Introduction
Skeletal muscle insulin resistance plays a major
role in the development of postprandial hyperglycemia (1). It is well established that excess
lipid accumulation in muscle leads to insulin resistance by
impairing insulin signaling (2,3).
As a key transcription factor regulating de novo
lipogenesis, the increased expression of sterol regulatory
element-binding protein-1c (SREBP-1c) can result in muscular
insulin resistance by promoting lipid accumulation (4–6).
Nonetheless, in a recent study of ours, we reported a new mechanism
through which SREBP-1c directly binds to the promoter region of
insulin receptor substrate-1 (IRS-1), suppressing IRS-1 expression
and, subsequently, the activation of the insulin signaling pathway
(7). AMP-activated protein kinase
(AMPK), a conserved serine/threonine protein kinase, is composed of
a catalytic subunit α and two regulatory subunits β and γ. The α
subunit contains a threonine residue (Thr172) that is necessary for
AMPK activation. AMPK is an intracellular energy sensor that plays
a key role in regulating cellular metabolism (8). In muscle, AMPK has been shown to
regulate insulin signaling and promote the translocation of glucose
transporter 4 (GLUT4), thereby stimulating glucose uptake (9). Multiple evidence has indicated that
the first-line antihyperglycemic drug, metformin, increases glucose
uptake in skeletal muscle mainly through AMPK activation (10–12).
It is known that SREBP-1c is negatively regulated by
AMPK. Previous studies have indicated that AMPK suppresses SREBP-1c
transcription (13,14) and inhibits SREBP-1c cleavage and
nuclear translocation by phosphorylating SREBP-1c (15) in hepatoma cell lines and a fatty
liver model, respectively. Studies have demonstrated that metformin
treatment reduces hepatic lipogenesis through the the AMPK/SREBP-1c
pathway (15,16). Furthermore, our recent study
indicated that metformin ameliorated IRS-1-associated insulin
signaling through its inhibitory effect on SREBP-1c in skeletal
muscle cells (7). Nonetheless,
whether AMPK is the molecular link through which metformin acts on
SREBP-1c and the subsequent insulin signaling requires
clarification.
Therefore, the present study was designed to
investigate the involvement of the AMPK/SREBP-1c pathway in the
beneficial effects of metformin on the insulin signaling pathway in
skeletal muscle cells. We found that metformin inhibited SREBP-1c
expression, which was coincident with AMPK activation in PA-treated
L6 myotubes. Further experiments using the AMPK inhibitor, compound
C, demonstrated that AMPK activation was required for the
inhibitory effects of metformin on SREBP-1c and subsequent insulin
signaling. These findings provide a more precise mechanism of
metformin regulation of insulin signaling in skeletal muscle
cells.
Materials and methods
Materials
L6 muscle cells were obtained from the Chinese
Academy of Sciences. All cell culture media and sera were purchased
from Invitrogen (Carlsbad, CA, USA). Metformin and palmitic acid
(PA) were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Compound C was obtained from EMD Millipore (Billerica, MA, USA).
2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-amino]-2-deoxy-D-glucose
(2-NBDG) was purchased from Cayman Chemical (Ann Arbor, MI, USA).
The anti-SREBP-1c antibody (sc-8984) was obtained from Santa Cruz
Biotechnology (Dallas, TX, USA). Antibodies against IRS-1 (3407),
phosphorylated-IRS-1 (p-IRS-1; Tyr608/612; 09–432) and
glyceraldehyde-3-phosphate dehydrogenase (GAPDH; MAB374) were
obtained from EMD Millipore. Antibodies against AMPKα (2603),
phosphorylated AMPKα (p-AMPKα; Thr172; 2535), Akt (9272) and
phosphorylated Akt (p-Akt; Ser473; 4060) were from Cell Signaling
Technology (Danvers, MA, USA). The anti-fatty acid synthase (FAS;
610962) antibody was obtained from BD Biosciences (Franklin Lakes,
NJ, USA).
Cell culture
The L6 muscle cells were cultured in DMEM
supplemented with 10% FBS (vol/vol) and differentiated into
myotubes in differentiation medium within 6 days, as previously
described (17). The monolayer of
myotubes was then serum-starved in DMEM for 8 h and used in the
following experiments. To examine the time-course effects of PA,
the L6 myotubes were incubated with 0.5 mM PA for 3, 6, 12, 18 and
24 h. To determine the effects of metformin and the involvement of
AMPK, the L6 myotubes were treated with 0.5 mM PA in the presence
or absence of metformin (1 or 10 mM) for 24 h or pre-treated with
the specific AMPK inhibitor, compound C (10 μM), for 30 min
and then exposed to 10 mM metformin for 24 h. The cells were then
harvested, and the protein was extracted for western blot
analysis.
Glucose uptake assay
The L6 cells were treated with the indicated
compounds for 24 h and then incubated in low-glucose and serum-free
DMEM containing 100 μM 2-NBDG for 1 h at 37°C in the dark.
After a 10-min treatment with 100 nM insulin, the cells were
collected and the fluorescence intensity was measured at an
excitation of 485 nm and an emission of 520 nm using a FACSCalibur
flow cytometer (BD Biosciences). The intensity of the fluorescence
reflected the 2-NBDG uptake of the cells.
