Introduction
Colorectal cancer, also known as colon cancer, is
the most common type of gastrointestinal cancer worldwide. Despite
advances in research and treatment modalities, colorectal cancer
still causes 500,000 deaths each year (1), and approximately 150,000 US
residents are diagnosed annually with colon cancer (2). It is one of the devastating cancers
and one third of patients with colon cancer will eventually succumb
to the disease (3). Over the past
3 decades, investigations into the molecular genetics of colon
cancer have revealed the regulation of cellular metabolism,
proliferation, differentiation and survival in colon cancer
(4). However, much work remains
to be done in order to fully understand and integrate the molecular
changes associated with the pathophysiology of colorectal
cancer.
Human nitrilase (NIT) proteins are members of the
nitrilase superfamily that contain a conserved nitrilase domain and
thiol enzymes involved in natural product biosynthesis and
post-translational modification in plants (5), animals, fungi and certain
prokaryotes (6,7). Based on the sequence analysis, the
NIT superfamily has been divided into 13 branches (6,8).
Branch 10 in the Brenner classification contains only 2 enzymes,
designated as NIT1 and NIT2, found in mammalian tissues (8–10).
The NIT2 gene is ubiquitously expressed in multiple types of
tissue and encodes the protein NIT2 mainly distributed in the
cytosol; it has been reported that NIT2 is a potential tumor
suppressor (11). However, to the
best of our knowledge, no study to date has reported the role of
NIT2 in colon cancer, and the cellular function of the
NIT2 gene remains elusive.
The RNA interference (RNAi) technique is a powerful
tool to carry out loss-of-function assays. It provides a new
approach to investigating cancer gene therapy (12,13). In the present study, we employed a
lentivirus-mediated RNAi system in order to achieve the highly
stable silencing of NIT2 in the colon cancer cell line,
HCT116. We evaluated the biological function of NIT2 and
aimed to reveal its contribution to the progression of colon
cancer. To the best of our knowledge, this is the first
presentation providing evidence that the knockdown of endogenous
NIT2 expression suppresses the oncogenic properties of colon
cells and induces apoptosis through the caspase-3 and
poly(ADP-ribose) polymerase (PARP) pathways.
Materials and methods
Cell culture
The human embryonic kidney cell line 293T (HEK293T)
and the human colon cancer cell line, HCT116, were obtained from
the Shanghai Institute of Cell Biology, the Chinese Academy of
Sciences, Shanghai, China. The HCT116 cells were maintained in
McCoy’s 5A medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented
with 10% fetal bovine serum (FBS) at 37°C in a humidified
atmosphere of 5% CO2. The HEK293T cells were maintained
in Dulbecco’s modified Eagle’s medium (DMEM) (HyClone, Logan, UT,
USA) supplemented with 10% FBS at 37°C in a humidified atmosphere
of 5% CO2.
Construction of recombinant
lentivirus
The following oligonucleotides were synthesized. The
short hairpin RNA (shRNA) sequence
(5′-ACATAATCAACTCCCTATTAACTCGAGTTAATAGGGAGTTGATTATGTTTTTT-3′,
sequence 1) against the human NIT2 gene (NM_020202) was
screened and validated to be a candidate shRNA. The negative
control shRNA was 5′-TTCTCCGAACGTGTCACGT-3′. The stem-loop-stem
oligos (shRNAs) were synthesized, annealed and ligated into the
AgeI/EcoRI-linearized pFH-L vector (Shanghai
Hollybio, Shanghai, China). The lentiviral-based shRNA-expressing
vectors were confirmed by DNA sequencing. The generated plasmids
were named pFH-L-shNIT2 and pFH-L-shCon.
The HEK293T cells (1.0×106) were seeded
into 10-cm dishes and cultured for 24 h to reach 70–80% confluence
as previously described (14).
Two hours prior to transfection, the medium was replaced with
serum-free DMEM. The plasmids, including 10 μg of
pFH-L-shNIT2 or pFH-L-shCon, 7.5 μg of the packaging vector
pHelper-1.0 and 5 μg of the expression plasmid pHelper-2.0
were added to 0.95 ml of Opti-MEM and 50 μl of Lipofectamine
2000. The mixture was added to the cells followed by incubation for
8 h before replacing the medium with 10 ml of complete DMEM medium
(supplemented with 10% PBS). Lentiviral particles were harvested 48
h after transfection. The HCT116 cells (8.0×104) were
cultured in 6-well plates and inoculated with recombinant
lentiviruses (Lv-shCon or Lv-shNIT2) at a multiplicity of infection
(MOI) of 15. As the lentivirus carries green fluorescence protein
(GFP), the infection efficiency was determined by counting the
numbers of GFP-expressing cells under a fluorescence microscope
(Olympus BX50; Olympus Corp., Tokyo, Japan) at 96 h after
infection.
