Introduction
Age-related macular degeneration (AMD) is a
progressive eye condition and the leading cause of severe vision
loss that affects the macula, which is the light-sensitive nerve
tissue lining at the back of the eye in the retina, in older
adults. AMD causes a number of complications, such as blurriness,
dark areas or distortion, with central vision (1). Choroidal neovascularization (CNV) is
a common symptom of the ‘wet’ form of AMD, which is the more
advanced type of AMD. CNV involves the sprouting of new blood
vessels that originate from the choroid under the macula that move
through a break in Bruch’s membrane and retinal pigment epithelium
(RPE) to reach the subretinal space (2). These blood vessels leak blood and
fluid that damages photoreceptor cells. The overexpression of
vascular endothelial growth factor (VEGF), a potent vascular
endothelial cell mitogen, appears to play a role in the development
of CNV. The RPE is a monolayer of pigmented cells of the retina.
Retinal pigment epithelial cells (RPE cells) play an important role
in the phagocytosis of shedded photoreceptor membranes, the
transport of nutrients from the vascular choroid, the formation of
the blood-retinal barrier and in the absorption of scattered light
(3).
Hypoxia, a condition in which the body is deprived
of a sufficient oxygen supply, is a potent inducer of VEGF
overexpression through an accumulation of hypoxia-inducible factors
(HIFs) in CNV (4). HIF-1 consists
of an oxygen-regulated inducible HIF-1α subunit and a
constitutively expressed HIF-1β subunit. Under hypoxic conditions,
HIF-1α is stabilized and accumulates. It then dimerizes with HIF-1β
and translocates to the nucleus, forming the active HIF complex
(5). Activated HIF then binds to
a hypoxia response element (HRE) and activates the transcription of
target genes, such as VEGF, which is a critical pathogenic factor
in CNV (4). Therefore, the
inhibition of the HIF pathway may be an attractive therapeutic
strategy for CNV.
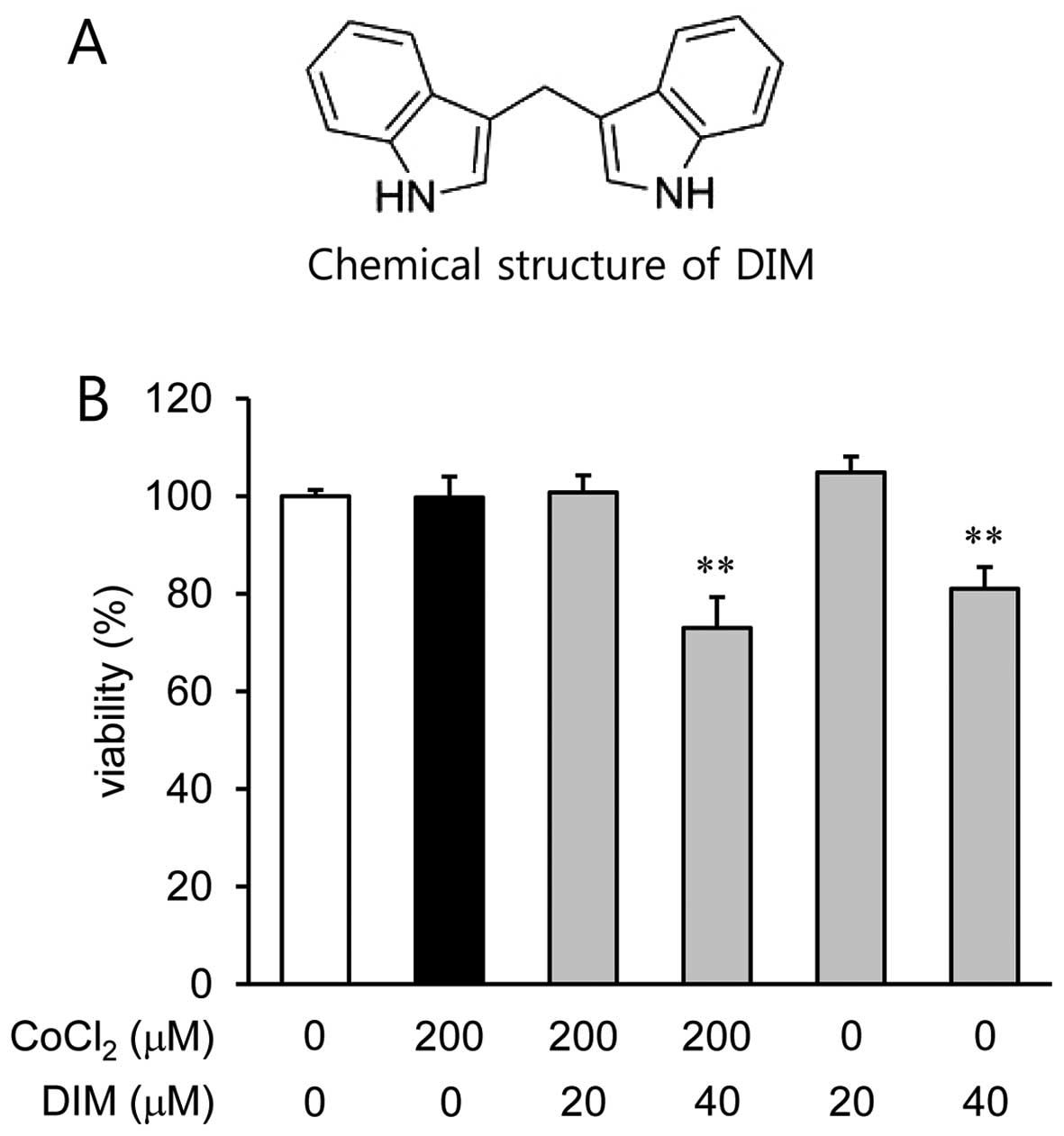
3,3′-Diindolylmethane (DIM) (Fig. 1A) is a major compound that is
derived from the digestion of indole-3-carbinol, which is found in
cruciferous vegetables, such as broccoli, Brussels sprouts,
cabbage, cauliflower and kale (6). DIM has been shown to have numerous
potential anti-cancer properties in breast, prostate and colorectal
cancer (7–9). Although DIM has been shown to exert
anti-angiogenic effects (10), to
the best of our knowledge, there are no available studies to date
on the effects of DIM on CNV. Thus, in the present study, we
investigated the anti-angiogenic effects of DIM and its effects on
signaling pathways using the human RPE cell line, APRE-19.