PA-induced cytotoxicity assay
Cell apoptosis was detected by Annexin V-FITC/PI
staining (Invitrogen). Annexin V-FITC is a fluorescence protein
that binds to phosphatidylserine of the plasma membrane during
early apoptosis. Propidium iodide (PI) is a fluorescent dye that
binds to nuclear DNA following the rupture of the plasma membrane.
L6 myotubes untreated or treated with 0.5 mM PA for 24 h were
resuspended in 400 μl Annexin Binding Buffer, and then
incubated in the dark for 15 min at room temperature with 4
μl of Annexin V-FITC and 4 μl of PI. Annexin V-FITC
and PI fluorescence were measured by flow cytometry. DNA
fragmentation was examined to determine late apoptosis. Total DNA
was extracted from the PA-treated L6 myotubes using a DNA
purification kit and analyzed on ethidium bromide (EtBr)-stained 2%
agarose gels.
Western blot analysis
The treated cells were washed twice with cold
phosphate-buffered saline and lysed in NP-40 lysis buffer
containing a Protease Inhibitor Cocktail (Roche, Basel,
Switzerland). The protein concentration was measured using the
bicinchoninic acid (BCA) method. Twenty micrograms of protein per
lane were loaded and separated by SDS-PAGE, transferred onto
polyvinylidene fluoride membranes, blocked with 7.5% non-fat dried
milk (wt/vol) in Tris-buffered saline with 0.1% Tween-20 (TBST) and
incubated with primary antibodies overnight at 4°C. The membranes
were washed with TBST and incubated with the appropriate secondary
antibody (goat anti-rabbit IgG, ZB-2301, ZSGB-BIO, Beijing, China
and goat anti-mouse IgG, GAM0072, MultiSciences Biotech, Suzhou,
China) at room temperature. The protein bands were then visualized
with enhanced chemiluminescence (EMD Millipore) and quantified by
densitometry (Quantity One software; Bio-Rad Laboratories,
Hercules, CA, USA).
Luciferase reporter assays
The reporter plasmid, pGL3-SREBP-1c, was constructed
to contain the rat SREBP-1c promoter region from −1,000 to +100 bp
and cloned into the KpnI/HindIII sites of the
pGL3-basic luciferase vector (Promega, Madison WI, USA). The L6
cells were co-transfected with a luciferase plasmid pGL3-SREBP-1c
(1,000 ng) and a pRL-TK Renilla plasmid (20 ng) using
Lipofectamine 2000 (Invitrogen). At 24 h post-transfection, the
cell medium was switched to a medium supplemented with the
indicated compounds. Twelve hours later, luciferase activity was
measured using the Dual-Luciferase reporter assay system (Promega).
The Renilla plasmid pRL-TK was used to normalize for
transfection efficiency.
Statistical analysis
All data are expressed as the means ± SEM.
Differences between groups were examined for statistical
significance using the Student’s t-test or one-way ANOVA. A value
of P<0.05 was considered to indicate a statistically significant
difference.
Results
Metformin promotes glucose uptake through
AMPK activation under PA-induced insulin-resistant states in L6
myotubes
It has been documented in a number of studies that
PA induces insulin resistance by decreasing glucose uptake
(18–20). First, in the present study, we
evaluated the role of PA in glucose uptake. The results revealed
that PA decreased glucose uptake in the L6 myotubes at doses from
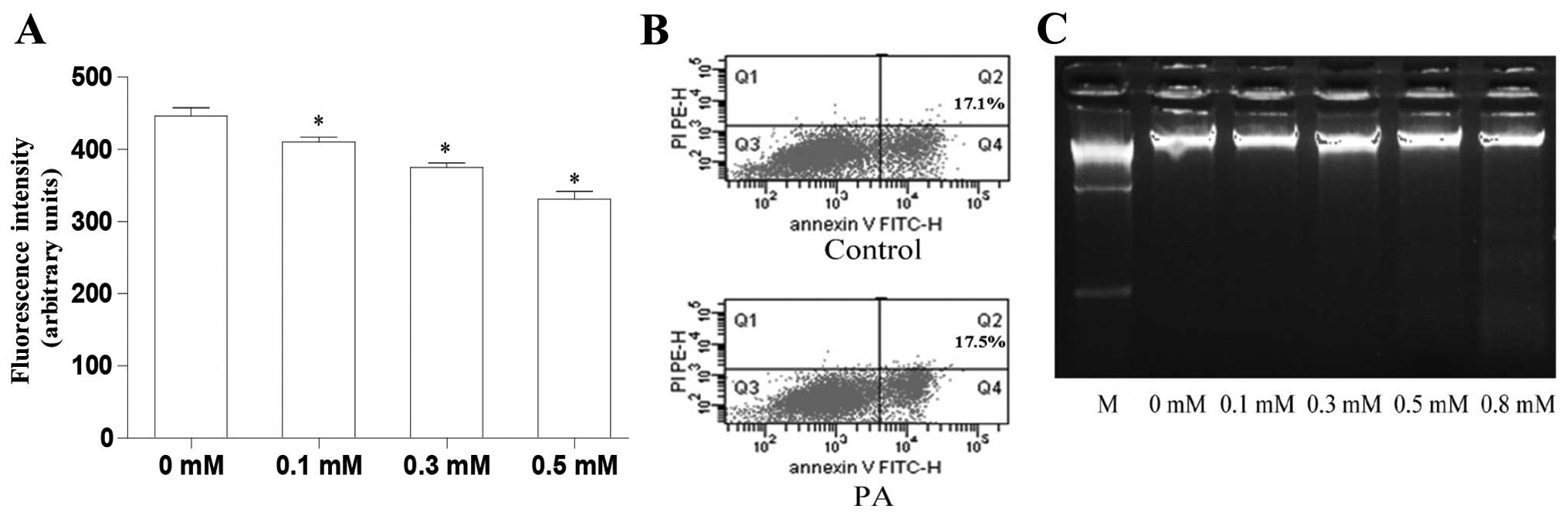
0.1 to 0.5 mM in a dose-dependent manner (Fig. 1A). Subsequently, we determined
whether treatment with 0.5 mM PA was toxic to the L6 myotubes
Annexin V-FITC/PI staining revealed that treatment with 0.5 mM PA
resulted in a similar amount of apoptosis to the control
(untreated) L6 myotubes (Fig.