RNA extraction and reverse
transcription-quantitative (real-time) PCR (RT-qPCR)
The human colon cancer cells, HCT116, were
pre-cultured and infected with the recombinant lentiviruses for 5
days as previously described (15). Total RNA was prepared using TRIzol
reagent (Gibco BRL, Grand Island, NY, USA) according to the
manufacturer’s instructions. Total RNA (5 mg) was used to
synthesize the first-strand cDNA using 200 U/ml SuperScript II
Reverse Transcriptase (RT) (Invitrogen, Carlsbad, CA, USA). The
NIT2 mRNA expression was evaluated by quantitative PCR on a BioRad
Connet Real-Time PCR platform (Bio-Rad Laboratories, Hercules, CA,
USA) with SYBR-Green PCR Core reagents. The qPCR reaction system
contains 10 μl 2X SYBR Premix Ex Taq, plus 0.8 μl
forward and reverse primers (2.5 μM), 5 μl cDNA
template and 4.2 μl ddH2O. β-actin was applied as
the internal reference. The following primers were synthesized and
applied: NIT2 forward, 5′-CGGGCTGTTGATAATCAGGT-3′ and reverse,
5′-TTCAGCCAGCTTCTTCAGGT-3′; and β-actin forward,
5′-GTGGACATCCGCAAAGAC-3′ and reverse, 5′-A AAGGGTGTAACGCAACTA-3′
(14). The reaction procedure was
initiated with denaturation at 95°C for 1 min followed by 40
repeated cycles (denaturation at 95°C for 5 sec and annealing
extension at 60°C for 20 sec). The results are presented as CT
values, defined as the threshold PCR cycle number at which an
amplified product is first detected. The average CT value was
calculated for both NIT2 and β-actin, and the ΔCT value was
determined as the mean of the triplicate CT values for NIT2 minus
the mean of the triplicate CT values for β-actin.
Western blot analysis
The HCT116 cells were cultured and infected with the
recombinant lentiviruses for 5 days. The cells were washed twice
with ice-cold PBS and lysed in 2X SDS sample buffer [100 mM
Tris-HCl (pH 6.8), 10 mM EDTA, 4% SDS and 10% glycine]. Equal
amount of proteins (30 μg) were loaded and separated by
electrophoresis (50 V, 3 h) on 10% SDS-PAGE gels and transferred
onto polyvinylidene difluoride (PVDF) membranes (Millipore,
Bedford, MA, USA) at 300 mA for 1.5 h. The membranes were blocked
and then probed with primary antibodies, rabbit anti-NIT2 (1:8,000
dilution, #31280; Signalway Antibody LLC, College Park, MD, USA) or
mouse anti-GAPDH (1:50,000 dilution, #10494-1-AP; Santa Cruz
Biotechnology, Inc., Dallas, Texas, USA) overnight at 4°C. After
washing, the blots were incubated with horseradish
peroxidase-conjugated secondary antibodies (1:5,000 dilution,
#SC-2054; Santa Cruz Biotechnology, Inc.) for 2 h at room
temperature and then visualized using Super ECL Detection reagent
(Applygen Technologies, Inc., Beijing, China).
Colony formation assay
The human colon cancer cells, HCT116, were cultured
in 6-well plates and treated with the recombinant lentiviruses.
After 96 h of incubation, the infected cells were washed,
re-cultured in the prepared 6-well plates at a density of 400
cells/well, and allowed to form natural colonies. After 7 days of
incubation, the treated cells were subjected to Crystal violet
staining. Subsequently, the cells were washed and fixed with
paraformaldehyde. The fixed cells were washed twice with PBS
solution, treated with Crystal violet for 10 min, washed 3 times
with ddH2O and then photographed using a digital camera
(D7000; Nikon Corp., Tokyo, Japan). The number of colonies (>50
cells/colony) was counted.
MTT assay for cell viability
The HCT116 cells were cultured in 6-well plates and
inoculated with the recombinant lentiviruses. After 96 h of
infection, the cells were washed and re-cultured in 96-well plates
at 2.5×103 cells/well. MTT solution was added to the
wells followed by incubation at 37°C for 4 h at different time
points after lentivirus infection (1, 2, 3, 4 and 5 days). The
converted dye was then solubilized in acidic isopropanol (10% SDS,
5% isopropanol and 0.01 M HCl) followed by incubation at 37°C for
10 min. The optical density was measured using a microplate reader
(Epoch; BioTek, Winooski, VT, USA) at the wavelength of 595 nm. The
experiment was repeated at least 3 times.
Flow cytometric analysis
The HCT116 cells were cultured in 6-well plates and
inoculated with the recombinant lentiviruses at a MOI of 20. After
3 days of infection, the cells were inoculated into 6-cm dishes at
a density of 1×105 cells/dish. After 40 h of incubation,
the cells in each well were harvested and the cell cycle was
determined by propidium iodide (PI) staining before the cell
density reached 80% confluency. Tests were performed in triplicate
for each sample, and analyses were performed using a FACScan flow
cytometer (Becton-Dickinson, San Jose, CA, USA) in accordance with
the manufacturer’s guidelines.
