Introduction
Transient forebrain ischemia induced by the
deprivation of brain blood flow leads to irreversible brain damage
in specific vulnerable areas of the brain (1–3).
Delayed neuronal death occurs several days after
ischemia-reperfusion injury in the stratum pyramidale (SP) of
region I of hippocampus proper (CA1) (4–7).
One of the mechanisms of delayed neuronal death is possibly
associated with a number of biochemical events triggered by
glutamate excitotoxicity (8–10).
Ischemic preconditioning (IPC) represents an
important adaptation of the central nervous system (CNS) to
sublethal ischemia, which can increase the ischemic tolerance of
the CNS to a subsequent longer or lethal period of ischemia
(11,12). IPC induces the expression of
diverse genes involved in cytoprotection and, in turn, encodes
proteins that lead to the enhancement of resistance to cerebral
ischemia (13). This phenomenon
is termed cerebral ‘ischemic tolerance’, although the basic
mechanisms underlying cerebral ischemic tolerance are not yet fully
understood (14).
Kynurenic acid (KYNA) is an endogenous metabolite of
the kynurenine pathway for tryptophan degradation and is produced
from its precursor L-kynurenine (KYN) by the enzyme,
kynurenine-aminotransferase (15). The formation of KYNA in
particular, has been shown to play an important role in the CNS at
physiological concentrations, as this metabolite selectively acts
as an antagonist of N-methyl-D-aspartate (NMDA) receptors by
blocking the co-agonist site for glycine (16,17), as well as a non-competitive
inhibitor of α7-nicotinic receptors for acetylcholine (18). Thus, KYNA protects neuronal cells
from excitotoxicity evoked by the overactivation of NMDA receptors.
Furthermore, KYNA plays a versatile role in pathological states,
including inflammatory (19),
vascular (20) and antioxidant
(21) processes. Indeed, the
exogenous administration of KYNA or its enhanced endogenous
synthesis has been proven to be a robust neuroprotectant in animal
models of forebrain ischemia (22–25). However, research on KYNA as a
neuroprotective agent is rather limited, as it hardly crosses the
blood-brain barrier (22,26).
To the best of our knowledge, the expression
patterns of endogenous KYNA immunoreactivity in the IPC-mediated
hippocampus following transient forebrain ischemia have not been
studied thus far. Therefore, the present study was carried out to
investigate the temporal changes and specific roles of endogenous
KYNA in the IPC-induced neuroprotective effects against transient
ischemic damage in the hippocampus of gerbils, which are considered
to be a good animal model for studying transient cerebral ischemia
(27,28).
Materials and methods
Experimental animals
We used the progeny of male Mongolian gerbils
(Meriones unguiculatus) obtained from the Experimental
Animal Center, Kangwon National University, Chuncheon, Korea. The
gerbils were used at 24 weeks of age (body weight, 65–75 g) and
were maintained under pathogen-free conditions with a temperature
of 23°C and a humidity of 60%. All the experimental protocols were
approved by the Institutional Animal Care and Use Committee (IACUC)
at Kangwon National University and adhered to guidelines that are
in compliance with the current international laws and policies
(Guide for the Care and Use of Laboratory Animals, The National
Academies Press, 8th edition, 2011).
Animal groups and induction of transient
forebrain ischemia
The animals were divided into 4 groups (n=7 at each
time point): i) group 1, the sham-operated group: the bilateral
common carotid arteries were exposed and the animals were not
subjected to ischemia (sham-operation); ii) group 2, the
ischemia-operated group: the animals were subjected to 5 min of
transient forebrain ischemia; iii) group 3, the IPC + sham-operated
group: the animals were subjected to 2 min of sublethal ischemia
prior to sham-operation; and iv) group 4, the IPC +
ischemia-operated group: the animals were subjected to 2 min of
sublethal ischemia prior to 5 min of transient ischemia. The IPC
paradigm has been proven to be very effective at protecting neurons
against ischemic damage in this ischemic model (29). The animals in groups 2 and 4 were
allowed to recover for different periods of time (sham, 1 day, 2
days and 5 days), as pyramidal neurons in the hippocampal CA1
region survive for 3 days and then begin to die 4–5 days following
ischemia-reperfusion.
Transient forebrain ischemia was developed according
to the method described in our previous study (30). In brief, the experimental animals
were anesthetized with a mixture of 2.5% isoflurane in 33% oxygen
and 67% nitrous oxide. Ischemia was induced by the occlusion of
arteries with non-traumatic aneurysm clips (Yasargil FE 723K;
Aesculap, Tuttlingen, Germany). After 2 or 5 min of occlusion, the
aneurysm clips were removed from the common carotid arteries. The
body (rectal) temperature under free-regulating or normothermic
(37±0.5°C) conditions was monitored with a rectal temperature probe
(TR-100; Fine Science Tools, Foster City, CA, USA) and maintained
using a thermometric blanket before, during and after surgery until
the animals had completely recovered from the anesthesia.
Thereafter, the animals were kept in a thermal incubator
(temperature, 23°C; humidity, 60%) (Mirae Medical Industry, Seoul,
Korea) to maintain the body temperature of the animals until they
were sacrificed (as described below).
Tissue processing for histological
analysis
For histological analysis, the gerbils (n=7 at each
time point) were deeply anesthetized with pentobarbital sodium and
perfused through the left ventricle with 0.1 M phosphate-buffered
saline (PBS, pH 7.4) followed by 4% paraformaldehyde in 0.1 M
phosphate-buffer (PB, pH 7.4). The brains were removed and
post-fixed in the same fixative for 6 h. The brain tissues were
embedded in tissue-freezing medium and serially sectioned into
30-μm coronal sections using a cryostat (Leica, Wetzlar,
Germany).
Cresyl violet (CV) staining and
Fluoro-Jade B (F-J B) histofluorescence
To investigate the delayed neuronal damage in the
hippocampus following ischemia-reperfusion, CV staining and F-J B
histofluorescence were performed as previously described (31). In brief, the sections were stained
with 1.0% (w/v) CV acetate (Sigma-Aldrich, St. Louis, MO, USA) and
dehydrated. They were then mounted with Canada balsam (Kanto
Chemical Co., Tokyo, Japan). For F-J B histofluorescence, the
sections were immersed in a 0.0004% F-J B staining solution
(Histochem Inc., Jefferson, AR, USA). After washing, the sections
were examined using an epifluorescent microscope (Carl Zeiss,
Göttingen, Germany) with blue (450–490 nm) excitation light and a
barrier filter.
