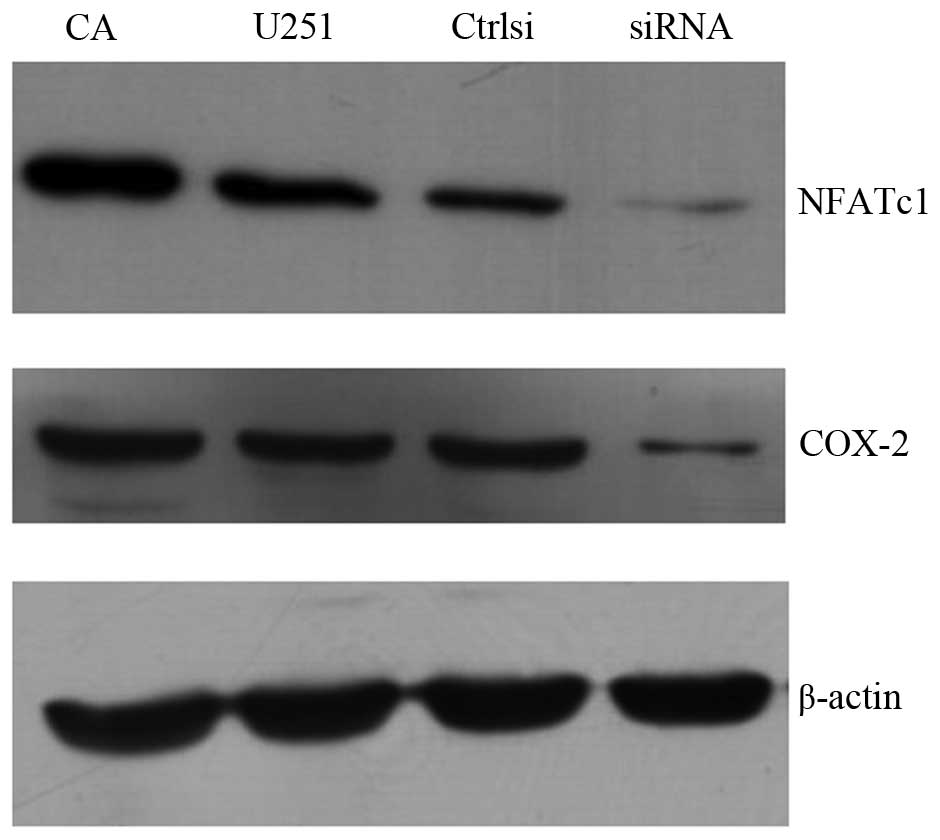
To address whether the induction of COX-2 mRNA
expression is paralleled by an increase in its protein expression,
western blot analysis was performed with extracts of U251 cells
treated with CA-NFATc1 and NFATc1 siRNA. The COX-2 protein levels
increased following treatment with CA-NFATc1, showing a pattern of
induction similar to that of its mRNA expression. The inhibition by
NFATc1 siRNA severely diminished the COX-2 protein levels (Fig. 6).