Materials and methods
Reagents
Cobalt chloride (CoCl2),
diphenyleneiodonium (DPI), N-acetyl-L-cysteine (NAC), U0126 [an
extracellular signal-regulated kinase (ERK)1/2 inhibitor] and DIM
were purchased from Sigma Chemical Co. (St. Louis, MO, USA). YCG063
[a mitochondrial reactive oxygen species (ROS) inhibitor] was
obtained from Millipore (Billerica, MA, USA). SB203580 [a p38
mitogen-activated protein kinase (MAPK) inhibitor] and SP600126 (a
JNK inhibitor) were purchased from Enzo Life Sciences, Inc.
(Farmingdale, NY, USA). PX-478 (an HIF-1α inhibitor) was purchased
from MedKoo Biosciences, Inc. (Chapel Hill, NC, USA). Mito PY1 was
purchased from Tocris Biosience (Bristol, UK). BAY 11-7082 and
parthenolide [nuclear factor (NF)-κB inhibitors] were purchased
from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). An
antibody against p65 was obtained from eBioscience (Cat. no.
14-6731; San Diego, CA, USA). An antibody against HIF-1α was
obtained from Novus Biologicals (Cat. no. NB100-105; Littleton, CO,
USA). Antibodies against phosphorylated (p)-ERK1/2 (Cat. no. 9106)
and p-p38 MAPK (Cat. no. 9211) were purchased from Cell Signaling
Technology (Beverly, MA, USA). Antibodies against ERK1/2 (Cat. no.
sc-94) and p38 MAPK (Cat. no. sc-535) were purchased from Santa
Cruz Biotechnology, Inc. Nitrocellulose membranes and an enhanced
chemiluminescence (ECL) kit were obtained from Amersham Pharmacia
Biotech (Uppsala, Sweden).
ARPE-19 cell culture
The ARPE-19 cells were obtained from the American
Type Culture Collection (Manassas, VA, USA) and cultured in
DMEM/F12 medium supplemented with 10% fetal bovine serum plus a 100
IU/ml penicillin and 100 µg/ml streptomycin mixture
(Gibco/BRL, Gaithersburg, MD, USA) in a humidified atmosphere (5%
CO2) at 37°C. The ARPE-19 cells were trypsinized, seeded
in 10-cm diameter dishes and incubated overnight until
attachment.
Cell viability assay
The viability of the ARPE-19 cells was determined
using the cell counting kit-8 (CCK-8) according to the
manufacturer’s instructions (Dojindo Laboratories, Kumamoto,
Japan). Briefly, the cells were seeded in triplicate at a density
of 1x104 cells/well into 96-well culture plates and
allowed to attach overnight. The cells were then incubated with the
indicated concentrations of DIM or 200 µM of
CoCl2 alone (to induce chemical hypoxia) or were treated
with DIM for 30 min prior to treatment with CoCl2. The
plates were incubated for 24 h and 10 µl of CCK-8 reagent
were added to each well. Following incubation for a further 2 h at
37°C, the plates were read at 450 nm using a microplate reader
(Model EL800; Bio-Tek, Winooski, VT, USA).
Depletion of HIF-1α by synthetic small
interfering RNA (siRNA)
HIF-1α-specific siRNA were purchased from Santa Cruz
Biotechnology, Inc. (Cat. no. sc-35561). Control siRNA-A (Cat. no.
sc-37007; Santa Cruz Biotechnology, Inc.) was used as a negative
control. In brief, 16 h after plating, the cells were trans-fected
with 20 nM HIF-1α-siRNA or control siRNA-A using siRNA transfection
reagent (Santa Cruz Biotechnology, Inc.) in accordance with the
manufacturer’s instructions. Following 6 h of incubation, an equal
volume of normal growth medium was added. Sixteen hours following
transfection, the cells were used for the experiments mentioned
below. The transfection efficiency was evaluated by determining
HIF-1α protein expression using western blot analysis.
Enzyme-linked immunosorbent assay
(ELISA)
VEGF levels in the cell culture medium were assessed
by ELISA. The cells were treated with various concentrations of DIM
for 30 min prior to CoCl2 stimulation. Following
incubation for 24 h under hypoxic conditions, the culture
supernatants were collected. The VEGF levels were measured using a
VEGF DuoSet ELISA Development kit (R&D Systems, Minneapolis,
MN, USA) according to the manufacturer’s instructions. The
absorbance at 450 nm was determined using a microplate reader
(Model EL800; Bio-Tek).
Western blot analysis
Western blot analysis was carried out as previously
described (11). The ARPE-19
cells were washed 3 times with PBS and lysed with lysis buffer
(Mammalian Cell-PE LB; G-Biosciences, St. Louis, MO, USA). Equal
amounts of protein were separated on 10% sodium dodecyl sulfate
(SDS)-polyacrylamide minigels and transferred onto nitrocellulose
transfer membranes. Following incubation with the appropriate
primary antibody, the membranes were incubated for 1 h at room
temperature with a secondary antibody conjugated to horseradish
peroxidase [goat anti-mouse IgG (Cat. no. sc-2031; Santa Cruz
Biotechnology, Inc.) and donkey anti-rabbit IgG (Cat. no. A16035,
Pierce, Rockford, IL, USA)]. Following 3 washes in Tris-buffered
saline Tween-20 (TBST), the immunoreactive bands were visualized
using the ECL detection system (Pierce).
Assay of mitochondrial ROS levels
The ARPE-19 cells (1×104 cells/well) were
seeded in 96-well plates in a humidified atmosphere containing 5%
CO2 at 37°C for 16 h. Following 16 h of incubation, the
cells were treated with 5 µM Mito PY1 for 30 min and further
incubated for 24 h with CoCl2. The cells were then
immediately evaluated. The mitochondrial ROS levels were measured
using a fluorescence microplate reader (SpetraMax M2; Molecular
Devices, Sunnyvale, CA, USA) at an excitation wavelength of 485 nm
and an emission wavelength of 538 nm.
