Introduction
Vascular remodeling, characterized by endothelial
dysfunction, vascular smooth muscle cell (VSMC) proliferation and
by the excessive accumulation of extracellular matrix (ECM), plays
an important role in the development of various cardiovascular
diseases, including hypertension, atherosclerosis and restenosis
following balloon injury (1,2).
Angiotensin II (Ang II), the main effector peptide of the
renin-angiotensin system, plays a critical role in the development
of vascular remodeling. Ang II-induced VSMC proliferation increaseS
the production of profibrotic factors, such as connective tissue
growth factor (CTGF) (3). CTGF
has been demonstrated as a strong profibrogenic factor in vascular
remodeling through the regulation of VSMC proliferation and
migration (4). In addition, CTGF
has been shown to be overexpressed in human atherosclerotic lesions
(5) and in the aorta of of Ang
II-infused rats and cultured VSMCs (6).
The molecular mechanisms involved in Ang II-induced
vascular remodeling, include the activation of several
intracellular signaling systems, such as mitogen-activated protein
kinases (MAPKs), redox processes and RhoA/Rho kinase (ROCK)
(7). Accumulating evidence has
demonstrated that Rho/ROCK are critically involved in Ang
II-induced vascular remodeling (8,9).
Thus, the activation of Rho/ROCK represents one of the key signal
transduction pathways that mediate the pathophysiological functions
of Ang II. Howerver, the molecular mechanisms through which Ang II
activates the Rho/ROCK pathway have not yet been fully
elucidated.
Farnesyl pyrophosphate synthase (FPPS) plays an
important role in the mevalonate pathway. It catalyzes isopentenyl
pyrophosphate and dimethylallyl pyrophosphate to form
geranylgeranyl pyrophosphate (GGPP) and farnesyl pyrophosphate
(FPP), and regulates the small GTPase-binding proteins, including
Ras and Rho which have been implicated in the pathogenesis of
various cardiovascular diseases (10). Recent studies have indicated that
FPPS participates in the pathogenesis of cardiovascular remodeling
through the modulation of the RhoA/ROCK and MAPK pathways (11–15). In a recent previous study of ours,
we demonstrated that the inhibition of FPPS prevented
norepinephrine (NE)-induced fibrotic responses in VSMCs obtained
from spontaneously hypertensive rats (SHRs) and that the underlying
mechanisms involved the Ras kinase and p38 pathways (16). However, the role of FPPS in Ang
II-induced fibrotic responses and the related molecular mechanisms
have not yet been elucicated. Thus, the aim of the present study
was to investigate the molecular mechanisms involved in the Ang
II-induced production of CTGF in the VSMCs associated with vascular
remodeling. We wished to examine the hypothesis that FPPS may be a
potent regulator of the Ang II-RhoA/Rho-kinase (ROCK) pathway and
CTGF functions.
Materials and methods
Materials
Ang II, ibandronte sodium (Iban), FTI-276 (a
selective inhibitor of farnesyltransferase), GGTI-286 [a selective
inhibitor of geranylgeranyl transferase-I (GGTase I)], farnesol
(FOH), geranylgeraniol (GGOH), p38 MAPK inhibitor (SB203580),
extracellular signal-regulated kinase (ERK)1/2 inhibitor (PD98059)
and c-Jun N-terminal kinase (JNK) inhibitor (SP600125) were
purchased from Sigma (St. Louis, MO, USA). Dulbecco’s modified
Eagle’s medium (DMEM), penicillin-streptomycin and fetal bovine
serum (FBS) and all other cell culture reagents were purchased from
Gibco (Life Technologies, Carlsbad, CA, USA). Unless otherwise
stated, the remaining agents used for cell treatment were prepared
in sterile saline and diluted to a working concentration in DMEM.
Rabbit anti-CTGF polyclonal antibody (ab6992) and rabbit anti-FPPS
polyclonal antibody (ab153805) were obtained from Abcam (Cambridge,
UK). MAPK antibodies, including phosphorylated ERK1/2 (p-ERK1/2;
4370), ERK1/2 (4695), phosphorylated p38 (p-p38; 4511) and p38
(8690) were obtained from Cell Signaling Technology (Danvers, MA,
USA). Phosphorylated JNK1/2/3 (p-JNK1/2/3; 3893-1) and JNK1
(3496-1) antibodies were obtained from Epitomics (Burlingame, CA,
USA). The RhoA activation assay kit was purchased from
Cytoskeleton, Inc. (Denver, CO, USA). Enhanced chemiluminescence
(ECL) reagent was from Amersham International (Bucks, UK).
Specific pathogen-free male Sprague-Dawley (SD)
rats, 4–6 weeks of age, were obtained from the Experimental Animal
Center, Zhejiang Academy of Sciences (Hangzhou, China). All
procedures were performed in accordance with the revised 1996
National Institutes of Health Guidelines for the Care and Use of
Laboratory Animals (NIH Publication no. 85-23) and the Animal Care
Committee of Zhejiang Hospital, Zhejiang, China.
VSMC culture and treatment
Primary cultures of VSMCs were isolated from the
thoracic aortas of SD rats as previously described using the
collagenase method (6,16). The cells were incubated in a
humidified incubator at 37°C and 5% CO2. Cells between
passages 3 and 5 were used in all the experiments. Cell quiescence
was established by first transferring the cells to 9-well culture
plates at 80% confluence followed by maintenance in quiescence
medium consisting of DMEM with 0.1% FBS for 48 h. The quiescent
cells were treated in the presence or absence of Ang II at 0.1
μM for 24 h as previously described (6,7).
In some experiments, the cells were pre-incubated for 2 h with Iban
10 μM, Iban plus GGOH (30 μM), Iban plus FOH (30
μM), GGTI-286 (a selective inhibitor of GGTase I, 10
μM) or FTI-276 (a selective inhibitor of
farnesyltransferase, 10 μM). Furthermore, in some other
experiments, in order to inhibit the MAPK pathway, PD98059 (an
ERK1/2 inhibitor, 50 μM), SB203580 (a p38 MAPK inhibitor, 10
μM) and SP600125 (a JNK1/2/3 inhibitor, 10 μM) were
added to the cells 2 h prior to stimulation with Ang II. All the
agents mentioned above were maintained in quiescence medium.
Cell proliferation assay
VSMC proliferation was measured using a by cell
counting kit (CCK-8) (Dojindo Laboratories, Tokyo, Japan) as
previously described (16) and
according to the manufacturer’s instructions.