Involvement of NFATc1 in the
transcriptional activation of the COX-2 promoter
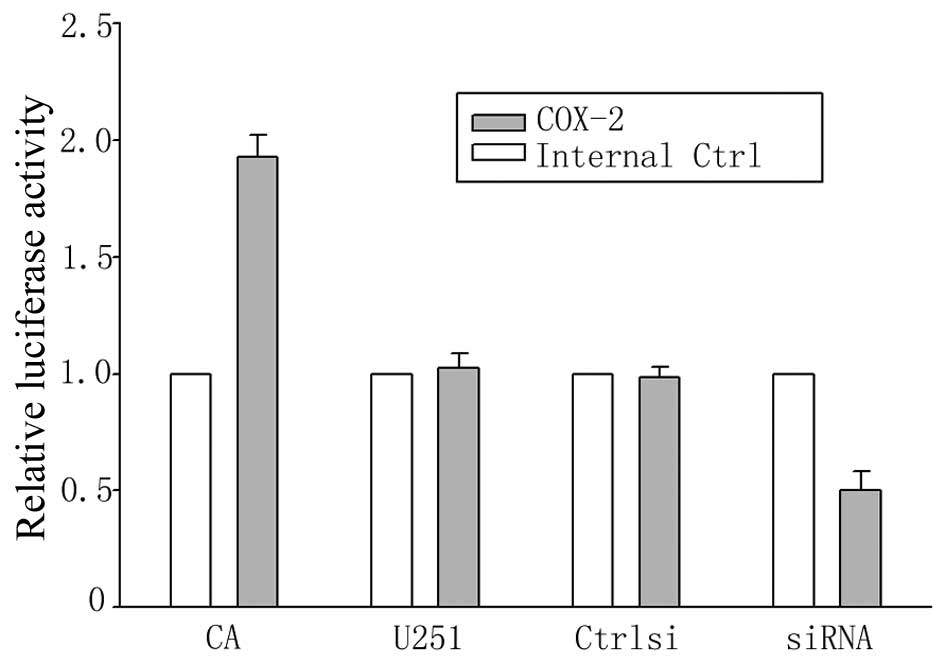
To determine whether the induction of COX-2 mRNA
expression by NFATc1 correlates with an increase in the
transcriptional activity mediated by the COX-2 promoter, the U251
cells were transiently transfected with COX-2 promoter. In
agreement with the regulation of COX-2 mRNA levels, CA-NFATc1
strongly increased transcription driven by a construct spanning
from −2,069 to −66 bp of the human COX-2 promoter (Fig. 7). To confirm the roles of NFATc1
in COX-2 expression, we also examined the effect of NFATc1
silencing by the siRNA approach in COX-2 promoter activity. The
transfection of siRNA targeting NFATc1 significantly inhibited
COX-2 promoter activity (Fig. 7).
However, the control siRNA did not influence COX-2 promoter
activity (Fig. 7). Taken
together, these results strongly suggest that NFATc1 is an
important mediator of COX-2 activity.
NFATc1 is involved in the regulation of
MMP-2 and MMP-9 expression
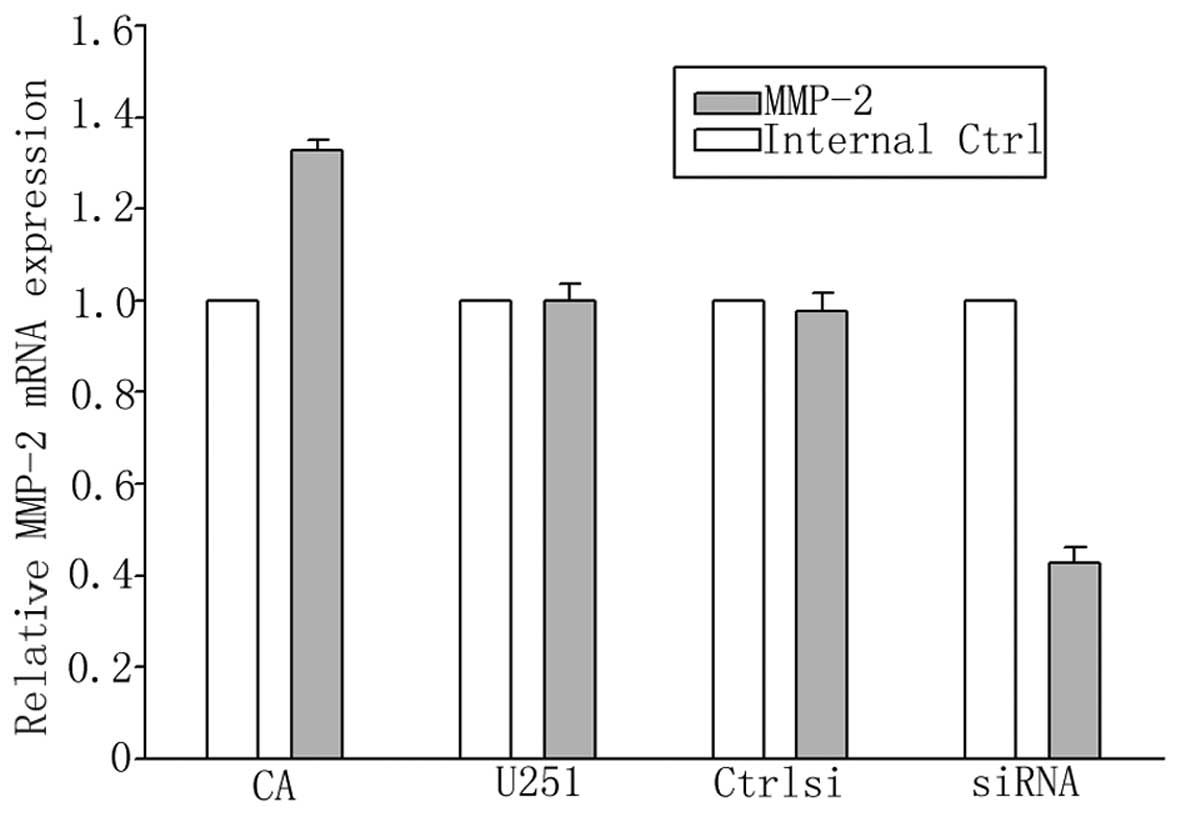
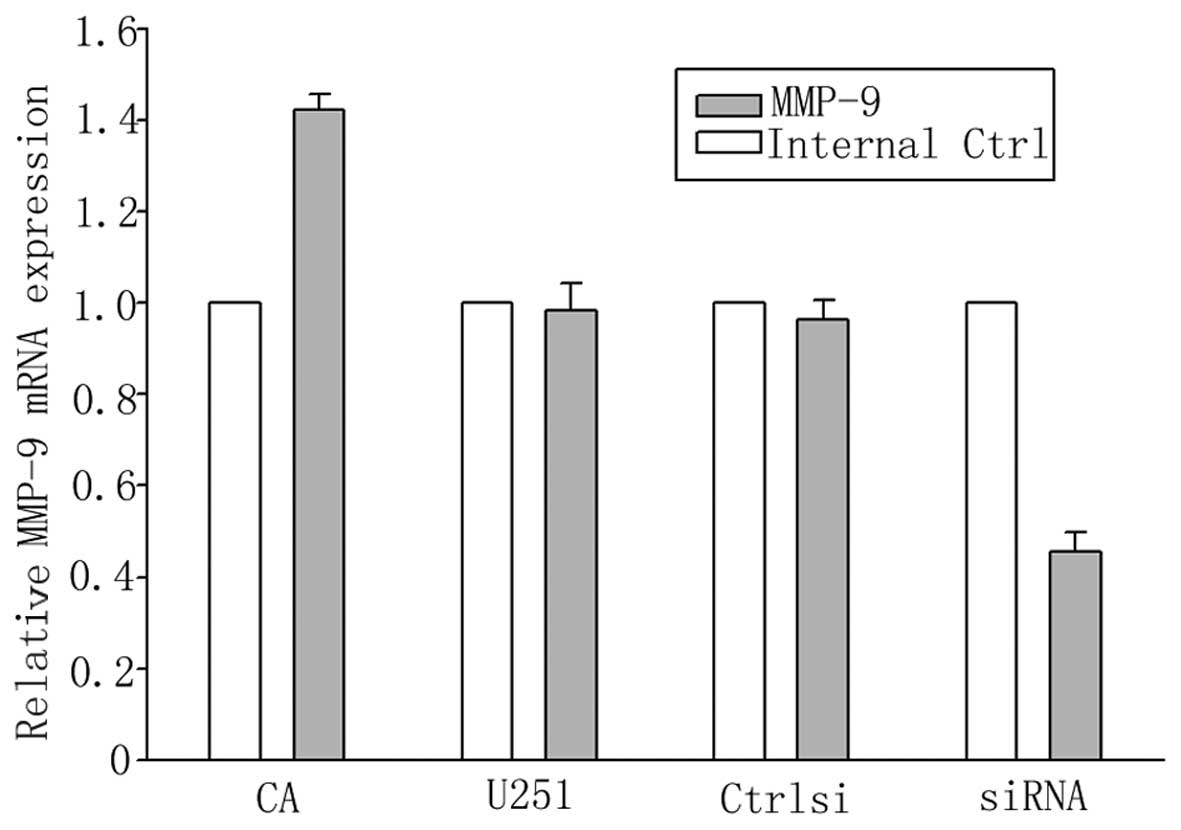
To examine whether NFATc1 regulates U251 cell
invasion by modulating MMP expression, we analyzed the mRNA levels
of MMP-2 and MMP-9 upon NFATc1 silencing and activation. The
results from qPCR revealed that the mRNA levels of MMP-2 and MMP-9
were decreased by >50% in the NFATc1-silenced cells (Figs. 8 and 9). The mRNA levels of MMP-2 and MMP-9 in