Immunohistochemistry for neuronal nuclei
(NeuN) and KYNA
For immunohistochemical staining, the sections were
prepared out according to the method described in our previous
study (31). The brain sections
were blocked with 10% normal goat serum in 0.05 M PBS followed by
staining with primary mouse anti-NeuN antibody (a neuron-specific
soluble nuclear antigen) (diluted 1:1,000; MAB377; Chemicon
International, Temecula, CA, USA) and rabbit anti-KYNA antibody
(diluted 1:200; ab37105; Abcam, Cambridge, MA, USA) overnight at
4°C. The sections were then incubated with the secondary antibodies
(BA-9200, BA-1000; Vector Laboratories Inc., Burlingame, CA, USA)
and were developed using the Vectastain ABC system (Vector
Laboratories Inc.). Subsequently, they were visualized with
3,3′-diaminobenzidine in 0.1 M Tris-HCl buffer. In order to
establish the specificity of the immunostaining, a negative control
test was carried out with pre-immune serum instead of primary
antibody. The negative control resulted in the absence of
immunoreactivity in any structures.
Western blot analysis
To obtain the accurate data for changes in the
protein levels of KYNA in the hippocampal CA1 region following
transient forebrain ischemia abd IPC, the animals (n=7 at each time
point) were sacrificed at designated time points (sham, 1, 2 and 5
days) following ischemia-reperfusion and the brain tissues were
used for western blot analysis. As previously described (31), after the animals were sacrificed,
the brain tissues were removed and transversely cut into sections
with a thickness of 400 μm using a vibratome (Leica), and
the hippocampal CA1 region was dissected with a surgical blade,
removing the hippocampus. The tissues were homogenized in 50 mM PBS
(pH 7.4) containing 0.1 mM ethylene glycolbis(2-aminoethyl
Ether)-N,N,N’,N’ tetraacetic acid (EGTA; pH 8.0), 0.2% Nonidet
P-40, 10 mM ethylendiaminetetraacetic acid (EDTA; pH 8.0), 15 mM
sodium pyrophosphate, 100 mM β-glycerophosphate, 50 mM NaF, 150 mM
NaCl, 2 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride
(PMSF) and 1 mM dithiothreitol (DTT). Following centrifugation (at
12,000 rpm for 10 min), the protein level was determined in the
supernatants using a Micro BCA protein assay kit with bovine serum
albumin as the standard (Pierce Chemical Co., Rockford, IL, USA).
Aliquots containing 20 μg of total protein were boiled in
loading buffer containing 150 mM Tris (pH 6.8), 3 mM DTT, 6% SDS,
0.3% bromophenol blue and 30% glycerol. Subsequently, each aliquot
was loaded onto a 12.5% polyacryamide gel. Following
electrophoresis, the gels were transferred onto nitrocellulose
transfer membranes (Pall Corp., East Hills, NY, USA). To reduce
background staining, the membranes were incubated with 5% non-fat
dry milk in PBS containing 0.1% Tween-20 for 45 min, and then with
rabbit anti-KYNA antibody (diluted 1:1,500; Santa Cruz
Biotechnology) and peroxidase-conjugated goat anti-rabbit IgG
(A0545; Sigma-Aldrich), and then subjected to enhanced
chemiluminescence using an ECL kit (Pierce Chemical Co.). Loading
controls were performed using antibodies against β-actin (Abcam,
Inc., Cambridge, MA, USA).
Data analysis
The brain tisue sections were selected according to
anatomical landmarks corresponding to target coordinates
[anteroposterior (AP) diameter from −1.4 to −1.8 mm] of the gerbil
brain atlas. The number of CV-positive, NeuN-immunoreactive and F-J
B-positive cells was counted in a 200×200 μm2
area, applied approximately at the center of the CA1 in the SP.
Cell counts were obtained by averaging the total cell numbers from
each animal per group.
In order to quantitatively analyze KYNA
immunoreactivity, we applied a method described in a previous study
of ours (27). In brief, the
density of immunoreactive structures was evaluated on the basis of
a relative optical density (ROD), which was obtained after the
transformation of the mean gray level using the following formula:
ROD = log (256/mean gray level).
According to a method described in a previous study
of ours (28), the results of
western blot analysis were quantified using Scion Image software
(Scion Corp., Frederick, MD, USA), which was used to count the ROD:
a ratio of the ROD was calibrated as a percentage, with the value
in the sham-operated group designated as 100%.
Statistical analysis
All data are presented as the means ± SEM. A
multiple-sample comparison was applied to examine the differences
between groups and days. The differences between groups on the same
day were assessed by one-way ANOVA and Tukey’s post-hoc test. For
the analysis of time-dependent differences between the groups,
two-way ANOVA with the Bonferroni post-hoc test were used. A
p-value ≤0.05 was considered to indicate a statistically
significant difference.
Results
IPC-mediated neuroprotection
CV-positive (CV+)
cells
We examined whether IPC is associated with a
decrease in neuronal damage/death in the gerbil hippocampus
following ischemia-reperfusion. CV+ cells were clearly
observed in all the subregions of the hippocampus in the
sham-operated group, and the neurons in the SP (pyramidal neurons)
had a slightly large, round or pyramid-like shaped morphology
(Fig. 1A and B). In the
ischemia-operated group, however, 5 days after
ischemia-reperfusion, the CV+ cells were significantly
damaged in the SP of the CA1 region, but not in the CA2/3 region,
compared with those of the sham-operated group (Fig. 1E); the damaged cells were shrunken
and contained dark and polygonal nuclei (arrows in Fig. 1F).