The VSMCs were transferred to a 96-well plate at a
density of 1×104 cells/well. After 24 h of treatment, 10
μl CCK-8 solution were added to each well followed by
incubation at 37°C for an additional 2 h. The absorbance of each
well at a wavelength of 450 nm was then measured using the Bio-Rad
680 micro-plate reader (Bio-Rad, Hercules, CA, USA). Cell
proliferation was expressed as the optical density.
Western blot analysis
Cellular protein was isolated by homogenization with
cell lysis buffer. The protein concentration was then determined
using the BCA method. Equal amounts of protein were denatured and
subjected to 10% sodium dodecyl sulphate-polyacrylamide gel
electrophoresis (SDS-PAGE). The membranes were incubated with
primary antibodies (against CTGF, FPPS, and phosphorylated and
total ERK1/2, p38 and JNK) overnight at 4°C. Following incubation
with secondary antibodies (HRP-labeled goat anti-rabbit: Cat. no.
GAR007; MultiSciences Biotech Co., Ltd, Hangzhou, China) for 1 h at
room temperature and washing with TBST, the proteins were detected
using an ECL Plus system (Amersham Bioscience, Piscataway, NJ,
USA). To ensure equal protein loading, glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) was used as an internal control. The results
of CTGF production were obtained from densitometric analysis and
expressed as the ratio CTGF/GAPDH as n-fold over the control.
RhoA activation assay
RhoA activity was determined using a pull-down assay
according to the manufacturer’s instructions. In brief, a BCA
protein assay was performed to equalize the total protein
concentration. RBD-bound Rho from the cell lysates was ‘pulled
down’ using agarose-conjugated Rhotekin-RBD and detected by western
blot analysis using specific anti-RhoA antibody [Cat. no. ARH03,
contained in the Rho Activation assay kit (BK036)]. A total of 20
μg of total cell lysate per sample was used to detect the
total amount of RhoA expression. In addition, GAPDH protein was
used as an endogenous control.
Statistical analysis
The results are presented as the means ± SEM and
expressed as the n-fold increase over the controls. One-way
analysis of variance (ANOVA) followed by the Bonferroni post hoc
test were used to determine significant differences among multiple
groups. The results were considered statistically significant at a
value of P<0.05.
Results
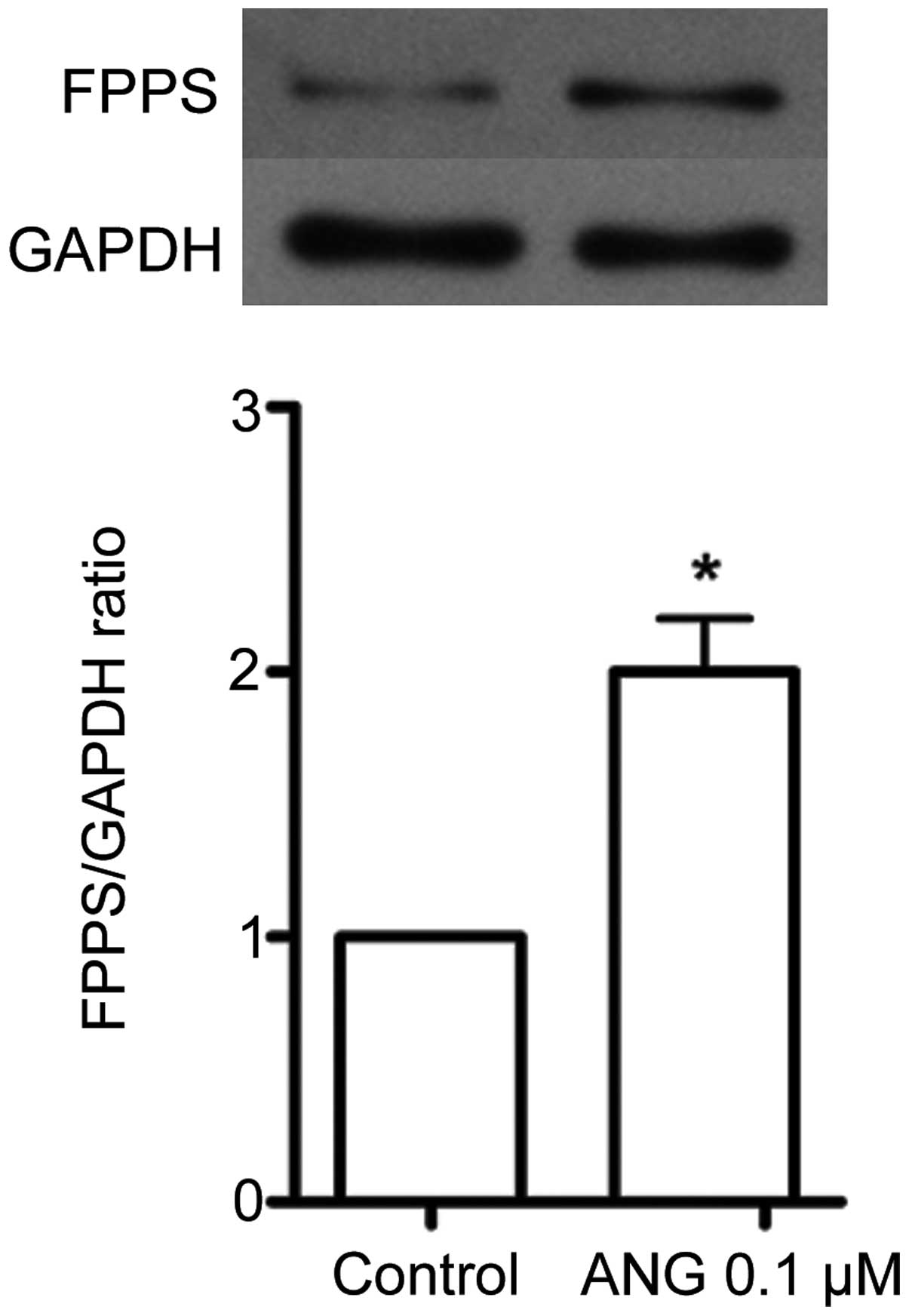
Effects of Ang II on FPPS expression in
cultured VSMCs
In the growth-arrested VSMCs, incubation with Ang II
(0.1 μM) for 24 h increased the protein expression of FPPS
(P<0.05; Fig. 1).