the cells transfected with the control siRNA were not altered under
these conditions (Figs. 8 and
9), indicating that the
downregulation of MMP-2 and MMP-9 expression by NFATc1 silencing is
specific. On the other hand, CA-NFATc1 treatment was found to
increase MMP-2 and MMP-9 mRNA expression (Figs. 8 and 9).
Discussion
NFAT was originally identified in immune cells as a
transcription factor required for the inducible expression of
cytokines critical for triggering the immune response (16,26). Previous studies have revealed that
NFAT is ubiquitously expressed and regulates a plethora of
transcriptional responses important for cell survival, angiogenesis
and cell growth in all cells and tissues (16,27). NFATc1 is a member of the NFAT
family. The constitutive activation of NFATc1 has been found in
approximately 70% of pancreatic carcinomas, and blocking NFATc1
activation with CsA has been shown to inhibit cell growth and
survival in a pancreatic cancer cell line (28). In a recent study, Oikawa et
al (29) found that the
constitutively active form of NFATc1 promoted cancer cell invasion
in association with changes in cell morphology in lung and breast
cancer cells. However, the mechanisms responsible for the malicious
activity of NFATc1 in GBM remains unclear. The present study
demonstrated that NFATc1 is constitutively activated in GBM tumors.
This finding indicates that NFATc1 is involved in the initiation