Preparation of nuclear extracts and
electrophoretic mobility shift assay (EMSA)
Nuclear extracts were prepared using NE-PER nuclear
extraction reagent (Pierce). As a probe for the gel retardation
assay, an oligonucleotide containing the immunoglobulin κ-chain
binding site (κB, 5′-GATCTCAGAGGGGACTTTCCGAGAGA-3′) was
synthesized. A typical double-stranded oligonucleotide for the
HIF-1α binding DNA sequence (5′-TCTGTACGTGACCACACTCACCTC-3′) was
purchased from Santa Cruz Biotechnology, Inc. (Cat. no. sc-2625). A
non-radioactive method in which the 3′ end of the probe was labeled
with biotin was used in these experiments (Pierce). The binding
reactions contained 5 µg of nuclear extract protein, buffer
(10 mM Tris, pH 7.5, 50 mM KCl, 5 mM MgCl2, 1 mM
dithiothreitol, 0.05% Nonidet P-40 and 2.5% glycerol), 50 ng of
poly(dI-dC) and 20 fM of the biotin-labeled DNA. The reactions were
incubated for 20 min at room temperature in a final volume of 20
µl. The competition reactions were conducted by the addition
of 100-fold excess of unlabeled NF-κB p65 and 25-fold excess of
unlabeled HIF-1α to the reaction mixture. The mixture was then
separated by electrophoresis on a 5% polyacrylamide gel in 0.5X
Tris-borate buffer and transferred onto nylon membranes. The
biotin-labeled DNA was detected using a LightShift chemiluminescent
EMSA kit (Pierce).
Statistical analysis
Data values represent the means ± standard error of
the mean (SEM). The statistical significance of the differences
between the data sets was evaluated using ANOVA. Data were analyzed
and plotted on graphs using GraphPad Prism software (GraphPad
Software, La Jolla, CA, USA). A value of p<0.05 was considered
to indicate a statistically significant difference.
Results
Effects of DIM on the viability of human
ARPE-19 cells
We examined the viability of human ARPE-19 cells
pre-treated with DIM (20 and 40 µM) by CCK-8 assay under
normoxic (no CoCl2) or hypoxic conditions
(CoCl2). DIM was not cytotoxic to the human ARPE-19
cells at concentrations of up to 20 µM; however, the cell
viability was reduced by 19% with the dose of 40 µM of DIM
(Fig. 1B) under both normoxic and
hypoxic conditions. Based on these results, a concentration lower
than 20 µM of DIM was selected for the subsequent
experiments.
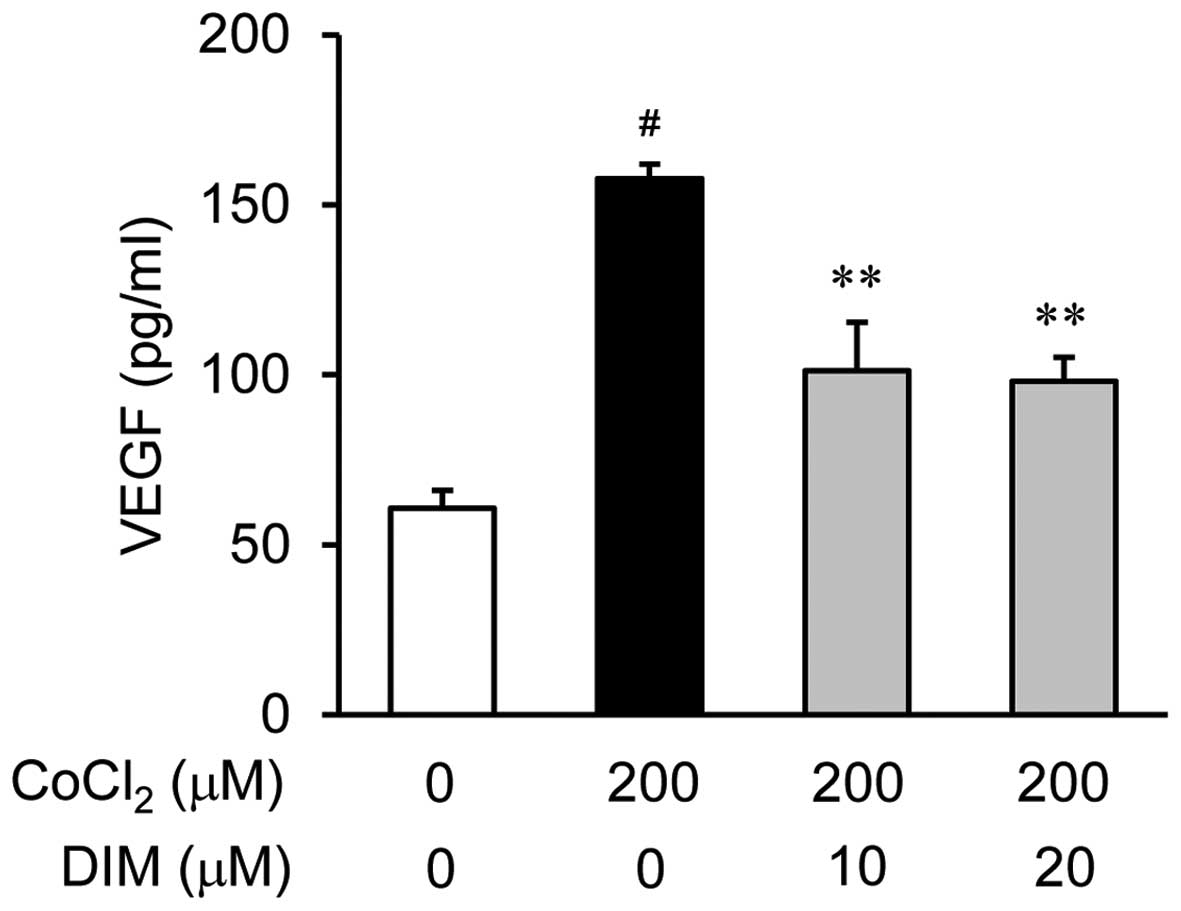
Effects of DIM on VEGF production under
hypoxic conditions
To examine the anti-angiogenic properties of DIM and
its effects on the CoCl2-induced production of VEGF in
the ARPE-19 cells, we measured the secretion of VEGF into the
culture medium using ELISA. The ARPE-19 cells were treated with
various concentrations of DIM (0, 10 or 20 µM) for 30 min
prior to stimulation with 200 µM CoCl2 for 24 h
(Fig. 2). As revealed by the VEGF
measurements obtained using ELISA, the VEGF levels were
significantly increased in the ARPE-19 cells following 24 h of
exposure to hypoxic conditions (CoCl2) compared to
normoxic conditions (no CoCl2; by approximately 3-fold);
this increase was reversed by treatment with DIM in a
dose-dependent manner.