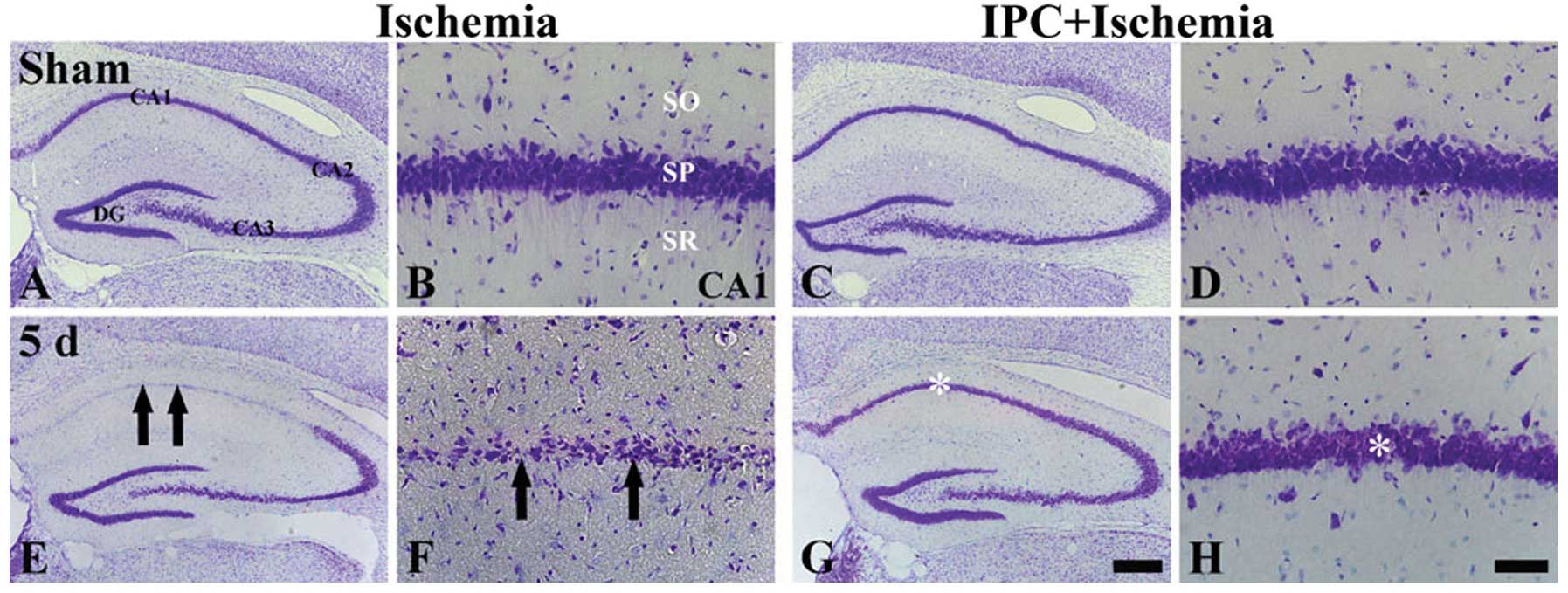 | Figure 1CV staining in the hippocampus of the
ischemia-operated (left 2, columns) and IPC + ischemia-operated
(right 2 columns) groups at (A-D) sham and (E-H) 5 days (5 d)
post-ischemia. CV+ cells (arrows) are damaged in the
stratum pyramidale (SP) of the CA1 region only at 5 days
post-ischemia in the ischemia-operated group; however,
CV+ cells (asterisk) in the IPC + ischemia-operated
group were similar to those in the sham-operated group. SO, stratum
oriens; SR, stratum radiatum; IPC, ischemic preconditioning; DG,
dentate gyrus; CA1, CA2 and CA3 represent regions I, II and III of
hippocampus proper, respectively; CV, cresyl violet. Scale bar: (A,
C, E and G) 800 μm and (B, D, F and H) 50 μm. |
In the IPC + sham-operated group, the CA1 pyramidal
neurons were evidently stained with CV (Fig. 1C and D), and in the IPC +
ischemia-operated group, the distribution pattern of the
CV+ cells in the SP was very similar to that in the IPC
+ sham-operated group at 5 days following ischemia-reperfusion
(Fig. 1G and H).
NeuN+ and F-J B+
cells
The assessment of the IPC-mediated neuroprotective
effects in the CA1 region was carried out using anti-NeuN
immunohistochemistry and F-J B histofluorescence staining (Fig. 2). In the sham-operated group, the
pyramidal neurons in the CA1 region were evidently stained with
NeuN, and no F-J B+ neurons were observed (Table I and Fig. 2A and B). In the ischemia-operated
group, however, the number of NeuN+ neurons was
significantly decreased in the SP of the CA1 region 5 days
following ischemia-reperfusion (Table
I and Fig. 2E), and, at this
time point, many F-J B+ cells were observed in the SP of
the CA1 region (Table I and
Fig. 2F).