and progression of gliomas. Moreover, we found that NFATc1
expression increased according to the histopathological grade of
the glioma.
The present study revealed that NFATc1 exerted
invasive effects in the majority of U251 cells. To address this
issue, we developed a GBM cell line with an elevated inducible
NFATc1 (CA-NFATc1) expression. As was expected, the inducible
expression of NFATc1 led to increased transcriptional activity with
an increase in invasion through Matrigel. We also evaluated COX-2
regulation in GBM cells by silencing NFATc1 expression using siRNA.
Transfection of the U251 cells with NFATc1 siRNA resulted in a
diminished effect of cell invasion, suggesting that NFATc1 is
required for the invasion of GBM cells.
In recent years, a remarkable discovery for oncology
is that the overexpression of COX-2 is closely associated with
tumor development. COX-2 is considered a new and important target
for the treatment of a variety of tumors since COX-2 inhibitors or
the knockout of the COX-2 gene has been shown to inhibit tumor
development and metastasis (30).
Previous studies (31–33) have demonstrated that COX-2
promotes tumor cell invasion and metastasis through various means,
including the regulation of downstream genes in gastrointestinal,
lung and breast cancer, as well as other types of cancer. Recently,
it has also been reported that COX-2 is involved in the invasion of
osteosarcoma cells (34).