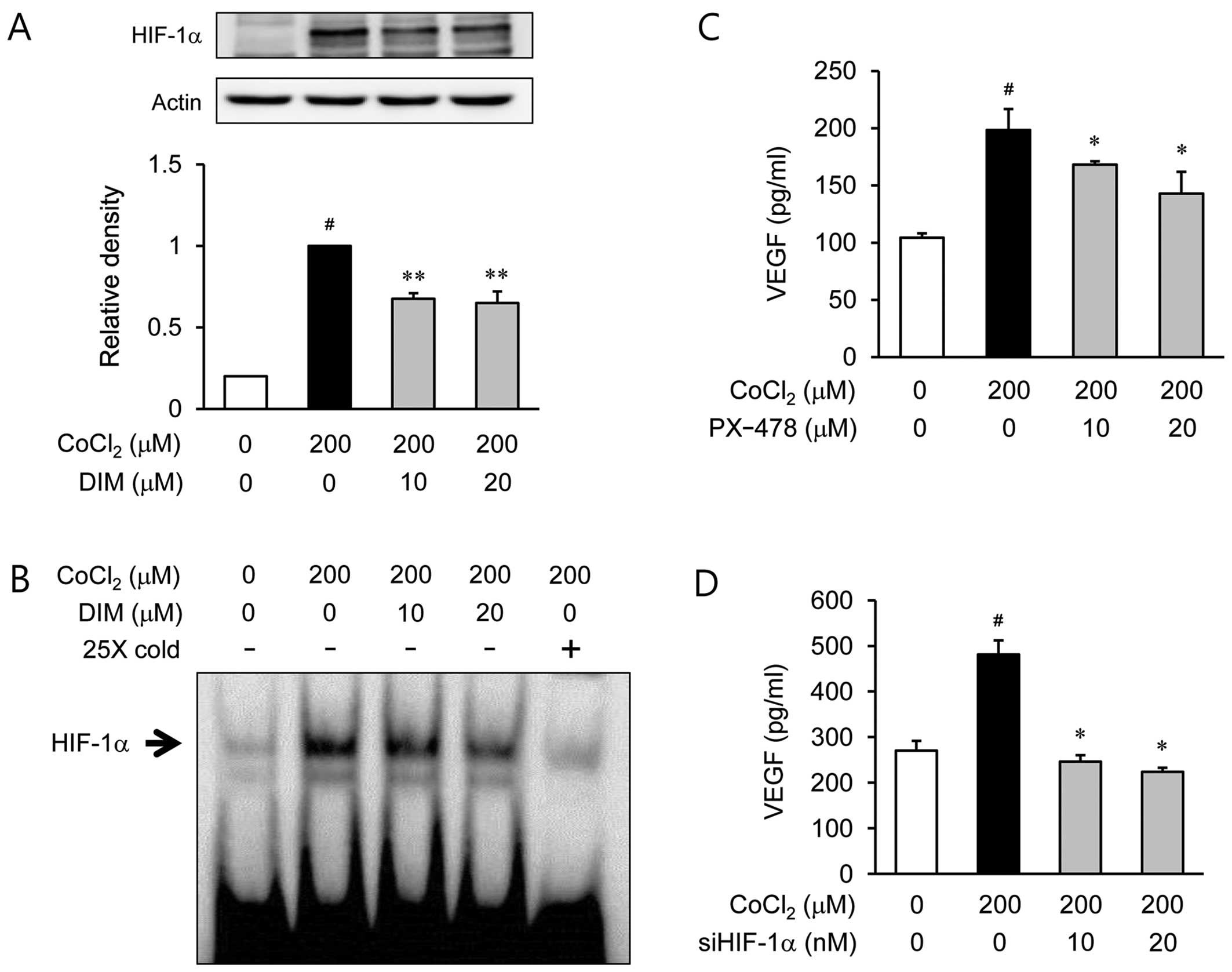
Effects of DIM on HIF-1α activation under
hypoxic conditions
To determine whether DIM inhibits HIF-1α
translocation, the ARPE-19 cells were incubated with DIM (10 or 20
µM) under hypoxic conditions for 6 h (Fig. 3A). The 200 µM
CoCl2-stimulated ARPE-19 cells elicited an almost 5-fold
increase in the translocation of HIF-1α to the nuclei compared to
the cells under normoxic conditions. By contrast, pre-treatment of
the cells with DIM (10 or 20 µM) significantly inhibited the
translocation of HIF-1α under hypoxic conditions. Further
experiments were carried out to determine whether the activation of
HIF-1α in the ARPE-19 cells is altered under hypoxic conditions by
pre-treatment with DIM. When the nuclear extract proteins from the
cells were probed with oligonucleotides within the VEGF promoter, a
subsequent gel shift analysis revealed that the ARPE-19 cells
displayed a marked increase in HIF-1α binding activity under
hypoxic conditions (Fig. 3B).
However, the induction of specific HIF-1α DNA binding activity
following stimulation with CoCl2 was inhibited by
treatment with DIM (10 or 20 µM). These results indicate
that DIM inhibits HIF-1α activity by preventing the translocation
of this transcription factor to the nucleus during hypoxic
conditions. To evaluate whether HIF-1α is a critical factor in the
expression of VEGF, PX-478 (10 or 20 µM), an HIF-1α
inhibitor (Fig. 3C), and siHIF-1α
(10 or 20 nM), siRNA against HIF-1α (Fig. 3D), were used. As expected, the
secretion of VEGF was significantly decreased following the
inhibition of HIF-1α.