Intracellular signaling array
Intracellular signaling array was carried out as
previously described (16). The
HCT116 cell lysates were analyzed using the PathScan Intracellular
Signaling Array kit (Cat. no. 7323; Cell Signaling Technology) for
the simultaneous detection of 18 important and well-characterized
phosphorylated signaling molecules. The lysate of HCT116 was
diluted to 200 mg/ml, and 75 ml of lysate was added to
nitrocellulose-coated glass slides pre-coated with the primary
antibodies. The plate was incubated overnight at 4°C, followed by
exposure to the detection antibody cocktail for 1 h at room
temperature. HRP-conjugated secondary antibody was then added and
the plate was incubated for 30 min at room temperature. Substrate
was then added and chemiluminescent signals were detected to screen
and compare the differences between cell lysates infected with
Lv-shCon and Lv-shNIT2.
Statistical analysis
All statistical analyses were performed using SPSS
version 13.0 software. The differences between groups were compared
using the Student’s test, and data are expressed as the means ± SD
of 3 independent experiments. A value of P<0.05 was considered
to indicate a statistical significant difference.
Results
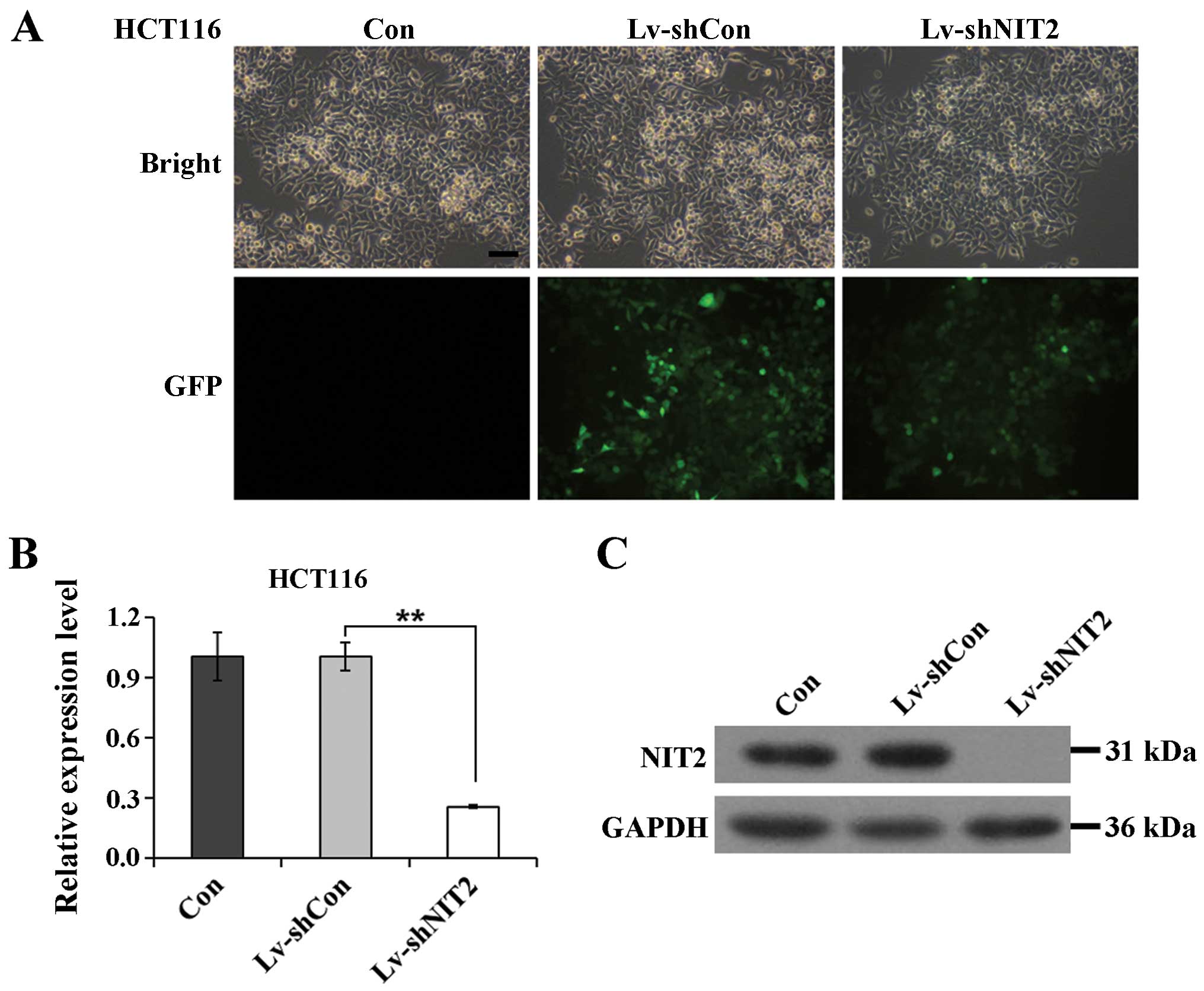
Efficacy of lentivirus-mediated RNAi
targeting of NIT2
In the present study, we constructed both a control
lentivirus (Lv-shCon) and a specific NIT2-targeting
lentivirus (Lv-shNIT2). The HCT116 cells were cultured and infected
with Lv-shCon or Lv-shNIT2. Non-infected cells were deemed as the
negative control (Con). To demonstrate the infection efficiency, we
employed a GFP tag which was embedded in the lentivirus to provide
visualized confirmation. As shown in Fig. 1A, >80% of the colon cancer
cells were GFP-positive, indicating that the transfection rate was
satisfactory. To further investigate the efficiency of the
NIT2 knockdown, we examined the expression levels of
NIT2 in the colon cancer cells following lentivirus
infection. The non-silencing lentivirus encoded with the irrelevant
sequence had a negligible effect on NIT2 expression, but the
NIT2-silencing lentivirus markedly downregulated the mRNA
expression of NIT2 by 74.6% (Fig. 1B). We further investigated the
protein expression level following infection with the constructed
lentiviruses. According to the NCBI gene database, the human
NIT2 gene encodes a deduced protein comprising of 276 amino
acids, with a calculated molecular mass of 30.6 kDa. As is shown in
Fig. 1C, the protein expression
of NIT2 in the HCT116 cells was noticeably depleted. Taken
together, the lentiviruses we constructed conferred us an efficient
vector system to knock down NIT2 expression in colon cancer
cells.