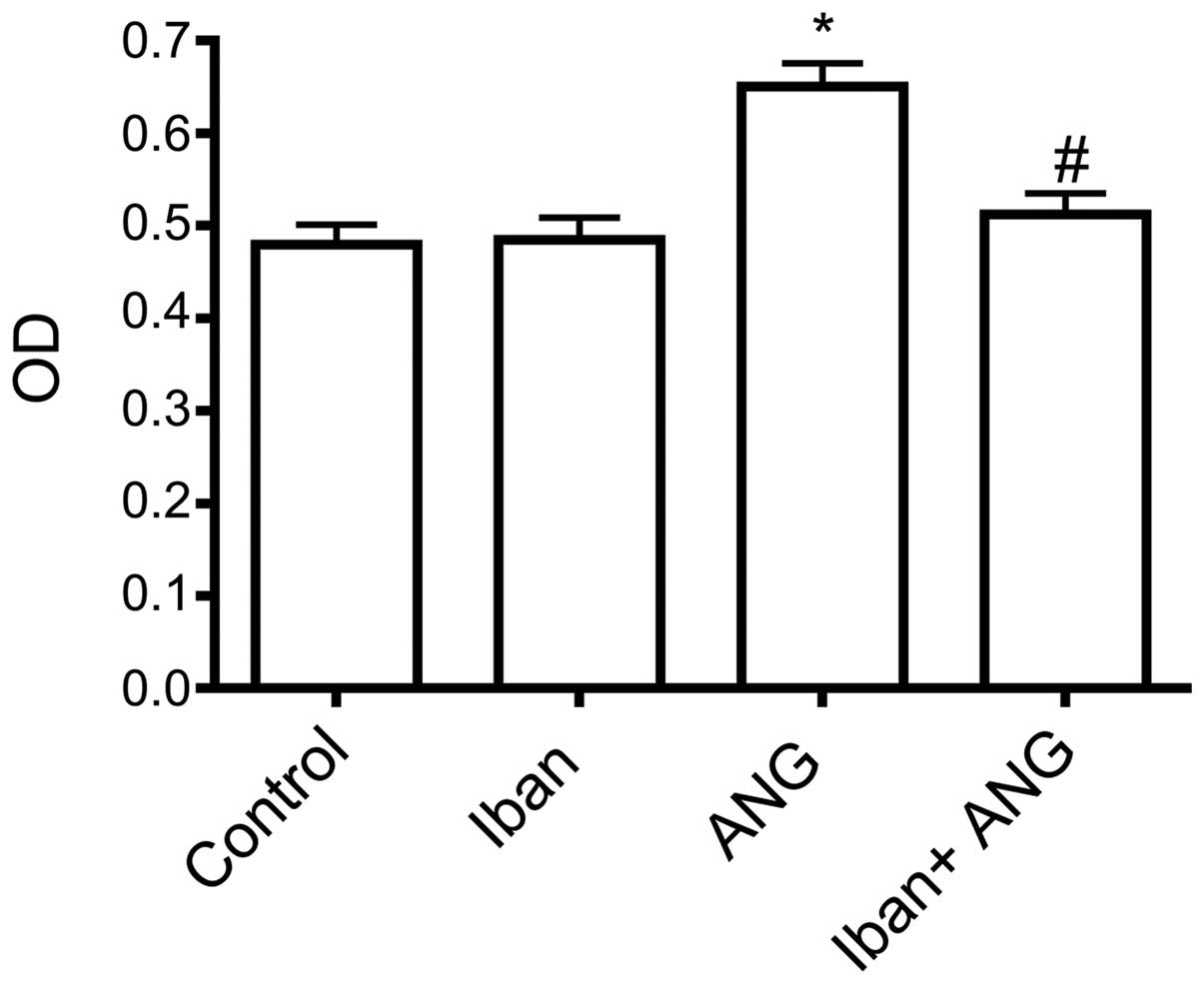
Inhibition of FPPS decreases the fibrotic
responses elicited by Ang II in cultured VSMCs
To investigate the potential role of FPPS in the
fibrotic responses elicited by Ang II, Iban, an FPPS inhibitor, was
tested using the Ang II-stimulated VSMCs. Pre-incubation with 10
μM Iban for 2 h reduced the Ang II-induced increase in cell
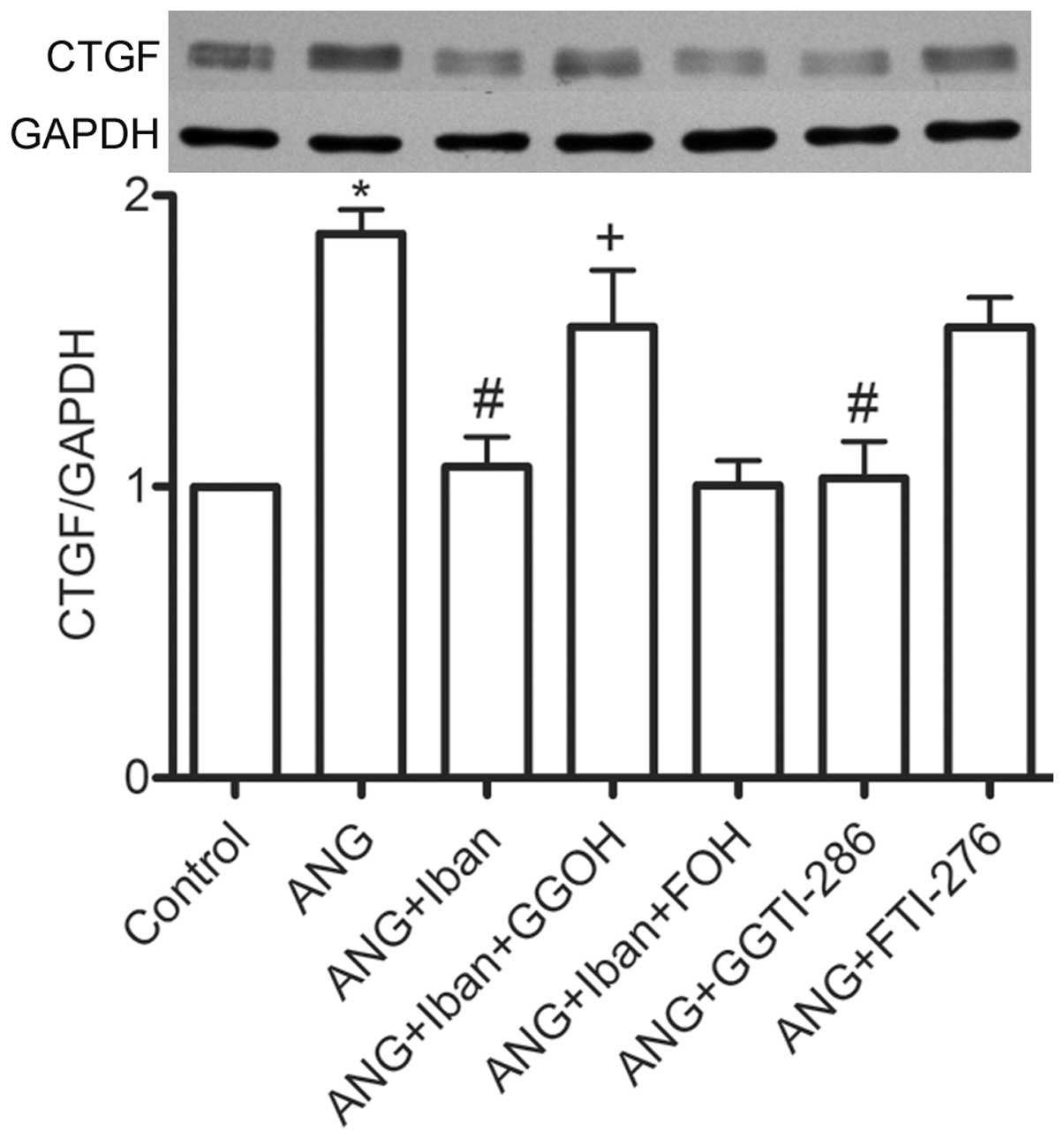
proliferation and CTGF protein expression (Figs. 2 and 3). The addition of GGOH, but not that of
FOH, partly reversed the inhibitory effects of Iban on the
expression of CTGF (Fig. 3). In
addition, pre-treatment with GGTI-286, but not with FTI-276,
substantially reduced the Ang II-induced incraase in the expression
of CTGF (Fig. 3). These results
suggest that Rho signaling is involved in this process.