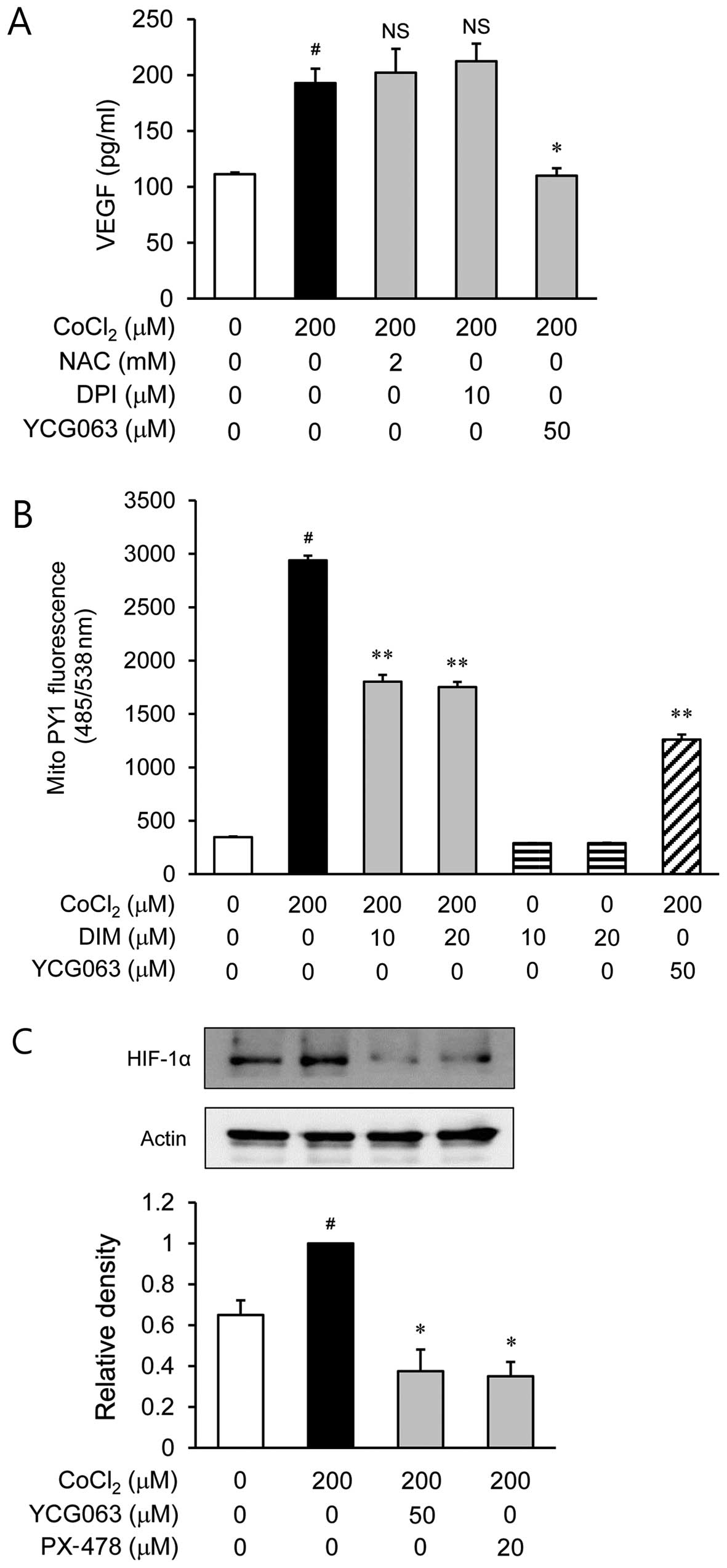
Involvement of mitochondrial ROS in the
secretion of VEGF through HIF-1α in CoCl2-stimulated
ARPE-19 cells
To examine the role of ROS in the
CoCl2-stimulated secretion of VEGF, the ARPE-19 cells
were treated with various inhibitors of ROS, such as NAC for ROS
scavengers, DPI as an NADPH oxidase inhibitor and YCG063 as a
mitochondrial ROS inhibitor, prior to CoCl2 stimulation.
NAC and DPI had no effect on VEGF production during hypoxia
(Fig. 4A). However, YCG063
reduced the production of VEGF which was induced by
CoCl2 stimulation. Based on this result, we investigated
whether DIM decreases the level of mitochondrial ROS generation in
CoCl2-stimulated ARPE-19 cells. As shown by our results,
DIM reduced the level of mitochondrial ROS (Fig. 4B). Subsequently, we examined the
effects of YCG063 on the accumulation of HIF-1α in
CoCl2-stimulated ARPE-19 cells (Fig. 4C). The accumulation of HIF-1α
increased in the nuclei of the CoCl2-stimulated ARPE-19
cells; this increase was significantly reduced by treatment with
YCG063.