1B). DNA gel electrophoresis revealed that PA at doses from 0.1
to 0.5 mM had no cytotoxic effects; however, cytotoxic effects were
observed when the concentration reached 0.8 mM, which resulted in a
dispersed DNA band (Fig. 1C). In
the following experiments all data were obtained using treatment
with 0.5 mM PA.
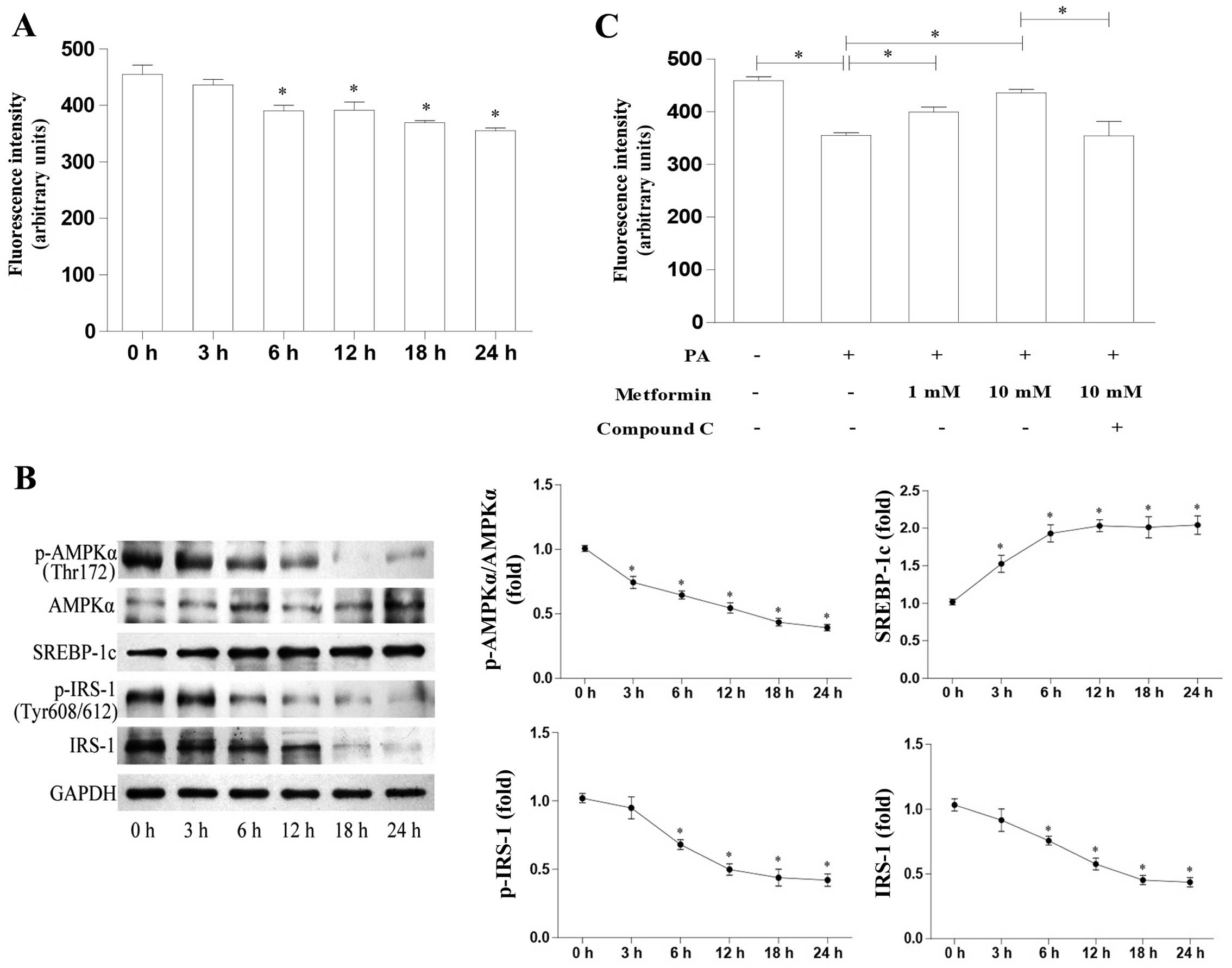
We investigated the time-course effects of PA on
glucose uptake in L6 myotubes and the molecular mechanisms involved
The results revealed that PA decreased glucose uptake in a
time-dependent manner (Fig. 2A).
We also evaluated AMPK and SREBP-1c expression in the L6 cells
treated with PA. As shown in Fig.