The induction of COX-2 by NFAT has been reported in
human T lymphocytes (14). This
issue has also been investigated in non-immune cells. For example,
NFAT induces the transcription of COX-2 in breast epithelial cells,
and this is required for the ability of NFAT to promote invasive
migration (35). In addition,
NFATc2 and NFAT5 proteins are expressed in invasive human ductal
breast carcinomas participating in promoting carcinoma migration
and invasion (3). It has also
been reported that the NFAT induction of COX-2 expression
contributes to tumor cell invasion and metastasis in a hepatitis
virus-induced hepatocellular carcinoma model (36). Similarly, a recent study also
provided evidence of COX-2 expression induced by NFAT in colon
carcinoma (15). However, to the
best of our knowledge, our study is the first to demonstrate the
induction of COX-2 by NFATc1 in GBM cells. More importantly, we
demonstrate that COX-2 is one of the genes responsible for
NFAT-driven Matrigel invasion.
To the best of our knowledge, our data also
demonstrate for the first time that activated NFATc1 is able to
transcriptionally upregulate COX-2 through direct interaction with
specific sequence elements within the COX-2 promoter. In GBM cell
lines, NFAT cooperates by binding to the COX-2 promoter, leading to
the expression of downstream regulators at the transcriptional
level, which, as we showed, was COX-2- and NFAT-dependent. The
inhibition of COX-2 in vivo has been found to attenuate the
metastatic potential of colorectal tumors in both humans (37) and mice (38), while the overexpression of COX-2
in intestinal cells modulates their adhesive properties (39) and increases MMP activity to
promote invasion (40).
One would also expect that for NFAT to function as a
pro-invasion transcription factor, it would induce the
transcription and secretion of MMPs that are required for efficient
basement membrane proteolysis during tumor invasion and metastasis
(41). While there is some
evidence that NFAT is required for MMP induction in myocytes and
mesangial cells, to date, no studies have addressed MMP regulation
by NFAT signaling in settings of carcinoma progression. Our
findings indicate that elevated expression levels of MMP-2 and
MMP-9 are associated with a higher incidence of invasion in human
GBM cells. MMP-2 and MMP-9 are upregulated by COX-2 and NFAT, thus
promoting the invasion of GBM cells.
In conclusion, our data add to the growing body of
evidence suggesting that NFATc1 transcription factors, in addition
to their well-defined roles as transcriptional regulators, have the
potential to control central aspects of invasion in GBM cells.
NFATc1 plays a functional role in the regulation of target genes
involved in the tumoral phenotype, such as COX-2. Moreover, our
study provides a rationale for future research into the use of
inhibitors of NFATc1 in adjuvant therapy for GBM.
Acknowledgments
This study was supported by a grant from the
National Natural Science Foundation of China (no. H1805)
References
|
1
|
Pan MG, Xiong Y and Chen F: NFAT gene
family in inflammation and cancer. Curr Mol Med. 13:543–554. 2013.
View Article : Google Scholar :
|
|
2
|
Daniel C, Gerlach K, Väth M, Neurath MF
and Weigmann B: Nuclear factor of activated T cells – a
transcription factor family as critical regulator in lung and colon
cancer. Int J Cancer. 134:1767–1775. 2014. View Article : Google Scholar
|
|
3
|
Jauliac S, Lopez-Rodriguez C, Shaw LM,
Brown LF, Rao A and Toker A: The role of NFAT transcription factors
in integrin-mediated carcinoma invasion. Nat Cell Biol. 4:540–544.
2002. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Medyouf H, Alcalde H, Berthier C, et al:
Targeting calcineurin activation as a therapeutic strategy for
T-cell acute lymphoblastic leukemia. Nat Med. 13:736–741. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang P, Cartwright CA, Li J, et al:
Arachidonic acid metabolism in human prostate cancer. Int J Oncol.