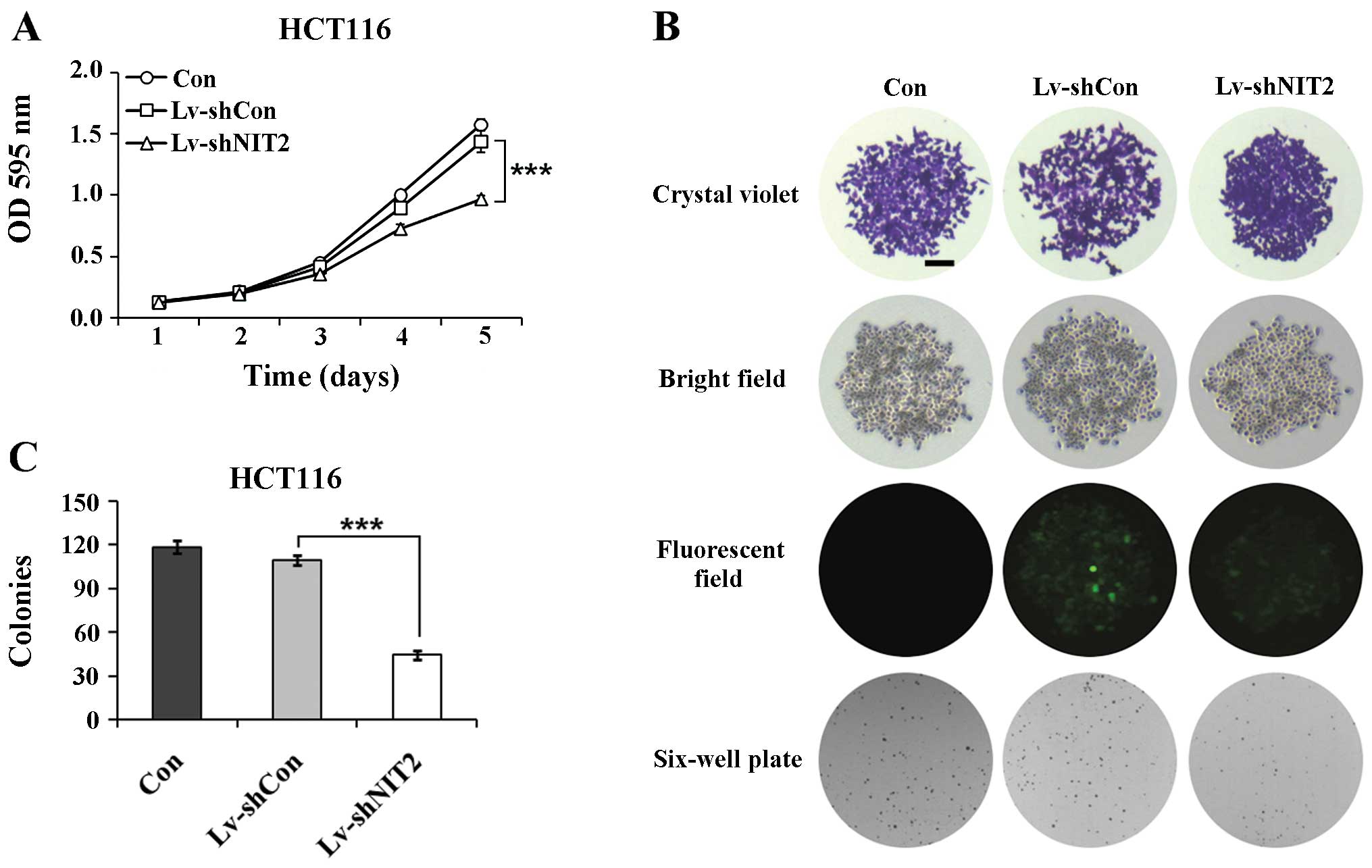
Depletion of NIT2 markedly inhibits the
proliferation of colon cancer cells
To better understand the role of NIT2 in
colon cancer tumorigenesis, we examined the variation tendency of
cell proliferation following lentivirus infection. We used MTT
assay due to its testing sensitivity and dynamic range and examined
the cell proliferation rate after 5 days of incubation. As
illustrated by the line chart in Fig.