2B, PA decreased the p-AMPKα protein levels in a time-dependent
manner, whereas the AMPKα protein levels were not affected.
SREBP-1c protein expression increased significantly after 3 h of
exposure to PA. In addition, IRS-1, as well as p-IRS-1 (Tyr608/612)
expression decreased significantly after 6 h of PA treatment. These
data suggest an inverse correlation between AMPK activation and
SREBP-1c expression. The AMPK/SREBP-1c pathway may thus play a key
role in PA-induced insulin resistance.
Metformin, a widely-used treatment for type 2
diabetes, has been reported to increase glucose uptake in skeletal
muscle, the mechanisms of which mainly involve AMPK activation. In
the present study, we investigated the role of metformin in glucose
uptake and found that metformin increased glucose uptake in a
dose-dependent manner compared with the PA-treated L6 cells
(Fig. 2C). Furthermore, the
metformin-induced enhancement of glucose uptake was attenuated
following treatment with the AMPK inhibitor, compound C (Fig. 2C). Taken together, these data
suggest that the metformin-induced upregulation of glucose uptake
is mediated through AMPK activation.
Metformin stimulates AMPK activity,
inhibits SREBP-1c expression and activates the IRS-1/Akt
pathway
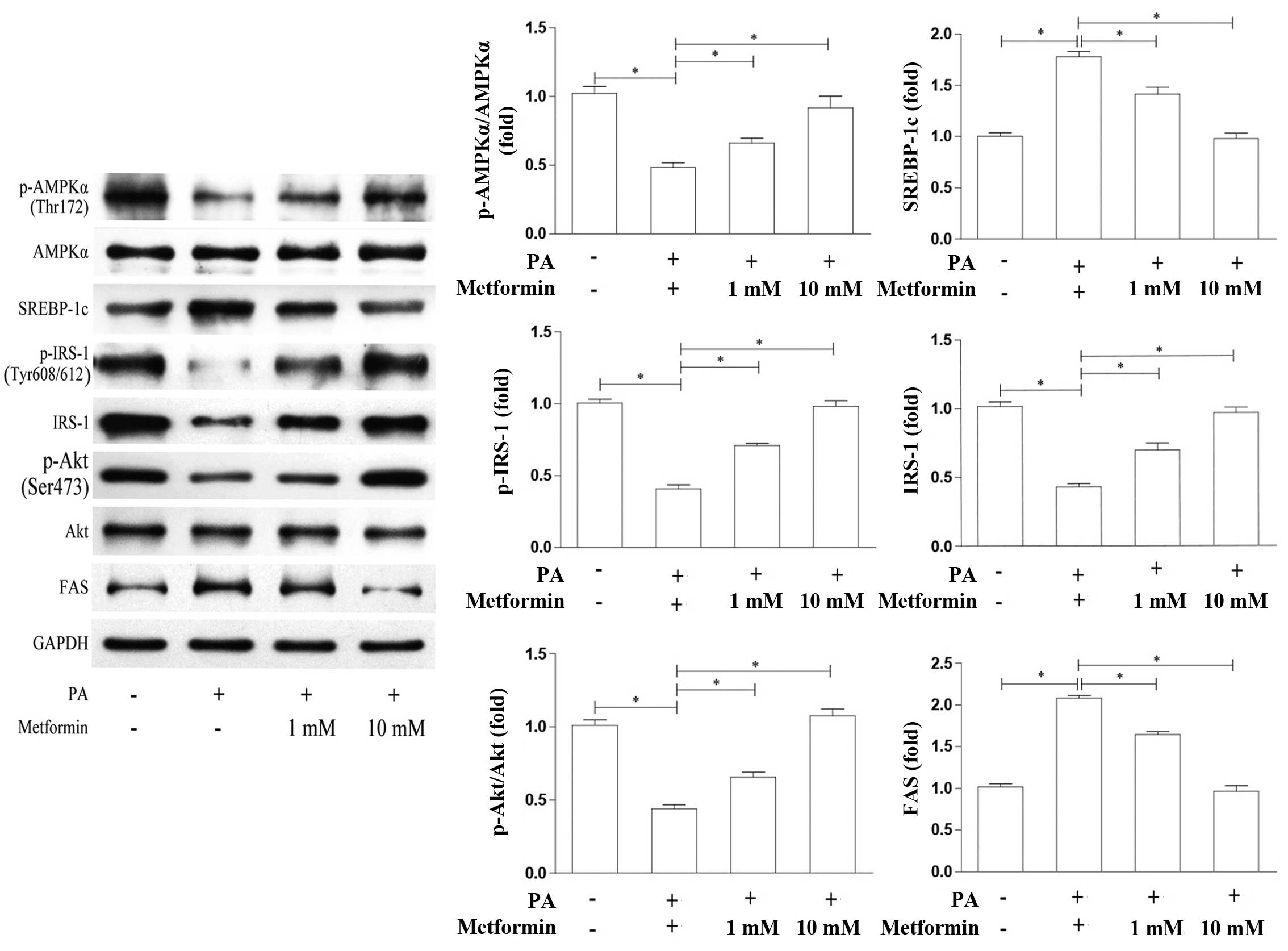
In order to further investigate the specific
mechanisms responsible for the metformin-induced increase in
glucose uptake in skeletal muscle cells, we detected the protein
levels of AMPKα and SREBP-1c in the PA-treated L6 cells which were
treated with various concentrations of metformin. In addition, we
also detected the expression of the SREBP-1c downstream protein,
FAS, and key proteins involved in insulin signaling in muscle
cells, including IRS-1 and Akt. As shown in Fig. 3, compared with the PA-treated
cells not treated with metformin, the levels of AMPKα
phosphorylation were increased in a dose-dependent manner in the
cells treated with metformin, while the AMPKα protein levels were
not affected. By contrast, the SREBP-1c and FAS protein levels were
decreased in a dose-dependent manner in the cells treated with
metformin. Correspondingly, the protein expression levels of IRS-1,
p-IRS-1 (Tyr608/612) and p-Akt (Ser473)/Akt were increased, similar
to AMPK activation. These results suggest that metformin inhibits
SREBP-1c expression by activating AMPK and then the IRS-1/Akt
pathway.