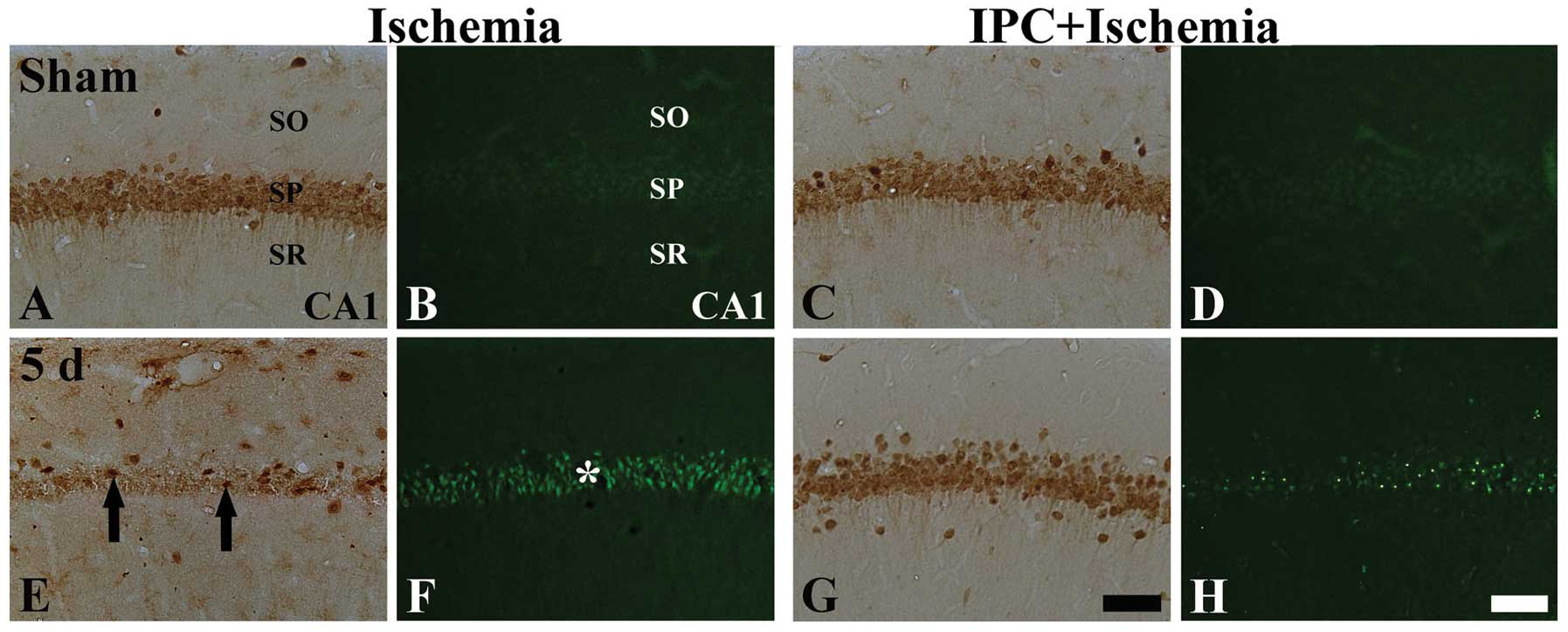 | Figure 2NeuN immunohistochemistry (the first
and third longitudinal columns) and F-J B histofluorescence
staining (the second and fourth longitudinal columns) in the CA1
region of the ischemia-operated (left 2 columns) and IPC +
ischemia-operated (right 2 columns) groups at (A-D) sham and (E-F)
5 days (5 d) following ischemia-reperfusion. In the sham-operated
group, many NeuN+ neurons, but no F-J B+
cells were observed in the stratum pyramidale (SP). In the
ischemia-operated group, a few NeuN+ (black arrows) and
many F-J B+ (asterisk) cells were detected in the SP at
5 days post-ischemia. However, in the IPC + ischemia-operated
group, abundant NeuN+ and few F-J B+ cells
were detected in the SP at 5 days post-ischemia. NeuN, neuronal
nuclei; F-J B, Fluoro-Jade B; CA1, region I of hippocampus proper;
IPC, ischemic preconditioning; SO, stratum oriens; SR, stratum
radiatum. Scale bar, 50 μm. |
 | Table IChanges in the mean number of
pyramidal neurons of the hippocampal CA1 region in the
ischemia-operated and IPC + ischemia-operated gerbils. |
Table I
Changes in the mean number of
pyramidal neurons of the hippocampal CA1 region in the
ischemia-operated and IPC + ischemia-operated gerbils.
| Time after IR | Ischemia-operated
group
| IPC +
ischemia-operated group
|
|---|
|
NeuN+ | F-J
B+ |
NeuN+ | F-J B+ |
|---|
| Sham | 354±14.14 | 0 | 344±12.36 | 0 |
| 1 day | 364±16.88 | 0 | 351±15.66 | 0 |
| 2 days | 359±13.65 | 5±3.72a | 357±16.71 | 0 |
| 5 days | 38±14.36a,b | 141±15.77a,b | 331±15.65a,b | 17±6.74a,b |
In the IPC + sham-operated group, the pyramidal
neurons in the CA1 region were also evidently stained with NeuN
(Fig. 2C), and no F-J
B+ cells were observed (Table I and Fig. 2D). In the IPC + ischemia-operated
group, the distribution patterns of the NeuN+ and F-J
B+ cells in the SP were not significantly altered
compared with those in the IPC + sham-operated group (Table I and Fig. 2G and H).
IPC-mediated effect on KYNA
immunoreactivity
CA1 region
KYNA immunoreactivity was easily detected in the SP
of the CA1 region in the sham-operated group (Table II and Fig. 3A, panels a-h). In the
ischemia-operated group, we found that KYNA immunoreactivity was
altered in the SP following ischemia-reperfusion. The
immunoreactivity was significantly increased 1 day following
ischemia-reperfusion and decreased at 2 days post-ischemia
(Table II and Fig. 3A, panels c and e). Five days after
ischemia, KYNA immunoreactivity was even more decreased in the SP
(Table II and Fig. 3A, panel g).