2A, the non-silenced cells showed no obvious difference
compared with the control cells (Lv-shCon vs. Con); however, a
significant inhibition of cell proliferation was observed in the
NIT2-silenced cells (Lv-shNIT2 vs. Lv-shCon;
P<0.001).
We subsequently cross evaluated the colony formation
capacity of the HCT116 cells. As shown in Fig. 2B and C, the downregulation of
NIT2 induced a significant decrease in the colony formation
ability of the cells. The colonies were markedly smaller and the
colony numbers were statistically fewer compared with the control
cells (P<0.001), whereas there were no noticeable differences
between the non-silenced cells and the control cells. Collectively,
the knockdown of NIT2 by RNAi markedly suppressed the
proliferation and colony formation ability of the colon cancer
cells.
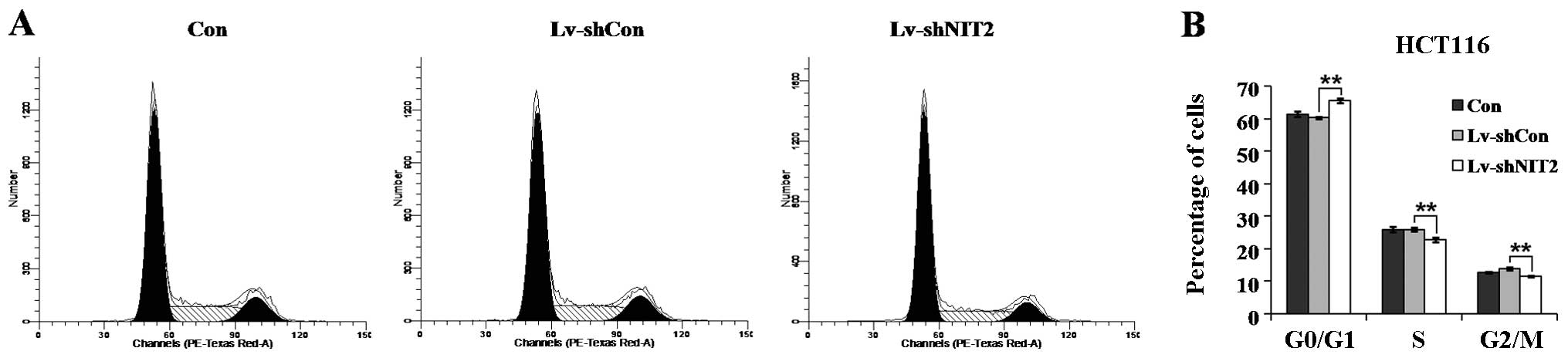
Cell cycle arrest is induced by the
downregulation of NIT2
To examine whether the knockdown of NIT2
suppresses the growth of colon cancer cells through the direct
regulation of the cell cycle, the effects of the suppression of
NIT2 on the cell cycle distribution were examined. We
explored the alterations in cell cycle distribution when the HCT116
cells were infected with the NIT2-silencing lentivirus. As
illustrated in Fig. 3, the
distribution of cells in the cell cycle (G0/G1, S and G2/M phase)
varied significantly between the 3 groups (Con, Lv-shCon and
Lv-shNIT2). In contrast to the control group, the cells infected
with Lv-shNIT2 were mostly distributed in the G0/G1 phase (65.61%)
and fewer cells were distributed in the S phase (22.78%) and the
G2/M phase (11.61%), indicating that the cell cycle was arrested in
the G0/G1 phase. Our results demonstrated that treatment with
Lv-shNIT2 markedly induced cell cycle arrest (P<0.01); however,
the cells infected with the non-silencing lentivirus did not show
any significant differences compared with the non-infected cells
(Lv-Con vs. Con). These findings are in agreement with those
observed for cell growth, which suggests that NIT2 modulates
colon cancer cell growth by controling the cell cycle.