The AMPK inhibitor, compound C, reverses
the suppressive effects of metformin on SREBP-1c and impairs the
activation of the IRS-1/Akt pathway
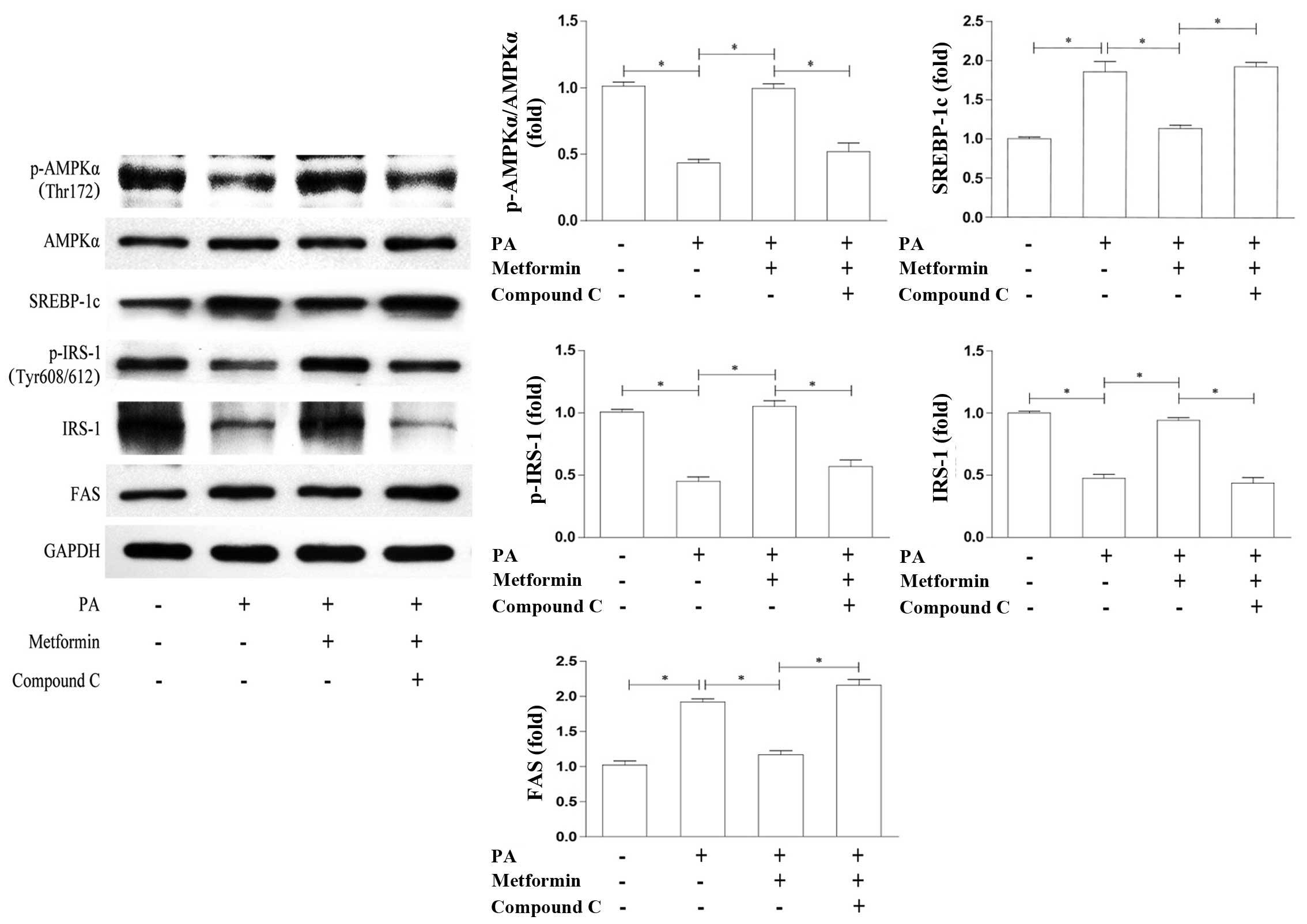
In order to examine whether the effects of metformin
on SREBP-1c are AMPK-dependent, we examined the relevant proteins
by co-incubation with the AMPK inhibitor, compound C. We found that
the metformin-induced enhancement of AMPKα phosphorylation and the
suppression of SREBP-1c expression were reversed by treatment with
compound C (Fig. 4). In
accordance with this, the protein expression of IRS-1 and p-IRS-1
(Tyr608/612) was reduced, whereas FAS expression was upregulated to
levels which were similar to those observed in the PA-treated cells
(Fig. 4). These results support
the notion that AMPK is required in the metformin-induced
suppression of SREBP-1c expression and contributes to the
activation of the IRS-1/Akt pathway.