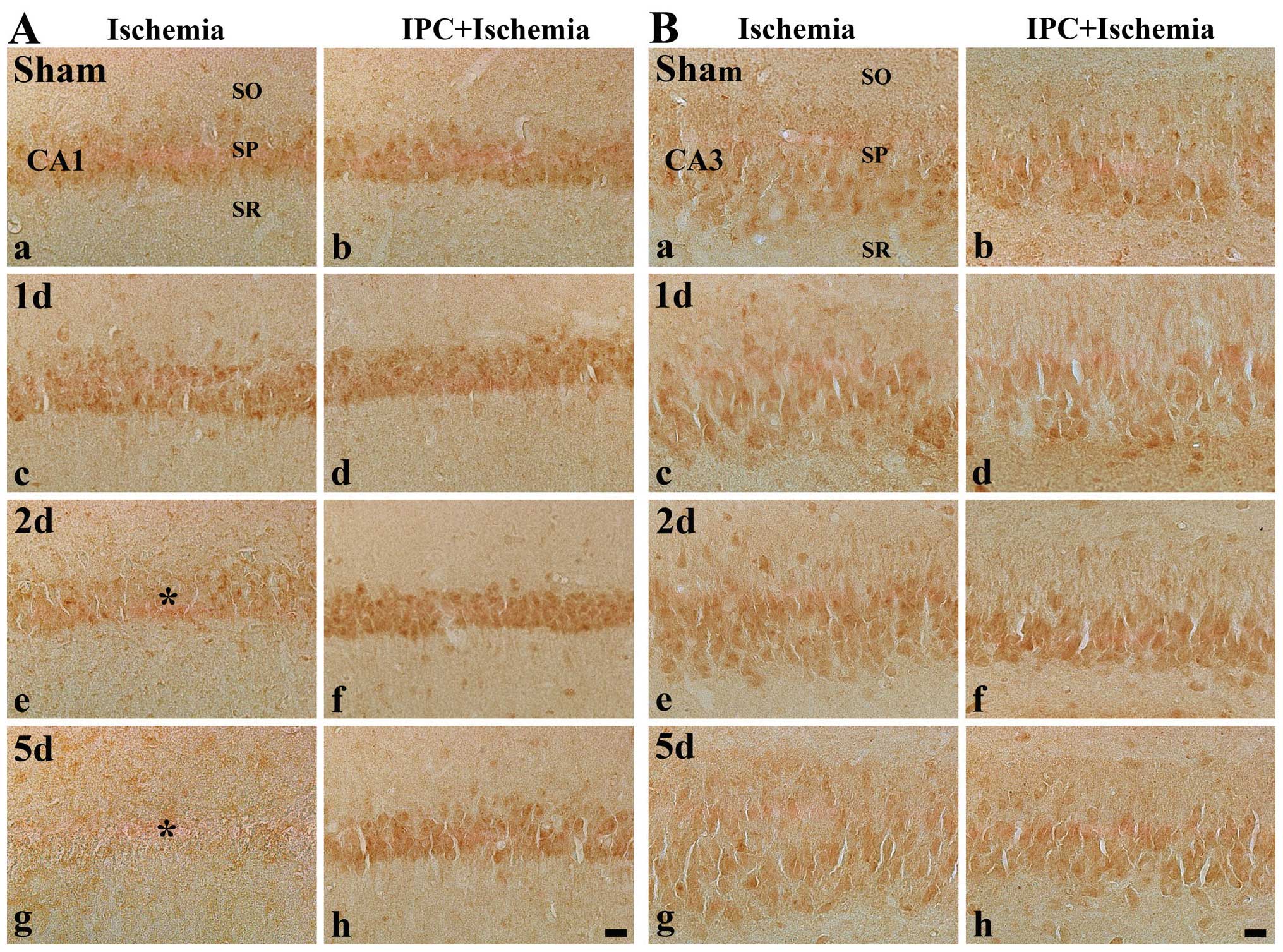 | Figure 3Immunohistochemistry for KYNA in (A)
the CA1 region and (B) CA2/3 region of the ischemia-operated (first
and third columns) and IPC + ischemia-operated (second and fourth
columns) groups at (a and b) sham, (c and d) 1 day (1 d), (e and f)
2 days (2 d) and (g and h) 5 days (5 d) following
ischemia-reperfusion. KYNA immunoreactivity was easily detected in
the stratum pyramidale (SP) in the sham-operated group. In the CA1
region, KYNA immunoreactivity in the SP (asterisk) was increased at
1 day post-ischemia, decreased at 2 days post-ischemia and markedly
decreased (reached its lowest level) at 5 days post-ischemia.
However, in the IPC + sham-operated group, KYNA immunoreactivity
was similar to that in the sham-operated group, and the
immunoreactivity was increased in the IPC + ischemia-operated
group. KYNA immunoreactivity in the SP of the CA2/3 region was not
significantly altered in all groups. KYNA, kynurenic acid; CA1,
region I of hippocampus propern; IPC, ischemic preconditioning; SO,
stratum oriens; SR, stratum radiatum. Scale bar, 50 μm. |
| Table IISemi-quantification of the
immunoreactivity of KYNA in pyramidal cells in the hippocampal CA1
and CA2/3 regions in the ischemia-operated and IPC +
ischemia-operated groups. |
Table II
Semi-quantification of the
immunoreactivity of KYNA in pyramidal cells in the hippocampal CA1
and CA2/3 regions in the ischemia-operated and IPC +
ischemia-operated groups.
| Antibody | Region | Groups | Category | Time after
ischemia-reperfusion
|
|---|
| Sham | 1 day | 2 days | 5 days |
|---|
| KYNA | CA1 | Ischemia | CSP | + | ++ | ± | ± |
| IPC + ischemia | CSP | + | ++ | ++ | ++ |
| CA3 | Ischemia | CSP | + | + | + | + |
| IPC + ischemia | CSP | + | + | + | + |
In the IPC + sham-operated group, KYNA
immunoreactivity was similar to that in the sham-operated group
(Table II and Fig. 3A, panel b). In the IPC +
ischemia-operated group, KYNA immunoreactivity in the SP was a
slightly increased at 1 day post-ischemia, and, thereafter, KYNA
immunoreactivity in the SP was significantly higher than that in
the IPC + sham-operated group (Table
II and Fig. 3A, panels d, f
and h).
CA2/3 region
In the CA2/3 region of the hippocampus of the brains
of the gerbils in the sham-operated group, moderate KYNA
immunoreactivity was detected in neurons of the SP (Table II and Fig. 3B, panel a). KYNA immunoreactivity
was not significantly altered in the SP following
ischemia-reperfusion (Table II
and Fig. 3B panels c, e and
g).
In the IPC + sham-operated and ischemia-operated
groups, KYNA immunoreactivity in the SP in the CA1 region was
similar to that in the sham-operated group (Table II and Fig. 3B panels b, d, f and h).