41:1495–1503. 2012.PubMed/NCBI
|
|
6
|
Castro-Sánchez L, Agra N, Llorente
Izquierdo C, et al: Regulation of 15-hydroxyprostaglandin
dehydrogenase expression in hepatocellular carcinoma. Int J Biochem
Cell Biol. 45:2501–2511. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Smith WL, DeWitt DL and Garavito RM:
Cyclooxygenases: structural, cellular, and molecular biology. Annu
Rev Biochem. 69:145–182. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tomozawa S, Tsuno NH, Sunami E, et al:
Cyclooxygenase-2 overexpression correlates with tumour recurrence,
especially haematogenous metastasis, of colorectal cancer. Br J
Cancer. 83:324–328. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Soslow RA, Dannenberg AJ, Rush D, et al:
COX-2 is expressed in human pulmonary, colonic, and mammary tumors.
Cancer. 89:2637–2645. 2000. View Article : Google Scholar
|
|
10
|
Denkert C, Kobel M, Pest S, et al:
Expression of cyclooxygenase 2 is an independent prognostic factor
in human ovarian carcinoma. Am J Pathol. 160:893–903. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Iniguez MA, Rodriguez A, Volpert OV,
Fresno M and Redondo JM: Cyclooxygenase-2: a therapeutic target in
angiogenesis. Trends Mol Med. 9:73–78. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chang SH, Liu CH, Conway R, et al: Role of
prostaglandin E2-dependent angiogenic switch in cyclooxygenase
2-induced breast cancer progression. Proc Natl Acad Sci USA.
101:591–596. 2004. View Article : Google Scholar :
|
|
13
|
Myung SJ and Kim IH: Role of
prostaglandins in colon cancer. Korean J Gastroenterol. 51:274–279.
2008.In Korean. PubMed/NCBI
|
|
14
|
Iniguez MA, Martinez-Martinez S, Punzon C,
Redondo JM and Fresno M: An essential role of the nuclear factor of
activated T cells in the regulation of the expression of the
cyclooxygenase-2 gene in human T lymphocytes. J Biol Chem.
275:23627–23635. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Duque J, Fresno M and Iniguez MA:
Expression and function of the nuclear factor of activated T cells
in colon carcinoma cells: involvement in the regulation of
cyclooxygenase-2. J Biol Chem. 280:8686–8693. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Crabtree GR and Olson EN: NFAT signaling:
choreographing the social lives of cells. Cell. 109(Suppl): S67–79.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Serfling E, Avots A, Klein-Hessling S,
Rudolf R, Vaeth M and Berberich-Siebelt F: NFATc1/αA: The other
face of NFAT factors in lymphocytes. Cell Commun Signal. 10:162012.
View Article : Google Scholar
|
|
18
|
Langworthy M, Zhou B, de Caestecker M,
Moeckel G and Baldwin HS: NFATc1 identifies a population of
proximal tubule cell progenitors. J Am Soc Nephrol. 20:311–321.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cao S, Yan Y, Zhang X, et al: EGF
stimulates cyclooxygenase-2 expression through the STAT5 signaling
pathway in human lung adenocarcinoma A549 cells. Int J Oncol.
39:383–391. 2011.PubMed/NCBI
|
|
20
|
Neal JW and Clipstone NA: A constitutively
active NFATc1 mutant induces a transformed phenotype in 3T3-L1
fibroblasts. J Biol Chem. 278:17246–17254. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang C, Cao S, Yan Y, et al: TLR9
expression in glioma tissues correlated to glioma progression and
the prognosis of GBM patients. BMC Cancer. 10:4152010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hernandez GL, Volpert OV, Iniguez MA, et
al: Selective inhibition of vascular endothelial growth
factor-mediated angiogenesis by cyclosporin A: roles of the nuclear
factor of activated T cells and cyclooxygenase 2. J Exp Med.
193:607–620. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sugimoto T, Haneda M, Sawano H, et al:
Endothelin-1 induces cyclooxygenase-2 expression via nuclear factor
of activated T-cell transcription factor in glomerular mesangial
cells. J Am Soc Nephrol. 12:1359–1368. 2001.PubMed/NCBI
|
|
24
|
Maier JA, Hla T and Maciag T:
Cyclooxygenase is an immediate-early gene induced by interleukin-1
in human endothelial cells. J Biol Chem. 265:10805–10808.