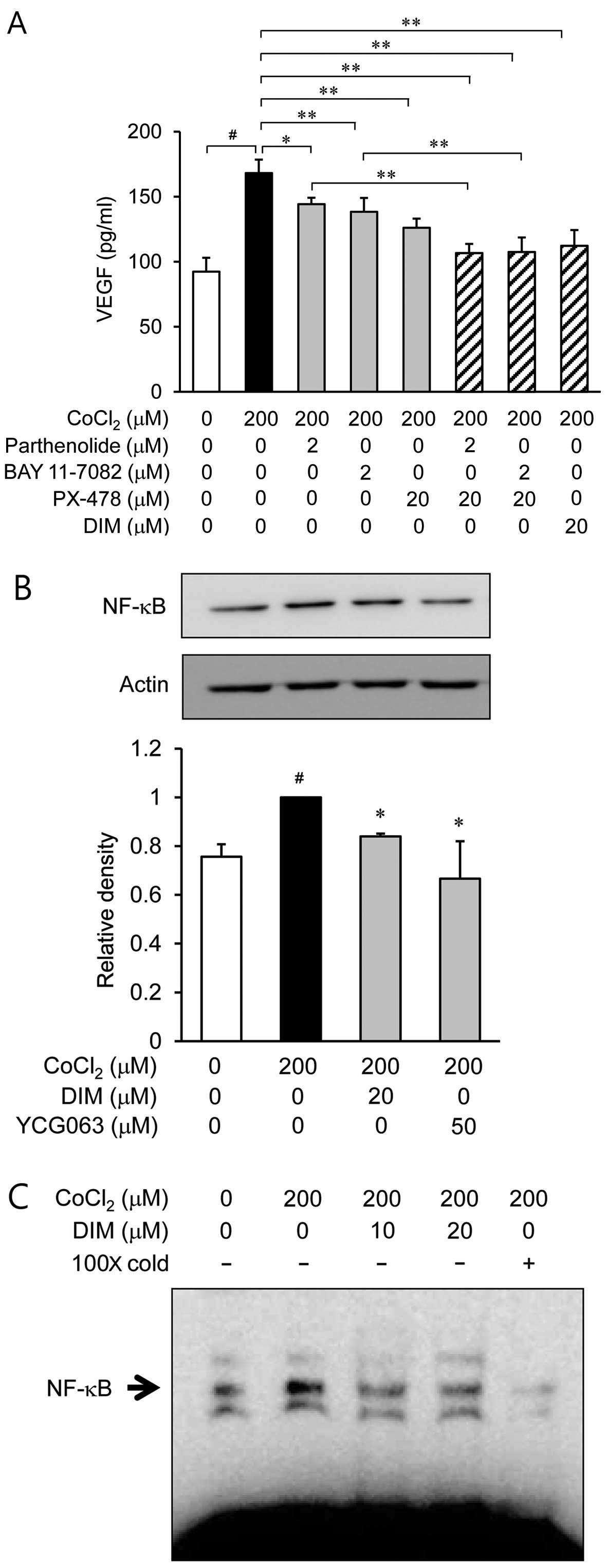
Inhibitory effects of DIM on the
secretion of VEGF through the inhibition of NF-κB activation
Since NF-κB is a transcription factor that regulates
the expression of VEGF (12), we
investigated whether DIM inhibits the translocation of NF-κB from
the cytosol to the nucleus or NF-κB binding activity. First, the
ARPE-19 cells were treated with the NF-κB inhibitors, parthenolide
and BAY 11-7082, prior to stimulation with CoCl2.
Parthenolide (2 µM) and BAY 11-7082 (2 µM) caused a
reduction in the production of VEGF induced by hypoxia
(CoCl2; Fig. 5A). The
HIF-1α inhibitor, PX-478, also caused a decrease in VEGF secretion.
Western blot analyses using nuclear fractions revealed that the
accumulation of NF-κB p65 in the nucleus was markedly increased
following treatment with CoCl2 alone; however,
pre-treatment with DIM (20 µM) reversed this trend (Fig. 5B). We then investigated the
effects of DIM on the DNA-binding activity of NF-κB using EMSA
(Fig. 5C). Stimulation with
CoCl2 caused a significant increase in the DNA-binding
activity of NF-κB, whereas treatment with DIM markedly reduced the
CoCl2-induced DNA-binding activity of NF-κB. Moreover,
the upregulated translocation of NF-κB p65 was significantly
inhibited by treatment with YCG063 (Fig. 5B). These results indicate that the
generation of ROS following exposure to hypoxia serves as an
upstream signal for the activation of NF-κB.
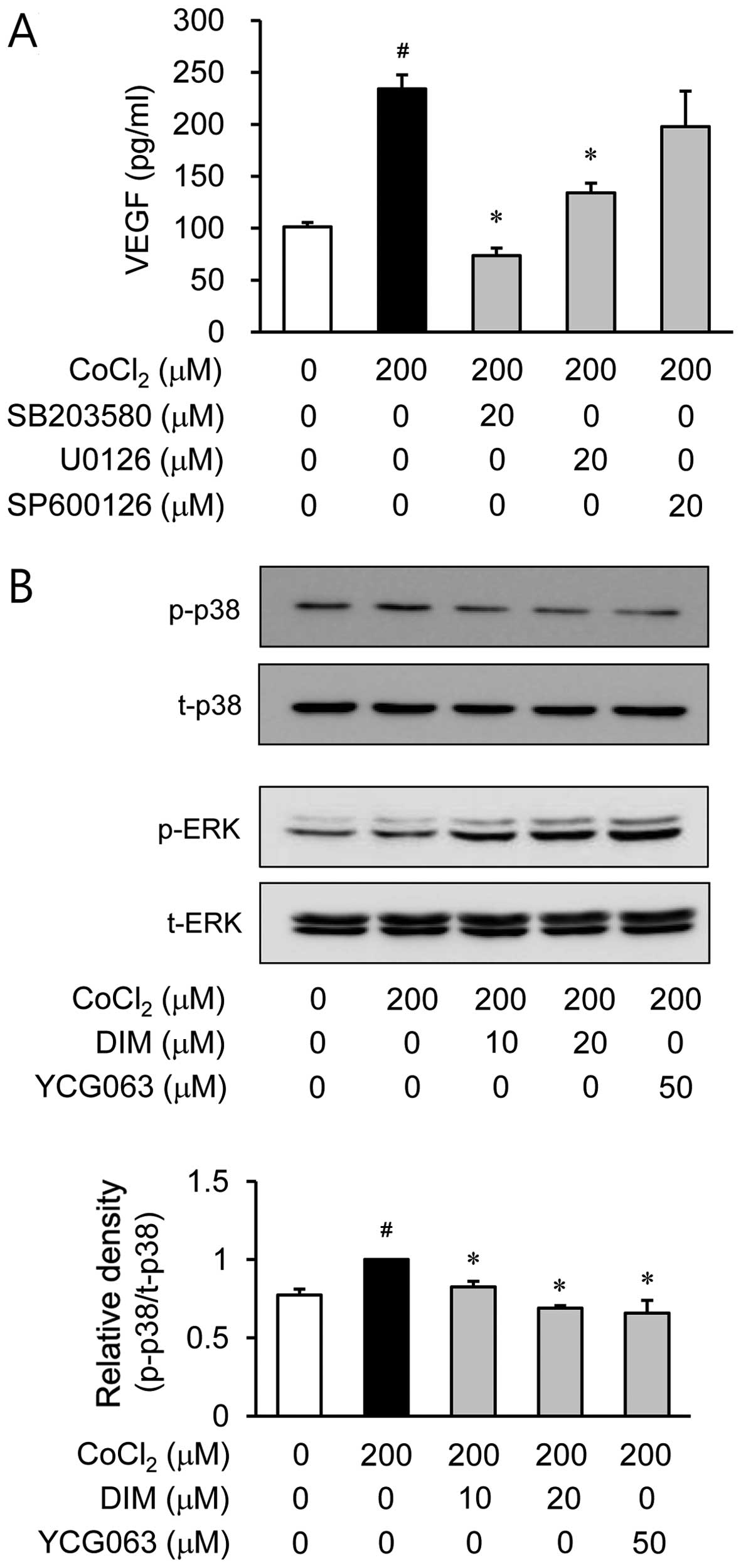
Effects of DIM on the phosphorylation
MAPKs in CoCl2-stimulated ARPE-19 cells
It is well known that MAPK signaling molecules are
able to regulate HIF-1α activation (13). Therefore, in this study, we
examined the effects of DIM on hypoxia-induced MAPK activation. The
ARPE-19 cells were pre-treated with various inhibitors of signal
transduction pathways, such as SB203580 for p38 MAPK, U0126 for ERK
and SP600126 for JNK, prior to CoCl2 stimulation. Among
these inhibitors, SP600126 did not affect VEGF production during
hypoxia. However, SB203580 and U0126 reduced VEGF production which
was induced by hypoxia (Fig. 6A).