IPC-mediated effect on KYNA
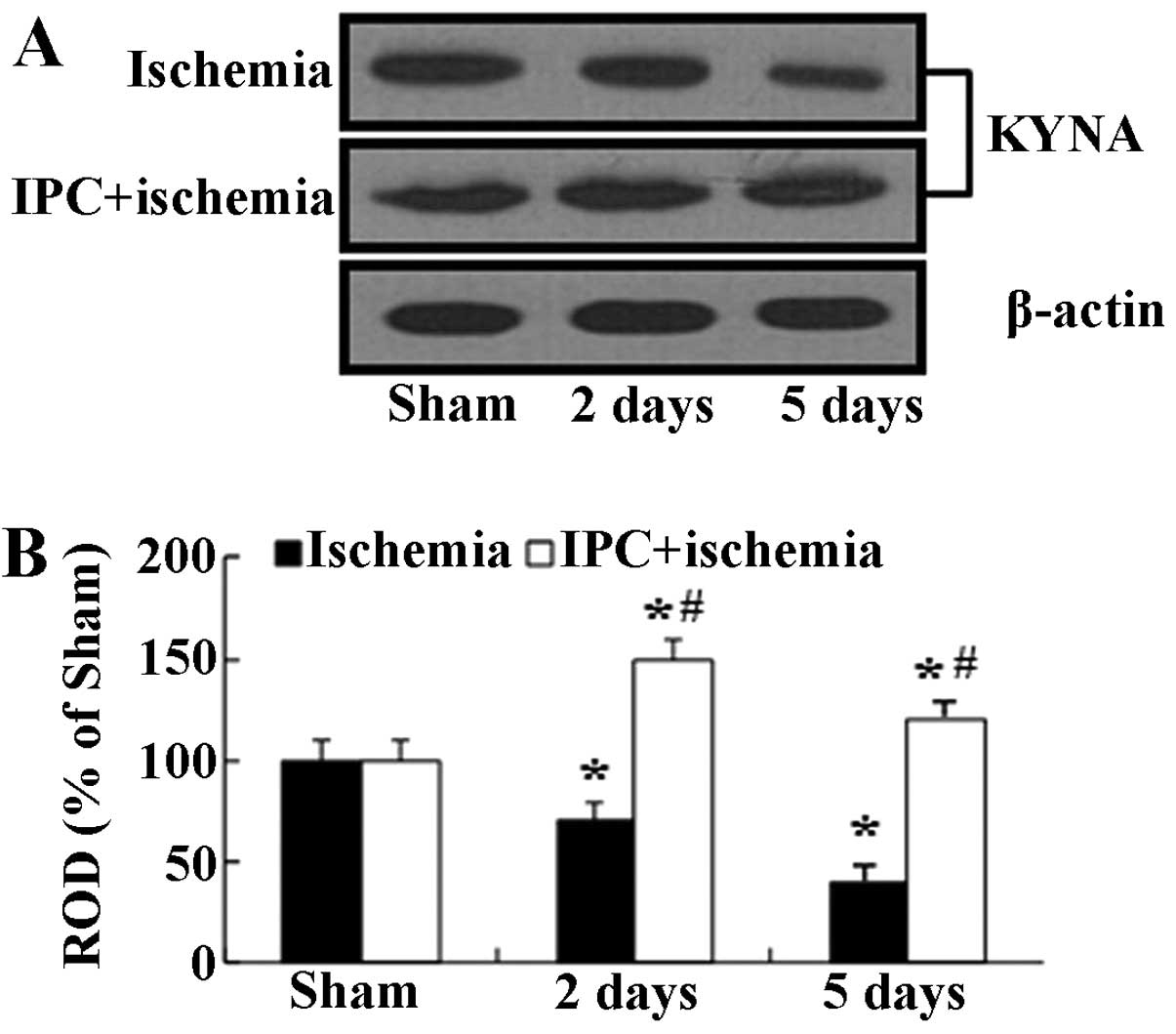
levels
In the present study, we examined KYNA protein
levels in the hippocampal CA1 homogenates at 2 and 5 days, when the
immunohistochemical data were significantly altered following
ischemia-reperfusion in both groups subjected to IPC. Western blot
analysis revealed that the changes in the protein expression
pattern of KYNA in the CA1 region following ischemia-reperfusion
were similar to those observed in the immunohistochemical data
(Fig. 4). In the
ischemia-operated group, the KYNA protein levels were significantly
decreased at 2 days and reached their lowest levels at 5 days
following ischemia-reperfusion. On the other hand, in the IPC +
sham-operated group, the KYNA protein levels were slightly
increased compared with the sham-operated group, and the levels in
the IPC + ischemia-operated-group were similar to those in the IPC
+ sham-operated group (Fig.
4).
Discussion
IPC is defined as a brief non-injurious episode of
ischemia that is able to protect the brain from a subsequent longer
ischemic insult (11,12). Kitagawa et al (32) reported the description of IPC in
the brain using a gerbil model of global ischemia. IPC shows a
tolerance, which has been termed ‘ischemic tolerance’ and is
activated at different time points following IPC; however, the
molecular mechanisms underlying ischemic tolerance are not yet
fully understood (33–35).
In the present study, we chose to induce a brief
period (2 min) of IPC to avoid histological tissue damage. This
brief IPC stimulus did not induce neuronal damage, as assessed by
CV and NeuN staining, and F-J B histofluorescence, which are
sensitive markers for the detection of acute neuronal injury
(11). We found that, at 5 days
post-ischemia, the CA1 pyramidal neurons showed typically neuronal
cell death, as shown by CV and NeuN staining, and F-J B
histofluorescence. However, the viable CA1 pyramidal neurons were
significantly protected from transient ischemic injury by IPC.
However, although IPC often provides strong neuroprotection against
ischemic brain injury, the exact mechanisms involved need to be
investigated in order to develop therapeutic strategies for
ischemic stroke.
KYNA, which is produced by astrocytes and neurons
(18), is an endogenous
metabolite of the kynurenine pathway for tryptophan degradation and
is an antagonist of both NMDA and α7-nicotinic acetylcholine
receptors (16–18). It is known that the level of
endogenous KYNA is altered in several neurodegenerative disorders
(36–38). However, data regarding the
concentration of endogenous KYNA following an ischemic insult are
limited. A few studies have demonstrated no change in the
hippocampal content of KYNA or its decrease at 4 days after
transient global ischemia (22,39,40). In the present study, we found that
KYNA immunoreactivity in pyramidal neurons was evidently altered
changed following ischemia and was hardly detectable in the
ischemia-operated group 5 days post-ischemia.
In the present study, we found that KYNA
immunoreactivity in the CA1 pyramidal neurons of the animals in the
IPC + ischemia-operated-group was evidently maintained or even
increased following transient ischemia. To the best of our
knowledge, no studies on the effects of IPC on KYN expression have
been published to date. Nevertheless, many researchers have
suggested that a sufficient elevation in the KYNA content in the
brain leads to a definite neuroprotective effect (41–44), although the therapeutic potential
of KYNA is limited, as KYNA is hardly able to cross the blood-brain
barrier (26).