1990.PubMed/NCBI
|
|
25
|
Herschman HR, Fletcher BS and Kujubu DA:
TIS10, a mitogen-inducible glucocorticoid-inhibited gene that
encodes a second prostaglandin synthase/cyclooxygenase enzyme. J
Lipid Mediat. 6:89–99. 1993.PubMed/NCBI
|
|
26
|
Hogan PG, Chen L, Nardone J and Rao A:
Transcriptional regulation by calcium, calcineurin, and NFAT. Genes
Dev. 17:2205–2232. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mancini M and Toker A: NFAT proteins:
emerging roles in cancer progression. Nat Rev Cancer. 9:810–820.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Buchholz M, Schatz A, Wagner M, et al:
Overexpression of c-myc in pancreatic cancer caused by ectopic
activation of NFATc1 and the Ca2+/calcineurin signaling
pathway. EMBO J. 25:3714–3724. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Oikawa T, Nakamura A, Onishi N, Yamada T,
Matsuo K and Saya H: Acquired expression of NFATc1 downregulates
E-cadherin and promotes cancer cell invasion. Cancer Res.
73:5100–5109. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cressey R, Pimpa S, Tontrong W,
Watananupong O and Leartprasertsuke N: Expression of
cyclooxygenase-2 in colorectal adenocarcinoma is associated with
p53 accumulation and hdm2 overexpression. Cancer Lett. 233:232–239.
2006. View Article : Google Scholar
|
|
31
|
Gately S and Li WW: Multiple roles of
COX-2 in tumor angiogenesis: a target for antiangiogenic therapy.
Semin Oncol. 31:2–11. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ogino S, Kirkner GJ, Nosho K, et al:
Cyclooxygenase-2 expression is an independent predictor of poor
prognosis in colon cancer. Clin Cancer Res. 14:8221–8227. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brown JR and DuBois RN: Cyclooxygenase as
a target in lung cancer. Clin Cancer Res. 10:4266s–4269s. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wu X, Cai M, Ji F and Lou LM: The impact
of COX-2 on invasion of osteosarcoma cell and its mechanism of
regulation. Cancer Cell Int. 14:272014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yiu GK and Toker A: NFAT induces breast
cancer cell invasion by promoting the induction of
cyclooxygenase-2. J Biol Chem. 281:12210–12217. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lara-Pezzi E, Gomez-Gaviro MV, Galvez BG,
et al: The hepatitis B virus X protein promotes tumor cell invasion
by inducing membrane-type matrix metalloproteinase-1 and
cyclooxygenase-2 expression. J Clin Invest. 110:1831–1838. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fenwick SW, Toogood GJ, Lodge JP and Hull
MA: The effect of the selective cyclooxygenase-2 inhibitor
rofecoxib on human colorectal cancer liver metastases.
Gastroenterology. 125:716–729. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yao M, Kargman S, Lam EC, et al:
Inhibition of cyclooxygenase-2 by rofecoxib attenuates the growth
and metastatic potential of colorectal carcinoma in mice. Cancer
Res. 63:586–592. 2003.PubMed/NCBI
|
|
39
|
Tsujii M and DuBois RN: Alterations in
cellular adhesion and apoptosis in epithelial cells overexpressing
prostaglandin endoperoxide synthase 2. Cell. 83:493–501. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tsujii M, Kawano S and DuBois RN:
Cyclooxygenase-2 expression in human colon cancer cells increases
metastatic potential. Proc Natl Acad Sci USA. 94:3336–3340. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mott JD and Werb Z: Regulation of matrix
biology by matrix metalloproteinases. Curr Opin Cell Biol.
16:558–564. 2004. View Article : Google Scholar : PubMed/NCBI
|