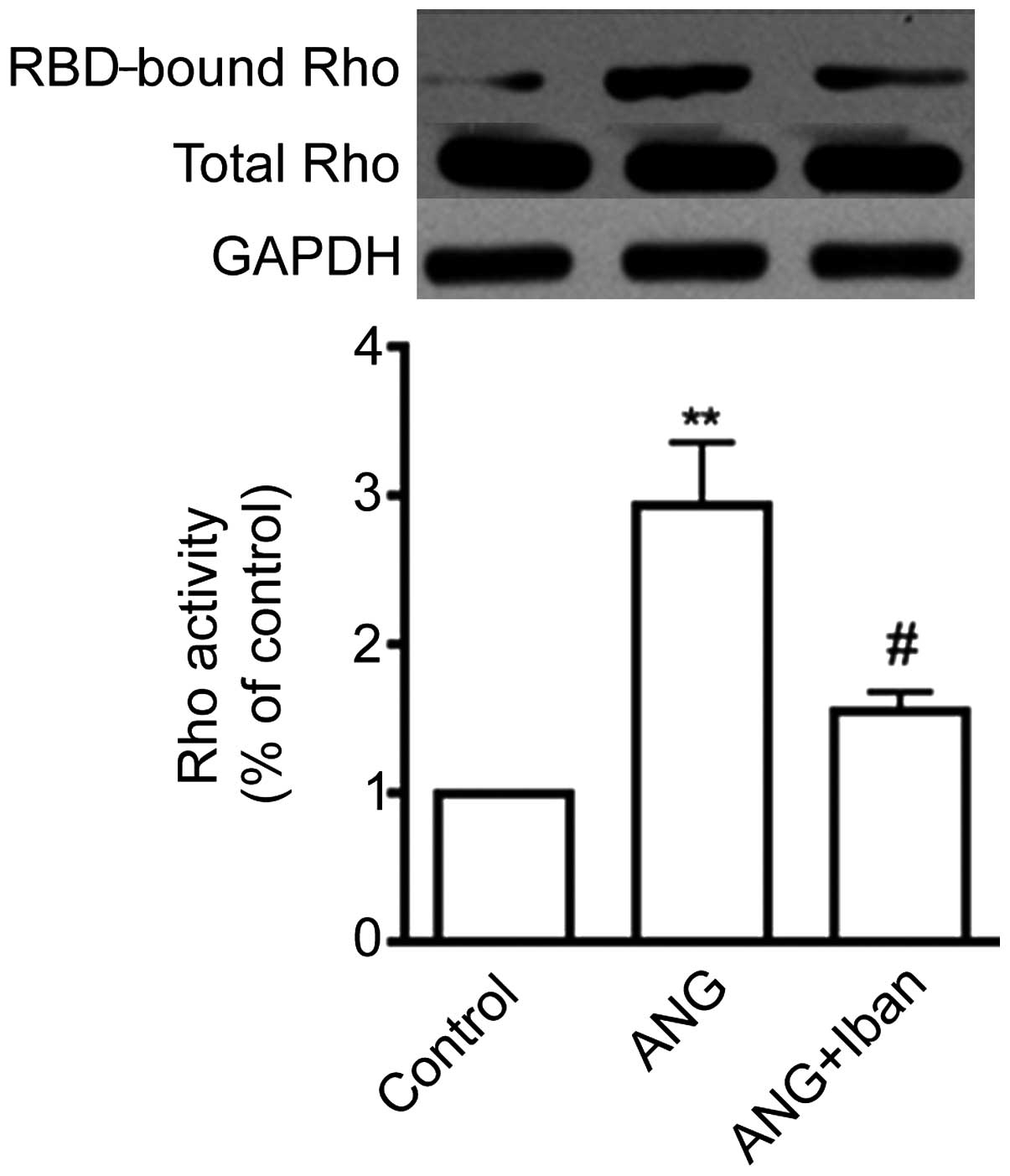
Inhibition of FPPS modulates the Ang
II-induced RhoA activation in cultured VSMCs
Stimulation with 0.1 μM Ang II for 15 min
increased the GTP-bound RhoA levels, as shown by pull-down assays
(Fig. 4), which is in accordance
with the findings of a previous study (7). We then wished to determine whether
Iban inhibits the activation of RhoA induced by Ang II.
Pre-treatment with 10 μM Iban for 24 h diminished the Ang
II-induced activation of RhoA, as shown by pull-down assays
(Fig. 4). These data demonstrate
that Iban inhibits RhoA activation induced by Ang II.