Pyramidal neurons in the hippocampal CA1 region are
particularly vulnerable to excitotoxic processes following
transient forebrain ischemia. Excitatory amino acids (EAAs) play an
important role in the pathogenesis of cerebral ischemia (45). Under ischemic conditions, the
release of excess EAAs contributes to the overactivation of
ionotropic NMDA receptors and
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)
receptors, which mediate the excessive entry of Ca2+,
initiating glutamate-induced excitotoxicity, and finally, these
excitotoxic processes eventually lead to neuronal death in the
hippocampal CA1 region (46,47). Therefore, it has been shown that
the blockade of the receptors for EAA (the antagonism of NMDA
receptors in particular) effectively reduces neuronal damage
following ischemia (48,49). Therefore, it is likely that IPC
elevates the KYNA concentration sufficiently enough to affect the
co-agonist site of the NMDA receptors.
In conclusion, the main findings of the present
study demonstrated that in the levels of KYNA in the pyramidal
neurons of the hippocampal CA1 region in animals subjected to IPC
were maintained or even increased following ischemia-reperfusion
and suggest that the increase in KYNA expression induced by IPC is
associated with the endogenous protective response of the brain to
ischemic injury.
Acknowledgments
The authors would like express their gratitude to
Mr. Seung Uk Lee for providing technical assistance. The present
study was supported by the Basic Science Research Program through
the National Research Foundation of Korea (NRF) funded by the
Ministry of Science, ICT and Future Planning
(NRF-2014R1A2A2A01005307), and by a Priority Research Centers
Program grant (NRF-2009-0093812) through the National Research
Foundation of Korea funded by the Ministry of Science, ICT and
Future Planning.
References
|
1
|
Kirino T: Delayed neuronal death in the
gerbil hippocampus following ischemia. Brain Res. 239:57–69. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lin CS, Polsky K, Nadler JV and Crain BJ:
Selective neocortical and thalamic cell death in the gerbil after
transient ischemia. Neuroscience. 35:289–299. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schmidt-Kastner R and Freund TF: Selective
vulnerability of the hippocampus in brain ischemia. Neuroscience.
40:599–636. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Abe K, Aoki M, Kawagoe J, et al: Ischemic
delayed neuronal death. A mitochondrial hypothesis. Stroke.
26:1478–1489. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Imon H, Mitani A, Andou Y, Arai T and
Kataoka K: Delayed neuronal death is induced without postischemic
hyperexcitability: continuous multiple-unit recording from ischemic
CA1 neurons. J Cereb Blood Flow Metab. 11:819–823. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shelat PB, Coulibaly AP, Wang Q, Sun AY,
Sun GY and Simonyi A: Ischemia-induced increase in RGS7 mRNA
expression in gerbil hippocampus. Neurosci Lett. 403:157–161. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vollenweider F, Bendfeldt K, Maetzler W,
Otten U and Nitsch C: GABA(B) receptor expression and cellular
localization in gerbil hippocampus after transient global ischemia.
Neurosci Lett. 395:118–123. 2006. View Article : Google Scholar
|
|
8
|
Hou ST and MacManus JP: Molecular
mechanisms of cerebral ischemia-induced neuronal death. Int Rev
Cytol. 221:93–148. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ientile R, Caccamo D, Marciano MC, et al:
Transglutaminase activity and transglutaminase mRNA transcripts in
gerbil brain ischemia. Neurosci Lett. 363:173–177. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu S and Cai J: Effects of aniracetam on
extracellular levels of transmitter amino acids in the hippocampus
of the conscious gerbils: an intracranial microdialysis study.
Neurosci Lett. 339:187–190. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schmued LC and Hopkins KJ: Fluoro-Jade B:
a high affinity fluorescent marker for the localization of neuronal
degeneration. Brain Res. 874:123–130. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lehotsky J, Burda J, Danielisova V,
Gottlieb M, Kaplan P and Saniova B: Ischemic tolerance: the
mechanisms of neuroprotective strategy. Anat Rec (Hoboken).
292:2002–2012. 2009. View
Article : Google Scholar
|
|
13
|
Gidday JM: Cerebral preconditioning and
ischaemic tolerance. Nat Rev Neurosci. 7:437–448. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kardesoglu E, Isilak Z, Uz O and Yiginer
O: Ischemic conditioning: a current concept in reducing reperfusion
injury. Chin Med J (Engl). 124:4802011.
|
|
15
|
Swartz KJ, During MJ, Freese A and Beal
MF: Cerebral synthesis and release of kynurenic acid: an endogenous
antagonist of excitatory amino acid receptors. J Neurosci.
10:2965–2973. 1990.PubMed/NCBI
|
|
16
|
Kemp JA, Foster AC, Leeson PD, et al:
7-Chlorokynurenic acid is a selective antagonist at the glycine
modulatory site of the N-methyl-D-aspartate receptor complex. Proc
Natl Acad Sci USA. 85:6547–6550. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kessler M, Terramani T, Lynch G and Baudry
M: A glycine site associated with N-methyl-D-aspartic acid
receptors: characterization and identification of a new class of
antagonists. J Neurochem. 52:1319–1328. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hilmas C, Pereira EF, Alkondon M,
Rassoulpour A, Schwarcz R and Albuquerque EX: The brain metabolite
kynurenic acid inhibits alpha7 nicotinic receptor activity and
increases non-alpha7 nicotinic receptor expression:
physiopathological implications. J Neurosci. 21:7463–7473.
2001.PubMed/NCBI
|
|
19
|
Moroni F, Cozzi A, Sili M and Mannaioni G:
Kynurenic acid: a metabolite with multiple actions and multiple
targets in brain and periphery. J Neural Transm. 119:133–139. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sas K, Csete K, Vecsei L and Papp JG:
Effect of systemic administration of L-kynurenine on
corticocerebral blood flow under normal and ischemic conditions of
the brain in conscious rabbits. J Cardiovasc Pharmacol. 42:403–409.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lugo-Huitron R, Blanco-Ayala T,
Ugalde-Muniz P, et al: On the antioxidant properties of kynurenic
acid: free radical scavenging activity and inhibition of oxidative
stress. Neurotoxicol Teratol. 33:538–547. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Salvati P, Ukmar G, Dho L, et al: Brain
concentrations of kynurenic acid after a systemic neuroprotective
dose in the gerbil model of global ischemia. Prog
Neuropsychopharmacol Biol Psychiatry. 23:741–752. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cozzi A, Carpenedo R and Moroni F:
Kynurenine hydroxylase inhibitors reduce ischemic brain damage:
studies with (m-nitrobenzoyl)-alanine (mNBA) and
3,4-dimethoxy-[-N-4-(nitrophenyl)thiazol-2yl]-benzenesulfonamide
(Ro 61-8048) in models of focal or global brain ischemia. J Cereb
Blood Flow Metab. 19:771–777. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Abo M, Yamauchi H, Suzuki M, Sakuma M and
Urashima M: Facilitated beam-walking recovery during acute phase by
kynurenic acid treatment in a rat model of photochemically induced
thrombosis causing focal cerebral ischemia. Neurosignals.