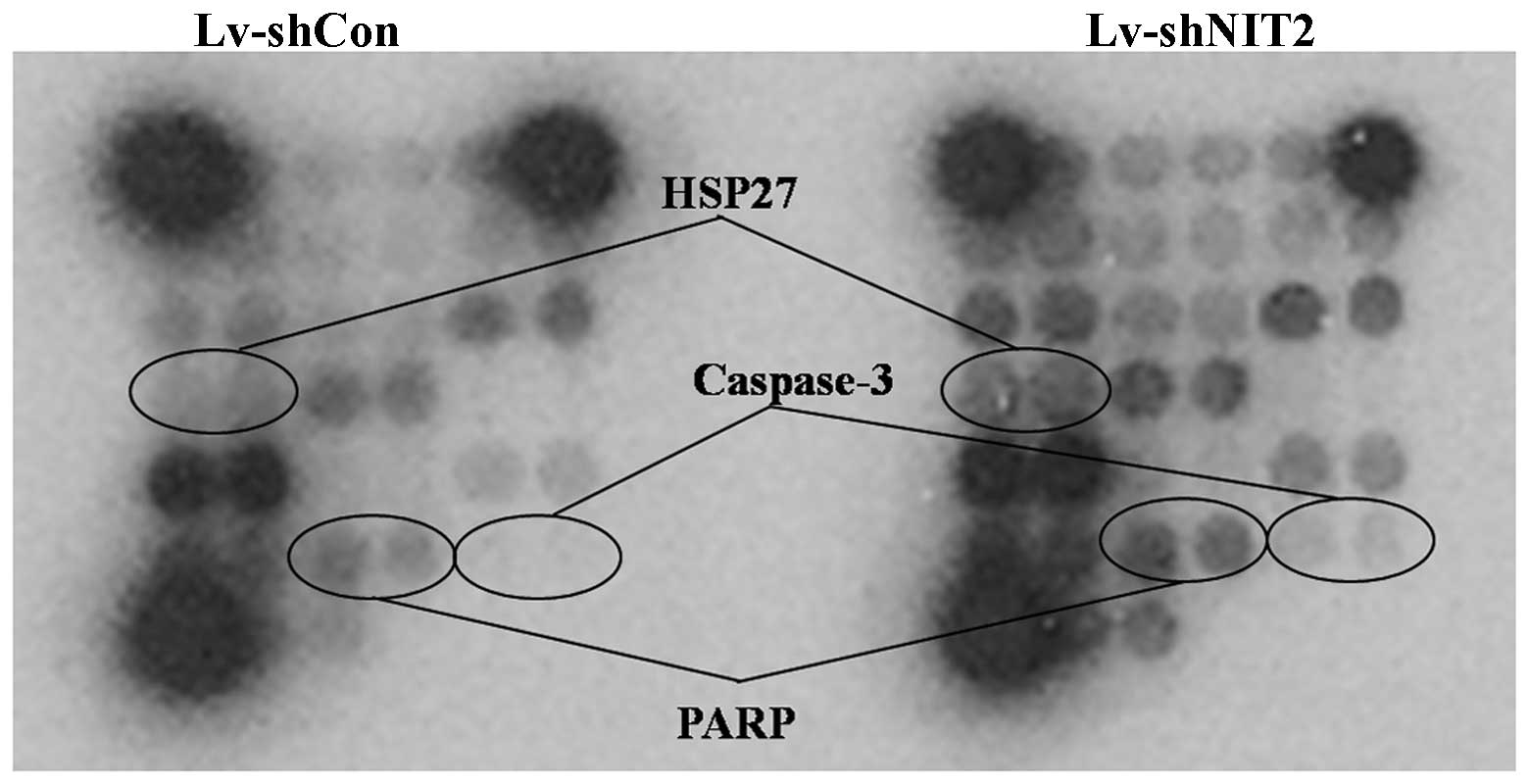
Cell cycle arrest and inhibition of
proliferation induced by the depletion of NIT2 are associated with
caspase-3 and PARP signaling
The PathScan Intracellular Signaling Array kit was
used to monitor the expression of 18 signaling molecules that are
phosphorylated or cleaved in response to signal transduction
pathway activation. Caspase-3 is a critical executor of apoptosis.
Caspase-3 is activated by endoproteolytic cleavage at Asp175 and
exerts its pro-apoptotic activity through the cleavage of multiple
cellular targets (17–20). PARP, an enzyme that is involved in
DNA repair, is one of the main substrates of activated caspase-3
(21,22). Increased levels of cleaved
caspase-3 and cleaved PARP are reliable indicators of apoptosis
(23). Heat shock protein 27
(HSP27), phosphorylated at Ser78 within the p38 MAPK pathway, is a
mediator of cell stress that confers resistance to adverse
environmental changes (24). In
the present study, we demonstrated that when the colon cancer
cells, HCT116, were treated with NIT2-silencing lentivirus,
the cleavage of PARP and caspase-3 were noticeably increased,
accompanied with an intracellular increase of phosphorylated HSP27
(Fig. 4).
Discussion
In mammals there are 2 metabolism routes for
glutamine. One is catalyzed by glutaminases K and L, followed by
the conversion of glutamate to α-ketoglutarate by transamination or
by the glutamate dehydrogenase reaction, whereas the glutaminase II
pathway employs ω-amidase catalyzed hydrolysis reaction from
α-ketoglutamate (KGM) to α-ketoglutarate (25–34). The glutaminase II pathway is one
of the clinical interests, particularly in certain inborn errors of
metabolism and cancers (35–38). This has been strengthened by the
finding of NIT2, which was demonstrated to be a putative tumor
suppressor (11) and identical to
ω-amidase (28,29,39). NIT2/ω-amidase plays a crucial role
in glutamine metabolism. However, its role in human colon cancer
remains unclear.
In the present study, we examined the biological
role of NIT2 in cell growth through an RNAi lentivirus
system using the colon cancer cell line, HCT116, in vitro.
We demonstrated that the downregulation of NIT2 suppressed
cell proliferation and colony formation. Moreover, cell cycle
arrest was observed by FACS analysis and apoptotic signaling
markers were detected by an intracellular signaling array. The
significant alterations in cell proliferation and cell cycle
progression prove that NIT2 plays a pivotal role in the growth of
colon cancer cells, which is in alignment with an increase in HSP27
phosphorylation at residue Ser78 and the cleavage of caspase-3 at
residue Asp175 and PARP at residue Asp214.