Inhibition of FPPS modulates the Ang
II-induced MAPK activation in cultured VSMCs
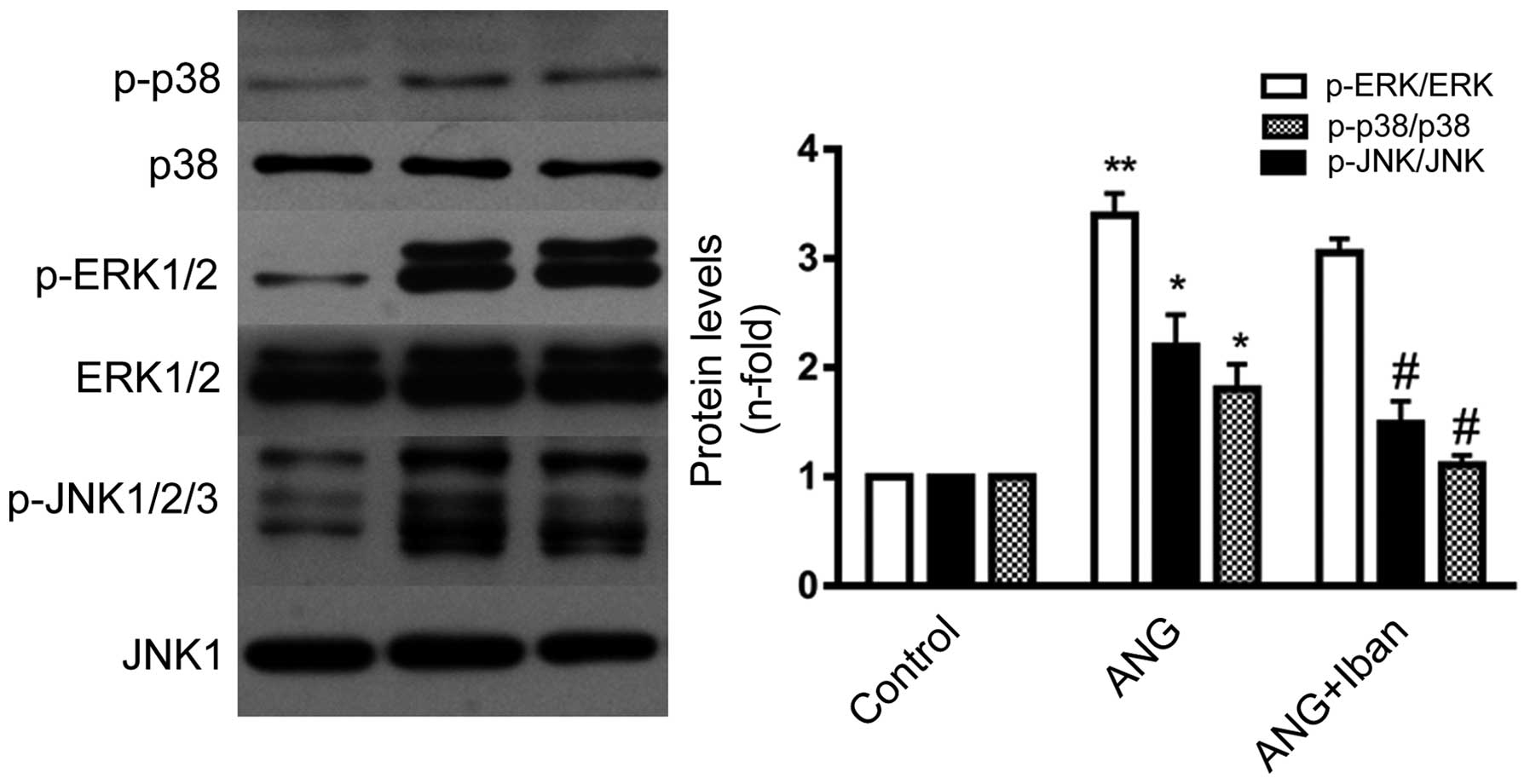
Treatment with Ang II 0.1 μM for 15 min
triggered the phosphorylation of p38, JNK and ERK1/2 in the VSMCs,
which is in accordance with the findings of a previous study
(7). To investigate whether the
activation of all 3 MAPKs is involved in the molecular mechanisms
underlying the ability of Iban to suppress the Ang II-induced
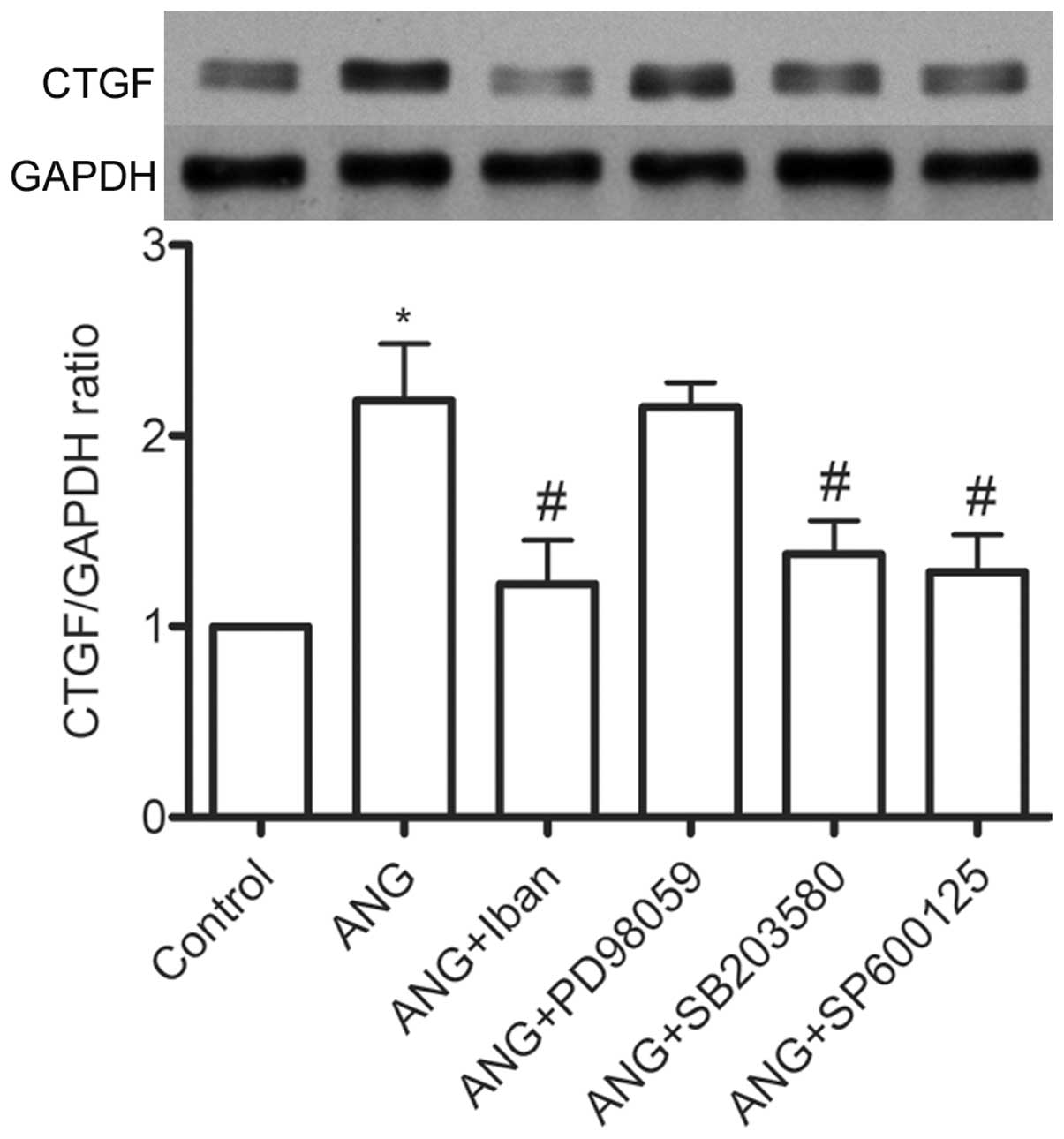
increase in the expression of CTGF, the VSMCs were treated with
specific inhibitors of MAPKs (SB203580 for p38, PD98059 for ERK1/2
and SP600125 for JNK) pior to stimulation with Ang II. Both the p38
inhibitor and JNK inhibitor diminished the Ang II-induced
production of CTGF (Fig. 5).