Based on these results, we investigated the effects of DIM on p38
MAPK and ERK activation induced by hypoxia. Pre-treatment with DIM
resulted in a significant inhibition of CoCl2-induced
p38 MAPK phosphorylation (Fig.
6B). However, DIM did not affect ERK phosphorylation during
hypoxia. These results demonstrated that the inhibition of VEGF
production by DIM in CoCl2-stimulated ARPE-19 cells was
associated with the downregulation of p38 MAPK phosphorylation. We
then investigated whether p38 MAPK phosphorylation is associated
with mitochondrial ROS generation. The ARPE-19 cells were treated
with CoCl2 for 2 h in the presence or absence of YCG063,
a mitochondrial ROS inhibitor. Pre-treatment with YCG063 resulted
in a significant attenuation of CoCl2-induced p38 MAPK
phosphorylation (Fig. 6B). These
results indicate that the generation of mitochondrial ROS induced
by exposure to hypoxia serves as an upstream signal for the
induction of VEGF production by p38 MAPK activation. Furthermore,
to elucidate the downstream signal responsible for the induction of
VEGF production by p38 MAPK activation, we investigated NF-κB
activation by western blot analysis of the cell nuclei. When the
ARPE-19 cells were treated with the p38 MAPK inhibitor, SB203580,
the translocation of NF-κB was reduced under hypoxic conditions
(Fig. 6C).
Discussion
Under hypoxic conditions, RPE cells release various
growth factors, resulting in angiogenesis, fibrovascular tissue
formation and retinal ablation (14). Among the hypoxia-stimulated growth
factors, VEGF is a pivotal regulator of vasculogenesis and
angiogenesis in CNV (4). In the
present study, we investigated whether DIM inhibits VEGF secretion
from ARPE-19 cells stimulated with CoCl2 (to mimic
hypoxic conditions). CoCl2 is commonly used as a
hypoxia-mimetic agent as, similar to hypoxia, it blocks the
degradation and thus induces the accumulation of HIF-1α protein
(15). As expected, the secretion
of VEGF significantly increased under chemical hypoxic conditions
without any cytotoxicity. However, the increased levels of VEGF
secretion were significantly inhibited by treatment with 10 and 20
µM DIM under hypoxic conditions (Fig. 2). This study demonstrates that DIM
inhibits the CoCl2-induced production of VEGF by
suppressing the activation of NF-κB and HIF-1α.
It has been reported that through the HIF
transcriptional complex, hypoxia induces the production of VEGF
(16,17). HIF-1α plays a critical role in
CNV. Therefore, the inhibition of the HIF pathway offers an
attractive therapeutic strategy for CNV. We thus investigated
whether DIM attenuates the translocation of HIF-1α to the nucleus
and binding to HRE of the VEGF promoter. Treatment with DIM
attenuated HIF-1α accumulation (Fig.
3A) and HIF-1α-dependent binding activity (Fig. 3B). To verify whether hypoxia
induces the production of VEGF through the HIF transcriptional
complex, we used an HIF-1α inhibitor and siRNA against HIF-1α under
chemical hypoxic conditions. As expected, our results revealed that
PX-478 (an inhibitor of HIF-1α) and siHIF-1α inhibited VEGF
production in the CoCl2-stimulated ARPE-19 cells,
suggesting that DIM may be useful for ocular neovascularization
through the downregulation of HIF-1α activation.
It has been demonstrated that ROS induces the
expression of VEGF (18) and is
crucial for the induction of angiogenesis (19). To deter, ome whether ROS levels
affect VEGF production, the CoCl2-stimulated ARPE-19
cells were pre-treated with NAC, DPI and YCG063 (Fig. 4A). Among these inhibitors of ROS,
NAC and DPI did not affect VEGF production during hypoxia. However,
YCG063 (an inhibitor of mitochondrial ROS) significantly decreased
the production of VEGF. Based on this result, we investigated
whether DIM decreases the level of mitochondrial ROS generation in
CoCl2-stimulated ARPE-19 cells (Fig. 4B). As shown by the results of the
fluorescence microplate analysis, DIM reduced the level of
mitochondrial ROS generation. Since ROS regulates VEGF production
through the induction of HIF-1α, we examined the effects of YCG063
against HIF-1α accumulation in CoCl2-stimulated ARPE-19
cells by western blot analysis (Fig.
4C). The accumulation of HIF-1α increased in the nuclei of the
CoCl2-stimulated ARPE-19 cells. However, this increase
was significantly reduced by treatment with YCG063. Taken together,
the inhibition of mitochondrial ROS generation by DIM is consistent
with inhibition of HIF-1α accumulation and VEGF expression, which
may result in reduced ocular neovascularization.