Semba et al (40) reported that aberrant NIT1
expression induced caspase-dependent apoptosis. In addition, an
apoptosis-inducing effect of NIT1 in plant wound and
herbicide-induced cell death has been proposed (41). In contrast to NIT1, much less is
known about the biological function of NIT2 in humans. To the best
of our knowledge, this is the first study to prove that the
depletion of NIT2 induces apoptosis through the caspase-3
and PARP pathways. Caspase-3 is a member of the cysteine-aspartic
acid protease family (42). It
interacts with caspase-8 and caspase-9 proteins and can trigger the
sequential activation of cell apoptosis. Caspase-3 zymogen has
virtually no activity until it is cleaved at Asp175 by an inhibitor
caspase (43). Together with its
main substrate PARP, both of them in the activated format can be
deemed as reliable indicators of apoptosis (23).
Looking back to the observation of cell cycle
arrest, all these data demonstrate that the depletion of
NIT2 may not only inhibit colon cancer cell growth, but may
also promote the apoptosis of cancer cells. The biochemical
significance of NIT2 has been demonstrated in the present
study, indicating that the depletion of NIT2 in colon cancer
cells leads to a significant reduction in cell growth. Therefore,
NIT2 may be a promising therapeutic target in colon cancer
and may also be an alternative option for the treatment of colon
cancer.
References
|
1
|
Merika E, Saif MW, Katz A, Syrigos K and
Morse M: Colon cancer vaccines: an update (Review). In Vivo.
24:607–628. 2010.PubMed/NCBI
|
|
2
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J and
Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 59:225–249.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cunningham D, Atkin W, Lenz HJ, et al:
Colorectal cancer. Lancet. 375:1030–1047. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fearon ER: Molecular genetics of
colorectal cancer. Annu Rev Pathol. 6:479–507. 2011. View Article : Google Scholar
|
|
5
|
Bartling D, Seedorf M, Mithofer A and
Weiler EW: Cloning and expression of an Arabidopsis nitrilase which
can convert indole-3-acetonitrile to the plant hormone,
indole-3-acetic acid. Eur J Biochem. 205:417–424. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pace HC and Brenner C: The nitrilase
superfamily: classification, structure and function. Genome Biol.
2:REVIEWS00012001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Horst RJ, Zeh C, Saur A, Sonnewald S,
Sonnewald U and Voll LM: The Ustilago maydis Nit2 homolog regulates
nitrogen utilization and is required for efficient induction of
filamentous growth. Eukaryot Cell. 11:368–380. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pace HC, Hodawadekar SC, Draganescu A, et
al: Crystal structure of the worm NitFhit Rosetta Stone protein
reveals a Nit tetramer binding two Fhit dimers. Curr Biol.
10:907–917. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pekarsky Y, Campiglio M, Siprashvili Z, et
al: Nitrilase and Fhit homologs are encoded as fusion proteins in
Drosophila melanogaster and Caenorhabditis elegans. Proc Natl Acad
Sci USA. 95:8744–8749. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Barglow KT, Saikatendu KS, Bracey MH, et
al: Functional proteomic and structural insights into molecular
recognition in the nitrilase family enzymes. Biochemistry.
47:13514–13523. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lin CH, Chung MY, Chen WB and Chien CH:
Growth inhibitory effect of the human NIT2 gene and its allelic
imbalance in cancers. FEBS J. 274:2946–2956. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim DH, Behlke MA, Rose SD, Chang MS, Choi
S and Rossi JJ: Synthetic dsRNA Dicer substrates enhance RNAi
potency and efficacy. Nat Biotechnol. 23:222–226. 2005. View Article : Google Scholar
|
|
13
|
Guo W, Zhang Y, Chen T, et al: Efficacy of
RNAi targeting of pyruvate kinase M2 combined with cisplatin in a
lung cancer model. J Cancer Res Clin Oncol. 137:65–72. 2011.
View Article : Google Scholar
|
|
14
|
Yang H, He X, Zheng Y, et al:
Down-regulation of asparagine synthetase induces cell cycle arrest
and inhibits cell proliferation of breast cancer. Chem Biol Drug
Des. 84:578–584. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shao X, Liu Y, Huang H, Zhuang L, Luo T
and Ge X: Down-regulation of G protein-coupled receptor 137 by RNA
interference inhibits cell growth of two hepatoma cell lines. Cell
Biol Int. Dec 9–2014.Epub ahead of print. PubMed/NCBI
|
|
16
|
Miyake M, Goodison S, Urquidi V, Gomes
Giacoia E and Rosser CJ: Expression of CXCL1 in human endothelial
cells induces angiogenesis through the CXCR2 receptor and the
ERK1/2 and EGF pathways. Lab Invest. 93:768–778. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Boatright KM and Salvesen GS: Mechanisms
of caspase activation. Curr Opin Cell Biol. 15:725–731. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shi Y: Caspase activation: revisiting the
induced proximity model. Cell. 117:855–858. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kurokawa M and Kornbluth S: Caspases and
kinases in a death grip. Cell. 138:838–854. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brognard J and Hunter T: Protein kinase
signaling networks in cancer. Curr Opin Genet Dev. 21:4–11. 2011.