Furthermore, pre-incubation with Iban inhibited the Ang II-induced
activation of p38 and JNK, but not that of ERK1/2 (Fig. 6), suggesting that the activation
of p38 and JNK is involved in the downregulation of CTGF by Iban.
These data indicate that the inhibition of FPPS modulates the Ang
II-induced production of CTGF through the regulation of the p38
MAPK and JNK pathways in cultured VSMCs.
Discussion
In the present study, we demonstrated that FPPS
expression is induced by Ang II in cultured VSMCs from SD rats.
Iban, an FPPS inhibitor, inhibited the Ang II-induced cell
proliferation and the production of CTGF in the cultured VSMCs. The
underlying mechanisms involved the inhibition of RhoA signaling and
the modulation of the p38 MAPK and JNK pathways.
Through the inhibition of osteoclastic activity and
bone resorption, nitrogen-containing bisphosphonates (N-BPs)
including alendronate, Iban and zoledronate are commonly used for
the treatment of bone-related diseases in clinical practice
(17,18). However, extensive research has
focused on their potential effects on cardiovascular diseases. Due
to their inhibitory effects on the mevalonate pathway and
particularly FPPS, they now act as a tool used to investigate the
role of FPPS in various diseases (19,20). FPPS has been implicated in the
development of various cardiovascular diseases, including
myocardial and vascular remodeling and endothelial dysfunction in
SHRs (12–16). The inhibition of FPPS by
alendronate or its knockdown by RNA interference has been shown to
prevent cardiac hypertrophy and fibrosis both in vivo and
in vitro (12–14). Moreover, the cardiac-specific
overexpression of FPPS has been shown to induce cardiac hypertrophy
and heart failure in mice (15).
In addition, in a recent study of ours, we demonstrated that the
inhibition of FPPS prevented NE-induced fibrotic responses,
including cell proliferation, the hydroxyproline content and CTGF
protein expression in SHR-VSMCs (16), indicating that FPPS may function
as a potent regulator of vascular remodeling.
The exaggerated VSMC activities and excessive ECM
protein accumulation induced by Ang II are thought to be key to the
devolepment of vascular remodeling (3–9). A
number of pharmacological agents, such as anti-thrombotics,
antiplatelet agents, angiotensin-converting enzyme inhibitors, as
well as mechanical and cellular approaches have been used in an
attempt to attenuate this pathophysiological process (21,22). A growing numbers of studies in the
literature have demonstrated that bisphosphonates may play an
important role in inhibiting the development of atherosclerosis and
neointimal hyperplasia in animal models. These effects are thought
to be mediated by the transient systemic inactivation of monocytes
and macrophages (23,24). However, in a balloon-injured rat
carotid artery model, both the systemic and local delivery of
zoledronate was shown to prevent intimal hyperplasia (25). Additionly, in a rabbit carotid
anastomosis model, zoledronic acid (ZA), a third generation of
N-BPs, was demonstrated to inhibit neointimal hyperplasia and
decrease VSMC intensity, suggesting that it may have a direct
inhibitory effect on VSMCs (26).
Indeed, etidronate, another bisphosphonate, has been reported to
exert an inhibitory effect on the growth of VSMCs from SHRs
(27). Similarly, in cultured rat
VSMCs, ZA has also been reported to inhibit the proliferation,
adhesion and migration of VSMCs without the induction of necrosis
or apoptosis (28). Recently, ZA
was reported to inhibit the growth of stimulated human arotic
smooth muscle cells, as well as their proliferation adhesion and
migration, but had no effect on quiescent cells. The addition of
GGOH significantly reversed the ZA-mediated alteration in cellular
viability and RAP1A/B prenylation, suggesting that ZA modulates the
mevalonate pathway and inhibits the prenylation of GTPase binding
proteins (29). Taken together,
the above data point out that BPs or N-BPs may play an important
role in vascular remodeling by modulating cell signaling, including
the prenylation of small signaling proteins (e.g., Ras, Rac, Rab
and Rho) through the mevalonate pathway. However, the underlying
mechanisms through which BPs or N-BPs inhibit VSMC activity and
prevent intimal hyperplasia in animal models induced by FPPS were
not elucidated in the above-mentioned studies.
In our previous study, we reported that Iban
inhibited NE-induced fibrotic responses in SHR-VSMCs through the
modulation of the Ras kinase and p38 pathways (16). In the present study, we further
demonstrated that Iban also inhibits fibrotic responses in VSMCs
induced by Ang II. As shown in Fig.