15:102–110. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Germano IM, Pitts LH, Meldrum BS,
Bartkowski HM and Simon RP: Kynurenate inhibition of cell
excitation decreases stroke size and deficits. Ann Neurol.
22:730–734. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fukui S, Schwarcz R, Rapoport SI, Takada Y
and Smith QR: Blood-brain barrier transport of kynurenines:
implications for brain synthesis and metabolism. J Neurochem.
56:2007–2017. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee CH, Park JH, Cho JH, et al: Changes
and expressions of Redd1 in neurons and glial cells in the gerbil
hippocampus proper following transient global cerebral ischemia. J
Neurol Sci. 344:43–50. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee CH, Park JH, Choi JH, Yoo KY, Ryu PD
and Won MH: Heat shock protein 90 and its cochaperone, p23, are
markedly increased in the aged gerbil hippocampus. Exp Gerontol.
46:768–772. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nakamura H, Katsumata T, Nishiyama Y,
Otori T, Katsura K and Katayama Y: Effect of ischemic
preconditioning on cerebral blood flow after subsequent lethal
ischemia in gerbils. Life Sci. 78:1713–1719. 2006. View Article : Google Scholar
|
|
30
|
Lee CH, Park JH, Yoo KY, et al: Pre- and
post-treatments with escitalopram protect against experimental
ischemic neuronal damage via regulation of BDNF expression and
oxidative stress. Exp Neurol. 229:450–459. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee JC, Kim IH, Cho GS, et al: Ischemic
preconditioning-induced neuroprotection against transient cerebral
ischemic damage via attenuating ubiquitin aggregation. J Neurol
Sci. 336:74–82. 2014. View Article : Google Scholar
|
|
32
|
Kitagawa K, Matsumoto M, Kuwabara K, et
al: ’Ischemic tolerance’ phenomenon detected in various brain
regions. Brain Res. 561:203–211. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dhodda VK, Sailor KA, Bowen KK and
Vemuganti R: Putative endogenous mediators of
preconditioning-induced ischemic tolerance in rat brain identified
by genomic and proteomic analysis. J Neurochem. 89:73–89. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stenzel-Poore MP, Stevens SL, King JS and
Simon RP: Preconditioning reprograms the response to ischemic
injury and primes the emergence of unique endogenous
neuroprotective phenotypes: a speculative synthesis. Stroke.
38:680–685. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Feng Z, Davis DP, Sasik R, Patel HH,
Drummond JC and Patel PM: Pathway and gene ontology based analysis
of gene expression in a rat model of cerebral ischemic tolerance.
Brain Res. 1177:103–123. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kaminski RM, Zielinska E, Dekundy A, van
Luijtelaar G and Turski W: Deficit of endogenous kynurenic acid in
the frontal cortex of rats with a genetic form of absence epilepsy.
Pol J Pharmacol. 55:741–746. 2003.
|
|
37
|
Stone TW: Kynurenines in the CNS: from
endogenous obscurity to therapeutic importance. Prog Neurobiol.
64:185–218. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zadori D, Klivenyi P, Szalardy L, Fulop F,
Toldi J and Vecsei L: Mitochondrial disturbances, excitotoxicity,
neuroinflammation and kynurenines: novel therapeutic strategies for
neurodegenerative disorders. J Neurol Sci. 322:187–191. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Saito K, Nowak TS Jr, Markey SP and Heyes
MP: Mechanism of delayed increases in kynurenine pathway metabolism
in damaged brain regions following transient cerebral ischemia. J
Neurochem. 60:180–192. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Saito K, Nowak TS Jr, Suyama K, et al:
Kynurenine pathway enzymes in brain: responses to ischemic brain
injury versus systemic immune activation. J Neurochem.
61:2061–2070. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Stone TW: Development and therapeutic
potential of kynurenic acid and kynurenine derivatives for
neuroprotection. Trends Pharmacol Sci. 21:149–154. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Schwarcz R and Pellicciari R: Manipulation
of brain kynurenines: glial targets, neuronal effects, and clinical
opportunities. J Pharmacol Exp Ther. 303:1–10. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Vamos E, Pardutz A, Klivenyi P, Toldi J
and Vecsei L: The role of kynurenines in disorders of the central
nervous system: possibilities for neuroprotection. J Neurol Sci.
283:21–27. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zadori D, Klivenyi P, Vamos E, Fulop F,
Toldi J and Vecsei L: Kynurenines in chronic neurodegenerative
disorders: future therapeutic strategies. J Neural Transm.
116:1403–1409. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lipton SA and Rosenberg PA: Excitatory
amino acids as a final common pathway for neurologic disorders. N
Engl J Med. 330:613–622. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Dirnagl U, Iadecola C and Moskowitz MA:
Pathobiology of ischaemic stroke: an integrated view. Trends
Neurosci. 22:391–397. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Endres M and Dirnagl U: Ischemia and
stroke. Adv Exp Med Biol. 513:455–473. 2002. View Article : Google Scholar
|
|
48
|
Lai TW, Zhang S and Wang YT:
Excitotoxicity and stroke: identifying novel targets for
neuroprotection. Prog Neurobiol. 115:157–188. 2014. View Article : Google Scholar
|
|
49
|
Hoyte L, Barber PA, Buchan AM and Hill MD:
The rise and fall of NMDA antagonists for ischemic stroke. Curr Mol
Med. 4:131–136. 2004. View Article : Google Scholar : PubMed/NCBI
|