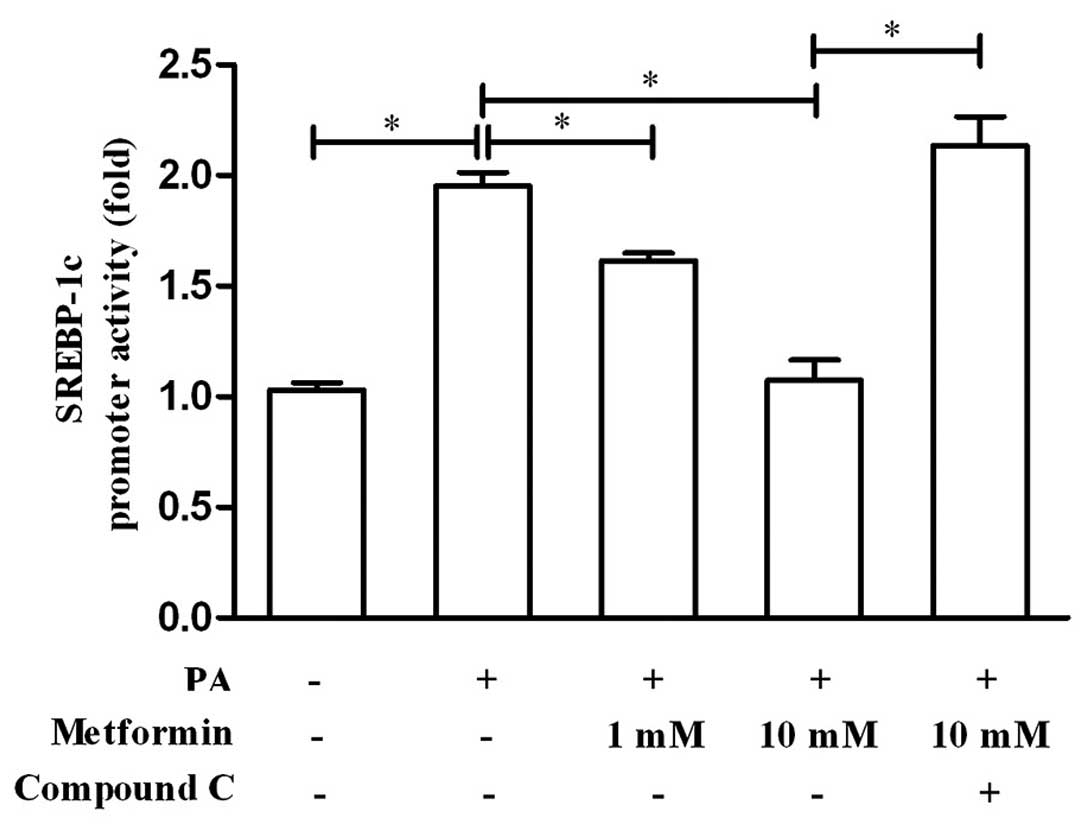
Metformin suppresses SREBP-1c promoter
activity in an AMPK-dependent manner
In ordre to confirm that metformin regulates
SREBP-1c transcription and whether its role is AMPK-dependent, we
examined the effects of metformin and compound C on SREBP-1c
promoter activity using a luciferase reporter assay. As shown in
Fig. 5, metformin suppressed the
promoter activity of SREBP-1c in a dose-dependent manner, while the
AMPK inhibitor, compound C, blocked this suppression of the
SREBP-1c promoter. These results demonstrate that metformin exerts
an inhibitory effect on SREBP-1c promoter activity and that this is
AMPK-dependent.
Discussion
Metformin has been recommended as the first-line
oral agent for the treatment of type 2 diabetes (21). There are many effects of metformin
on glucose metabolism, including a decrease in hepatic glucose
output, an increase in glucose uptake in muscle and fat, an
increase in glucose utilization in the gut and an enhancement in
incretin signaling (22).
However, the precise molecular mechanisms of action of metformin
require further clarification.
Studies on the liver have indicated that
AMPK-dependent and AMPK-independent mechanisms may both account for
metformin action on liver cells [reviewed in (23)]. However, studies on skeletal
muscle have suggested that metformin exerts its glucose-lowering
efficacy primarily through the activation of AMPK (24,25). In the present study, our results
support the hypothesis that metformin activates AMPK and improves
glucose uptake in PA-treated L6 cells and that this beneficial
effect is abolished by the AMPK inhibitor, compound C.
In addition to an increase in muscle glucose uptake,
a considerable amount of evidence has indicated that the activation
of AMPK by metformin also results in the inhibition of lipogenesis
(26,27). Studies have also established that
AMPK activation leads to a reduction in lipogenesis by suppressing
SREBP-1c expression (15,16,28,29). Recently, the expanding roles of
SREBP-1c in insulin signaling have been revealed. Studies have
suggested that SREBP-1c directly inhibits IRS-2 expression and the
subsequent insulin signaling pathway in the liver (30,31). In a previous study of ours, it was
demonstrated that SREBP-1c transcriptionally suppresses IRS-1
expression, inhibits IRS-1-associated insulin signaling and thereby
decreases glucose uptake in muscle (7). In the present study, we observed
that the effects of metformin treatment on insulin signaling were
mediated through AMPK activation and SREBP-1c inhibition when the
cells were under an insulin-resistant state induced by PA.