1 and in our previous study (16), Iban alone did not influence cell
proliferation and CTGF expression in quiescent VSMCs, but markedly
reduced the Ang II-induced increase in cell proliferation and CTGF
expression (Figs. 2 and 3). Besides, Ang II upregulated FPPS in
the cultured VSMCs. To determine whether the anti-fibrotic effects
of Iban depend on the modulation of FPPS, analogues or antagonists
of the mevalonate pathway were used in this study. The results
revealed that addition of GGOH, but not that of FOH, partly
reversed the anti-fibrotic effects of Iban (Fig. 3). In addition, GGTI-286, a
specific inhibitor of GGTase I, but not FTI-276 (a selective
inhibitor of farnesyltransferase), mimicked the anti-fibrotic
effects of Iban (Fig. 3). Since
RhoA is a geranylgeranylated protein (30), our results suggest that RhoA
signaling is involved in this process.
In a previous study, it was clearly demonstrated
that pretreatment with RhoA and ROCK inhibitors diminished the Ang
II-induced overexpression of CTGF, indicating that Ang II increases
the production of CTGF in VSMCs through the RhoA/ROCK pathway
(7). In the present study, the
inhibition of FPPS with Iban diminished the Ang II-induced RhoA
activation in cultured VSMCs, suggesting that FPPS may be involved
in the modulation of Ang II-induced CTGF production through the
RhoA/ROCK pathway. This is consistent with the results of other
studies on the effects of in vivo and in vitro
alendronate treatment with hypertrophic response induced by Ang II
in cultured neonatal ventricular myocytes and animal models
(12,13). However, the levels of isoprenoid
intermediates, such as FOH or GGOH were not measured after the
suppression of FPPS. The knockdown of FPPS by RNA interference in
VSMCs is another critical issue to be confirmed in view of the role
of FPPS in Ang II-mediated vascular remodeling. Besides, the in
vivo effect of the vascular-specific interference of FPPS in
vascular remodeling needs to be validated in animal models. These
limitations require further investigation.
It is well known that MAPKs are a widely distributed
group of enzymes. The consisting of 3 isoforms (ERK, p38 and JNK)
and play an important role in Ang II-mediated vascular remodeling
(7,8,31).
To determine whether Iban modulates the Ang II-induced
overexpression of CTGF by altering the activity of the MAPK
pathway, VSMCs were pre-treated with the specific inhibitor of the
MAPK pathway prior to stimulation with Ang II. As shown in Fig. 5, both the JNK and p38 MAPK
inhibitor diminished the Ang II-induced production of CTGF. This is
consistent with the findings of a pregvious study on the effects of
HMG-CoA reductase inhibitors (statins) on the modulation of Ang
II-mediated vascular responses (7). Furthermore, as previously described
(7) and shown in Fig. 6 in the present study, treatment
with Ang II triggered the phosphorylation of all 3 MAPKs. However,
only the phosphorylation of JNK and p38 was diminished by treatment
with Iban. These data indicate that JNK and p38 MAPK may be
involved in the Iban-mediated inhibition of CTGF overexpression
induced by Ang II in VSMCs.
In conclusion, in this study, we provide evidence
that FPPS expression is elevated in Ang II-stimulated VSMCs. The
ihibibition of FPPS by Iban attenuates the Ang II-induced increase
in cell proliferation and CTGF expression in VSMCs, and the
underlying mechanisms, at least in part, involve the modulation of
RhoA activity, and the p38 and JNK pathways.
Acknowledgments
This study was supported by the grants from the
Natural Science Foundation of Zhejiang province (Project for Young
Scientists, no. LQ13H020004).
Abbreviations:
|
VSMCs
|
vascular smooth muscle cells
|
|
SHR
|
spontaneously hypertensive rats
|
|
Ang II
|
angiotensin II
|
|
FPP
|
farnesyl pyrophosphate
|
|
GGPP
|
geranylgeranyl pyrophosphate
|
|
GGOH
|
geranylgeraniol
|
|
FOH
|
farnesol
|
|
Iban
|
ibandronate sodium
|
|
CTGF
|
connective tissue growth factor
|
|
FPPS
|
farnesyl pyrophosphate synthase
|
|
ECM
|
extracellular matrix
|
|
MAPK
|
mitogen-activated protein kinase
|
|
DMEM
|
Dulbecco’s modified Eagle’s medium
|
|
FBS
|
fetal bovine serum
|
|
SDS-PAGE
|
sodium dodecyl sulphate-polyacrylamide
gel electrophoresis
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
JNK
|
c-Jun N-terminal kinase
|
|
GAPDH
|
glyceraldehyde-3-phosphate
dehydrogenase
|
References
|
1
|
Rizzoni D, Muiesan ML, Porteri E, De
Ciuceis C, Boari GE, Salvetti M, Paini A and Rosei EA: Vascular
remodeling, macro-and microvessels: Therapeutic implications. Blood
Press. 18:242–246. 2009. View Article : Google Scholar
|
|
2
|
Inoue T and Node K: Molecular basis of
restenosis and novel issues of drug-eluting stents. Circ J.
73:615–621. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Touyz RM and Schiffrin EL: Signal
transduction mechanisms mediating the physiological and
pathophysiological actions of angiotensin II in vascular smooth
muscle cells. Pharmacol Rev. 52:639–672. 2000.PubMed/NCBI
|
|
4
|
Fan WH, Pech M and Karnovsky MJ:
Connective tissue growth factor (CTGF) stimulates vascular smooth
muscle cell growth and migration in vitro. Eur J Cell Biol.
79:915–923. 2000. View Article : Google Scholar
|
|
5
|
Oemar BS, Werner A, Garnier JM, Do DD,
Godoy N, Nauck M, März W, Rupp J, Pech M and Lüscher TF: Human
connective tissue growth factor is expressed in advanced
atherosclerotic lesions. Circulation. 95:831–839. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rupérez M, Lorenzo O, Blanco-Colio LM,
Esteban V, Egido J and Ruiz-Ortega M: Connective tissue growth
factor is a mediator of angiotensin II-induced fibrosis.
Circulation. 108:1499–1505. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rupérez M, Rodrigues-Díez R, Blanco-Colio
LM, Sánchez-López E, Rodríguez-Vita J, Esteban V, Carvajal G, Plaza
JJ, Egido J and Ruiz-Ortega M: HMG-CoA reductase inhibitors
decrease angiotensin II-induced vascular fibrosis: Role of
RhoA/ROCK and MAPK pathways. Hypertension. 50:377–383. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ohtsu H, Mifune M, Frank GD, Saito S,
Inagami T, Kim-Mitsuyama S, Takuwa Y, Sasaki T, Rothstein JD,
Suzuki H, et al: Signal-crosstalk between Rho/ROCK and c-Jun
NH2-terminal kinase mediates migration of vascular smooth muscle
cells stimulated by angiotensin II. Arterioscler Thromb Vasc Biol.