View Article : Google Scholar :
|
|
21
|
Isabelle M, Moreel X, Gagne JP, et al:
Investigation of PARP-1, PARP-2, and PARG interactomes by
affinity-purification mass spectrometry. Proteome Sci. 8:222010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Piskunova TS, Yurova MN, Ovsyannikov AI,
et al: Deficiency in poly(ADP-ribose) polymerase-1 (PARP-1)
accelerates aging and spontaneous carcinogenesis in mice. Curr
Gerontol Geriatr Res. 7541902008.PubMed/NCBI
|
|
23
|
Yu SW, Andrabi SA, Wang H, et al:
Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR)
polymer-induced cell death. Proc Natl Acad Sci USA.
103:18314–18319. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Parcellier A, Schmitt E, Gurbuxani S, et
al: HSP27 is a ubiquitin-binding protein involved in I-kappaBalpha
proteasomal degradation. Mol Cell Biol. 23:5790–5802. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Meister A and Otani TT: ω-Amide and
ω-amino acid derivatives of alpha-ketoglutaric and oxalacetic
acids. J Biol Chem. 224:137–148. 1957.PubMed/NCBI
|
|
26
|
Meister A, Levintow L, Greenfield RE and
Abendschein PA: Hydrolysis and transfer reactions catalyzed by
omega-amidase preparations. J Biol Chem. 215:441–460.
1955.PubMed/NCBI
|
|
27
|
Meister A: Preparation of enzymatic
reactions of the keto analogues of asparagine and glutamine. J Biol
Chem. 200:571–589. 1953.PubMed/NCBI
|
|
28
|
Krasnikov BF, Chien CH, Nostramo R, et al:
Identification of the putative tumor suppressor Nit2 as
omega-amidase, an enzyme metabolically linked to glutamine and
asparagine transamination. Biochimie. 91:1072–1080. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jaisson S, Veiga-da-Cunha M and Van
Schaftingen E: Molecular identification of omega-amidase, the
enzyme that is functionally coupled with glutamine transaminases,
as the putative tumor suppressor Nit2. Biochimie. 91:1066–1071.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hersh LB: Rat liver-amidase. Kinetic
evidence for an acyl-enzyme intermediate. Biochemistry.
11:2251–2256. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hersh LB: Rat liver omega-amidase.
Purification and properties. Biochemistry. 10:2884–2891. 1971.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Duffy TE, Cooper AJ and Meister A:
Identification of alpha-ketoglutaramate in rat liver, kidney, and
brain. Relationship to glutamine transaminase and ω-amidase
activities. J Biol Chem. 249:7603–7606. 1974.PubMed/NCBI
|
|
33
|
Cooper AJ and Meister A: Isolation and
properties of a new glutamine transaminase from rat kidney. J Biol
Chem. 249:2554–2561. 1974.PubMed/NCBI
|
|
34
|
Cooper AJ: Asparagine transaminase from
rat liver. J Biol Chem. 252:2032–2038. 1977.PubMed/NCBI
|
|
35
|
Kuhara T, Inoue Y, Ohse M, Krasnikov BF
and Cooper AJ: Urinary 2-hydroxy-5-oxoproline, the lactam form of
α-ketoglutaramate, is markedly increased in urea cycle disorders.
Anal Bioanal Chem. 400:1843–1851. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Szeliga M and Obara-Michlewska M:
Glutamine in neoplastic cells: focus on the expression and roles of
glutaminases. Neurochem Int. 55:71–75. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang JB, Erickson JW, Fuji R, et al:
Targeting mitochondrial glutaminase activity inhibits oncogenic
transformation. Cancer Cell. 18:207–219. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Erickson JW and Cerione RA: Glutaminase: a
hot spot for regulation of cancer cell metabolism? Oncotarget.
1:734–740. 2010.
|
|
39
|
Krasnikov BF, Nostramo R, Pinto JT and
Cooper AJ: Assay and purification of omega-amidase/Nit2, a
ubiquitously expressed putative tumor suppressor, that catalyzes
the deamidation of the α-keto acid analogues of glutamine and
asparagine. Anal Biochem. 391:144–150. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Semba S, Han SY, Qin HR, et al: Biological
functions of mammalian Nit1, the counterpart of the invertebrate
NitFhit Rosetta stone protein, a possible tumor suppressor. J Biol
Chem. 281:28244–28253. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cutler SR and Somerville CR: Imaging plant
cell death: GFP-Nit1 aggregation marks an early step of wound and
herbicide induced cell death. BMC Plant Biol. 5:42005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Alnemri ES, Livingston DJ, Nicholson DW,
et al: Human ICE/CED-3 protease nomenclature. Cell. 87:1711996.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Walters J, Pop C, Scott FL, et al: A
constitutively active and uninhibitable caspase-3 zymogen
efficiently induces apoptosis. Biochem J. 424:335–345. 2009.
View Article : Google Scholar : PubMed/NCBI
|