Moreover, we found that the above-mentioned effects were
AMPK-dependent. These results demonstrate that metformin increases
IRS-1-associated insulin signaling partly through the AMPK/SREBP-1c
pathway.
We have described the specific signaling pathway of
metformin action on glucose uptake at the cellular and molecular
level; however, further studies using tissues from animals or
patients with insulin resistance are required in the future. In the
present study, the percentage of apoptotic cells was at a high
level. We speculated that the cells had a low nutritional status
during the process of differentiation. In line with the majority of
in vitro studies, the metformin concentration used in this
study was above its physiological concentration. In this regard, a
possible reason for this disparity is the differential expression
of organic cation transporter (OCT) between skeletal muscle and
immortalized cell lines. OCT facilitates metformin uptake into
cells, and is required for the efficient action of metformin
(32). Although the present study
had certain limitations, our data do contribute to a better
understanding of metformin action.
In conclusion, our data demonstrated that metformin
enhanced IRS-1-associated insulin signaling preferentially by
activating AMPK and suppressing SREBP-1c activation in L6 cells
with PA-induced insulin-resistance, which led to the promotion of
glucose uptake. In addition, FAS as the downstream molecule of
SREBP-1c was also inhibited, which reduced lipogenesis and further
contributed to the improvement in muscular insulin resistance.
Therefore, the mechanism of actions of metformin revealed in the
present study suggest that AMPK and the SREBP-1c pathway may serve
as effective targets for the treatment of insulin resistance.
Acknowledgments
We would like to thank Professor Gong Dawei from the
University of Maryland for his constructive advice during the
preparation of the manuscript. The present study was sponsored by
grants from the National Natural Science Foundation of China Grant
Award (81270906, 81370947, 81070636), the Project of National Key
Clinical Division, the China Postdoctoral Science Foundation
(2012M521050), the Jiangsu Postdoctoral Science Foundation, Jiangsu
Province’s Key Provincial Talents Program (RC2011011), Jiangsu
Province’s Key Discipline of Medicine (XK201105), and the Key
Project of Nanjing Medical Science and Technology Development
Foundation (ZKX11017).
Abbreviations:
|
2-NBDG
|
2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-amino]-2-deoxy-D-glucose
|
|
AMPK
|
AMP-activated protein kinase
|
|
PA
|
palmitic acid
|
|
SRE
|
sterol regulatory element
|
|
SREBP-1c
|
sterol regulatory element binding
protein-1c
|
|
IRS-1
|
insulin receptor substrate-1
|
|
GLUT4
|
glucose transporter 4
|
|
FAS
|
fatty acid synthase
|
|
GAPDH
|
glyceraldehyde-3-phosphate
dehydrogenase
|
References
|
1
|
DeFronzo RA and Tripathy D: Skeletal
muscle insulin resistance is the primary defect in type 2 diabetes.
Diabetes Care. 32:S157–S163. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bosma M, Kersten S, Hesselink MK and
Schrauwen P: Re-evaluating lipotoxic triggers in skeletal muscle:
relating intramyocellular lipid metabolism to insulin sensitivity.
Prog Lipid Res. 51:36–49. 2012. View Article : Google Scholar
|
|
3
|
Muoio DM: Revisiting the connection
between intramyocellular lipids and insulin resistance: a long and
winding road. Diabetologia. 55:2551–2554. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mingrone G, Rosa G, Greco AV, Manco M,
Vega N, Nanni G, Castagneto M and Vidal H: Intramyocitic lipid
accumulation and SREBP-1c expression are related to insulin
resistance and cardiovascular risk in morbid obesity.
Atherosclerosis. 170:155–161. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shimano H: SREBP-1c and TFE3, energy
transcription factors that regulate hepatic insulin signalling. J
Mol Med. 85:437–444. 2007. View Article : Google Scholar
|
|
6
|
Raghow R, Yellaturu C, Deng X, Park EA and
Elam MB: SREBPs: the crossroads of physiological and pathological
lipid homeostasis. Trends Endocrinol Metab. 19:65–73. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bi Y, Wu W, Shi J, et al: Role for sterol
regulatory element binding protein-1c activation in mediating
skeletal muscle insulin resistance via repression of rat insulin
receptor substrate-1 transcription. Diabetologia. 57:592–602. 2014.
View Article : Google Scholar
|
|
8
|
Mantovani J and Roy R: Re-evaluating the
general(ized) roles of AMPK in cellular metabolism. FEBS Lett.
585:967–972. 2011. View Article : Google Scholar
|
|
9
|
Friedrichsen M, Mortensen B, Pehmøller C,
Birk JB and Wojtaszewski JF: Exercise-induced AMPK activity in
skeletal muscle: role in glucose uptake and insulin sensitivity.
Mol Cell Endocrinol. 366:204–214. 2013. View Article : Google Scholar
|
|
10
|
Musi N, Hirshman MF, Nygren J, et al:
Metformin increases AMP-activated protein kinase activity in
skeletal muscle of subjects with type 2 diabetes. Diabetes.