25:1831–1836. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kobayashi N, Nakano S, Mita S, Kobayashi
T, Honda T, Tsubokou Y and Matsuoka H: Involvement of Rho-kinase
pathway for angiotensin II-induced plasminogen activator
inhibitor-1 gene expression and cardiovascular remodeling in
hypertensive rats. J Pharmacol Exp Ther. 301:459–466. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Roskoski R Jr: Protein prenylation: A
pivotal posttranslational process. Biochem Biophys Res Commun.
303:1–7. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li L, Hu SJ, Dong HT, Kang L, Chen NY and
Fang YQ: Alterations in gene expression of series key enzymes in
mevalonic acid pathway detected by RNA array in spontaneously
hypertensive rats. Chin J Pathophysiol. 24:54–59. 2008.
|
|
12
|
Ye Y, Hu SJ and Li L: Inhibition of
farnesylpyrophosphate synthase prevents angiotensin II-induced
hypertrophic responses in rat neonatal cardiomyocytes: Involvement
of the RhoA/Rho kinase pathway. FEBS Lett. 583:2997–3003. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ye Y, Mou Y, Bai B, Li L, Chen GP and Hu
SJ: Knockdown of farnesylpyrophosphate synthase prevents
angiotensin II-mediated cardiac hypertrophy. Int J Biochem Cell
Biol. 42:2056–2064. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li L, Chen GP, Yang Y, Ye Y, Yao L and Hu
SJ: Chronic inhibition of farnesyl pyrophosphate synthase
attenuates cardiac hypertrophy and fibrosis in spontaneously
hypertensive rats. Biochem Pharmacol. 79:399–406. 2010. View Article : Google Scholar
|
|
15
|
Yang J, Mou Y, Wu T, Ye Y, Jiang JC, Zhao
CZ, Zhu HH, Du CQ, Zhou L and Hu SJ: Cardiac-specific
overexpression of farnesyl pyrophosphate synthase induces cardiac
hypertrophy and dysfunction in mice. Cardiovasc Res. 97:490–499.
2013. View Article : Google Scholar
|
|
16
|
Du CQ, Yang L, Yang J, Han J, Hu XS, Wu T
and Hu SJ: Inhibition of farnesyl pyrophosphate synthase prevents
norepinephrine-induced fibrotic responses in vascular smooth muscle
cells from spontaneously hypertensive rats. Hypertens Res.
37:26–34. 2014. View Article : Google Scholar
|
|
17
|
Owens JM, Fuller K and Chambers TJ:
Osteoclast activation: Potent inhibition by the bisphosphonate
alendronate through a nonresorptive mechanism. J Cell Physiol.
172:79–86. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Papapoulos SE: Ibandronate: A potent new
bisphosphonate in the management of postmenopausal osteoporosis.
Int J Clin Pract. 57:417–422. 2003.PubMed/NCBI
|
|
19
|
van Beek E, Pieterman E, Cohen L, Löwik C
and Papapoulos S: Farnesyl pyrophosphate synthase is the molecular
target of nitrogen-containing bisphosphonates. Biochem Biophys Res
Commun. 264:108–111. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kavanagh KL, Guo K, Dunford JE, Wu X,
Knapp S, Ebetino FH, Rogers MJ, Russell RG and Oppermann U: The
molecular mechanism of nitrogen-containing bisphosphonates as
antiosteoporosis drugs. Proc Natl Acad Sci USA. 103:7829–7834.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lefkovits J and Topol EJ: Pharmacological
approaches for the prevention of restenosis after percutaneous
coronary intervention. Prog Cardiovasc Dis. 40:141–158. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kiernan TJ, Yan BP, Cruz-Gonzalez I,
Cubeddu RJ, Caldera A, Kiernan GD and Gupta V: Pharmacological and
cellular therapies to prevent restenosis after percutaneous
transluminal angioplasty and stenting. Cardiovasc Hematol Agents
Med Chem. 6:116–124. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ylitalo R, Oksala O, Ylä-Herttuala S and
Ylitalo P: Effects of clodronate (dichloromethylene bisphosphonate)
on the development of experimental atherosclerosis in rabbits. J
Lab Clin Med. 123:769–776. 1994.PubMed/NCBI
|
|
24
|
Danenberg HD, Golomb G, Groothuis A, Gao
J, Epstein H, Swaminathan RV, Seifert P and Edelman ER: Liposomal
alendronate inhibits systemic innate immunity and reduces in-stent
neointimal hyperplasia in rabbits. Circulation. 108:2798–2804.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu L, Zhu L, Shi WH, Yu B and Cai D:
Zoledronate inhibits intimal hyperplasia in balloon-injured rat
carotid artery. Eur J Vasc Endovasc Surg. 41:288–293. 2011.
View Article : Google Scholar
|
|
26
|
Güzeloğlu M, Gül M, Reel B, Yürekli I,
Aykut K and Hazan E: The effects of zoledronic acid on neointimal
hyperplasia: A rabbit carotid anastomosis model. Anadolu Kardiyol
Derg. 11:93–100. 2011. View Article : Google Scholar
|
|
27
|
Su JZ, Fukuda N, Kishioka H, Hu WY and
Kanmatsuse K: Etidronate influences growth and phenotype of rat
vascular smooth muscle cells. Pharmacol Res. 46:7–13. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu L, Zhu L, Shi WH, Zhang J, Ma D and Yu
B: Zoledronate inhibits the proliferation, adhesion and migration
of vascular smooth muscle cells. Eur J Pharmacol. 602:124–131.
2009. View Article : Google Scholar
|
|
29
|
Albadawi H, Haurani MJ, Oklu R, Trubiano
JP, Laub PJ, Yoo HJ and Watkins MT: Differential effect of
zoledronic acid on human vascular smooth muscle cells. J Surg Res.
182:339–346. 2013. View Article : Google Scholar :
|
|
30
|
Van Aelst L and D’Souza-Schorey C: Rho
GTPases and signaling networks. Genes Dev. 11:2295–2322. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Miyata Y and Nishida E: Distantly related
cousins of MAP kinase: Biochemical properties and possible
physiological functions. Biochem Biophys Res Commun. 266:291–295.
1999. View Article : Google Scholar : PubMed/NCBI
|