51:2074–2081. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou G, Myers R, Li Y, et al: Role of
AMP-activated protein kinase in mechanism of metformin action. J
Clin Invest. 108:1167–1174. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sajan MP, Bandyopadhyay G, Miura A, et al:
AICAR and metformin, but not exercise, increase muscle glucose
transport through AMPK-, ERK-, and PDK1-dependent activation of
atypical PKC. Am J Physiol Endocrinol Metab. 298:E179–E192. 2010.
View Article : Google Scholar :
|
|
13
|
Yang J, Craddock L, Hong S and Liu ZM:
AMP-activated protein kinase suppresses LXR-dependent sterol
regulatory element-binding protein-1c transcription in rat hepatoma
McA-RH7777 cells. J Cell Biochem. 106:414–426. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yap F, Craddock L and Yang J: Mechanism of
AMPK suppression of LXR-dependent Srebp-1c transcription. Int J
Biol Sci. 7:645–650. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li Y, Xu S, Mihaylova MM, et al: AMPK
phosphorylates and inhibits SREBP activity to attenuate hepatic
steatosis and atherosclerosis in diet-induced insulin-resistant
mice. Cell Metab. 13:376–388. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jung EJ, Kwon SW, Jung BH, Oh SH and Lee
BH: Role of the AMPK/SREBP-1 pathway in the development of orotic
acid-induced fatty liver. J Lipid Res. 52:1617–1625. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rachek LI, Musiyenko SI, LeDoux SP and
Wilson GL: Palmitate induced mitochondrial deoxyribonucleic acid
damage and apoptosis in L6 rat skeletal muscle cells.
Endocrinology. 148:293–299. 2007. View Article : Google Scholar
|
|
18
|
Dimopoulos N, Watson M, Sakamoto K and
Hundal HS: Differential effects of palmitate and palmitoleate on
insulin action and glucose utilization in rat L6 skeletal muscle
cells. Biochem J. 399:473–481. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hirabara SM, Silveira LR, Abdulkader F,
Carvalho CR, Procopio J and Curi R: Time-dependent effects of fatty
acids on skeletal muscle metabolism. J Cell Physiol. 210:7–15.
2007. View Article : Google Scholar
|
|
20
|
Zhao HL, Liu LZ, Sui Y, Ho SK, Tam SK, Lai
FM, Chan JC and Tong PC: Fatty acids inhibit insulin-mediated
glucose transport associated with actin remodeling in rat L6 muscle
cells. Acta Diabetol. 47:331–339. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bailey T: Options for combination therapy
in type 2 diabetes: comparison of the ADA/EASD position statement
and AACE/ACE algorithm. Am J Med. 126:S10–S20. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cho YM and Kieffer TJ: New aspects of an
old drug: metformin as a glucagon-like peptide 1 (GLP-1) enhancer
and sensitiser. Diabetologia. 54:219–222. 2011. View Article : Google Scholar
|
|
23
|
Rena G, Pearson ER and Sakamoto K:
Molecular mechanism of action of metformin: old or new insights?
Diabetologia. 56:1898–1906. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee JO, Lee SK, Jung JH, Kim JH, You GY,
Kim SJ, Park SH, Uhm KO and Kim HS: Metformin induces Rab4 through
AMPK and modulates GLUT4 translocation in skeletal muscle cells. J
Cell Physiol. 226:974–981. 2011. View Article : Google Scholar
|
|
25
|
Lee SK, Lee JO, Kim JH, et al: Metformin
sensitizes insulin signaling through AMPK-mediated PTEN
down-regulation in preadipocyte 3T3-L1 cells. J Cell Biochem.
112:1259–1267. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Barbero-Becerra VJ, Santiago-Hernandez JJ,
Villegas-Lopez FA, Mendez-Sanchez N, Uribe M and Chavez-Tapia NC:
Mechanisms involved in the protective effects of metformin against
nonalcoholic fatty liver disease. Curr Med Chem. 19:2918–2923.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fullerton MD, Galic S, Marcinko K, et al:
Single phosphorylation sites in Acc1 and Acc2 regulate lipid
homeostasis and the insulin-sensitizing effects of metformin. Nat
Med. 19:1649–1654. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Quan HY, Kim do Y, Kim SJ, Jo HK, Kim GW
and Chung SH: Betulinic acid alleviates non-alcoholic fatty liver
by inhibiting SREBP1 activity via the AMPK-mTOR-SREBP signaling
pathway. Biochem Pharmacol. 85:1330–1340. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee HI, Yun KW, Seo KI, Kim MJ and Lee MK:
Scopoletin prevents alcohol-induced hepatic lipid accumulation by
modulating the AMPK-SREBP pathway in diet-induced obese mice.
Metabolism. 63:593–601. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shimomura I, Matsuda M, Hammer RE,
Bashmakov Y, Brown MS and Goldstein JL: Decreased IRS-2 and
increased SREBP-1c lead to mixed insulin resistance and sensitivity
in livers of lipodystrophic and ob/ob mice. Mol Cell. 6:77–86.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ide T, Shimano H, Yahagi N, et al: SREBPs
suppress IRS-2-mediated insulin signalling in the liver. Nat Cell
Biol. 6:351–357. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen S, Zhou J, Xi M, Jia Y, Wong Y, Zhao
J, Ding L, Zhang J and Wen A: Pharmacogenetic variation and
metformin response. Curr Drug Metab. 14:1070–1082. 2013. View Article : Google Scholar : PubMed/NCBI
|