It has been reported that hypoxia induced by
CoCl2 leads to increased ROS production, the activation
of NF-κB and the induction of VEGF in human RPE cells (20). Therefore, in this study, we
examined the effects of the NF-κB inhibitors, parthenolide and BAY
11-7082, on the CoCl2-induced secretion of VEGF by
ARPE-19 cells. Our results revealed that pre-treatment with
parthenolide and BAY 11-7082 inhibited the secretion of VEGF under
hypoxic conditions (Fig. 5A).
However, pre-treatment with the HIF-1α inhibitor, PX-478, had a
greater inhibitory effect than pre-treatment with the NF-κB
inhibitors on CoCl2-stimulated VEGF secretion. Of note,
the inhibition of both the HIF-1α and NF-κB pathways with a
combination (Fig. 5A:
parthenolide + PX-478 and BAY 11-7082 + PX-478, respectively)
pre-treatment had the greatest inhibitory effect on VEGF secretion
in the CoCl2-stimulated ARPE-19 cells. These results
suggest that both signaling pathways, HIF-1α and NF-κB, play
critical and synergistic roles in the production of VEGF. Based on
the above results of the inhibitory effects of NF-κB on VEGF
secretion, we investigated whether DIM modulates NF-κB activity in
CoCl2-stimulated ARPE-19 cells. Since activated free
NF-κB p65 enters the nucleus and induces VEGF expression, we
investigated the nuclear translocation of the NF-κB subunit, p65.
Western blot analysis revealed that stimulation with
CoCl2 induced the translocation of NF-κB p65 into the
nuclear compartment (Fig. 5B).
However, the CoCl2-induced the nuclear translocation of
NF-κB p65 was inhibited in the presence of DIM. According to the
EMSA data, CoCl2 stimulation increased the DNA-binding
activity of NF-κB. However, pre-treatment with DIM suppressed the
CoCl2-induced DNA-binding activity of NF-κB (Fig. 5C). These data indicate that DIM
inhibits NF-κB activity in CoCl2-stimulated ARPE-19
cells by suppressing the nuclear translocation of p65 and,
consequently, attenuates the production of VEGF.
A previous study indicated that MAPK signaling
pathways are involved in regulating the expression of HIF-1α and
VEGF under hypoxic conditions (21). Based on this result, we
hypothesized that anti-neovascularization mechanisms are related to
MAPK pathways. To elucidate the regulatory mechanism of DIM in
these processes, we investigated whether the MAPK signaling
pathways are involved in regulating VEGF production. p38 MAPK- and
ERK-specific inhibitors suppressed the secretion of VEGF. However,
the JNK-specific inhibitor did not affect VEGF production during
hypoxia (Fig. 6A). Subsequently,
to investigate whether DIM regulates these pathways, we used
western blot analyses to examine the phosphorylation of ERK and p38
MAPK. CoCl2 stimulation increased the phosphorylation of
p38 MAPK (Fig. 6B). However,
pre-treatment with DIM markedly reduced the phosphorylation of p38
MAPK in the CoCl2-stimulated ARPE-19 cells. Moreover, to
examine the downstream pathway of p38 MAPK, we investigated whether
the p38 MAPK inhibitor, SB203580, regulates NF-κB activation in
CoCl2-stimulated ARPE-19 cells. According to the results
of western blot analyses using nuclear fractions, we demonstrated
that the accumulation of NF-κB p65 in the nucleus was increased
following treatment with CoCl2 alone. However,
pre-treatment with SB203580 (a MAPK inhibitor) markedly reversed
this trend. Of note, the MAPK inhibitor did not affect HIF-1α
accumulation during hypoxia (data not shown). Moreover, the
phosphorylation of p38 MAPK was suppressed by pre-treatment with
the mitochondrial ROS inhibitor, YCG063. Collectively, these
results demonstrate that DIM inhibits the CoCl2-induced
production of VEGF by regulating the mitochondrial ROS/p38
MAPK/NF-κB signaling pathways in ARPE-19 cells. The MAPK pathways
are upstream of NF-κB, but not HIF-1α.
In conclusion, the findings of this study
demonstrated that hypoxia induced by CoCl2 increased the
release of VEGF from cultured ARPE-19 cells. Pre-treatment with DIM
significantly attenuated the production of VEGF in ARPE-19 cells
under hypoxic conditions by suppressing the activation of the
mitochondrial ROS/HIF-1α and mitochondrial ROS/p38 MAPK/NF-κB
pathways. These data suggest that DIM is effective in preventing
the hypoxia-induced secretion of VEGF in RPE cells and may be
developed into a novel anti-angiogenic agent for AMD.
Acknowledgments
This study was supported the Basic Science Research
Program through the National Research Foundation of Korea (NRF)
funded by the Ministry of Education, Science and Technology
(2013-R1A1A4A01011649). This study was also supported by the 2011
Research Grant from Kangwon National University.
Abbreviations:
|
AMD
|
age-related macular degeneration
|
|
CoCl2
|
cobalt chloride
|
|
CNV
|
choroidal neovascularization
|
|
VEGF
|
vascular endothelial growth factor
|
|
DIM
|
3,3′-diindolylmethane
|
|
RPE
|
retinal pigment epithelium
|
|
HIF-1α
|
hypoxia-inducible factor-1α
|
|
MAPKs
|
mitogen-activated protein kinases
|
|
ROS
|
reactive oxygen species
|
References
|
1
|
Costagliola C, Agnifili L, Arcidiacono B,
Duse S, Fasanella V, Mastropasqua R, Verolino M and Semeraro F:
Systemic thromboembolic adverse events in patients treated with
intravitreal anti-VEGF drugs for neovascular age-related macular
degeneration. Expert Opin Biol Ther. 12:1299–1313. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Campochiaro PA: Ocular neovascularization.
J Mol Med Berl. 91:311–321. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Simó R, Villarroel M, Corraliza L,
Hernández C and Garcia-Ramírez M: The retinal pigment epithelium:
Something more than a constituent of the blood-retinal barrier -
implications for the pathogenesis of diabetic retinopathy. J Biomed
Biotechnol. 2010:1907242010. View Article : Google Scholar
|
|
4
|
Yang XM, Wang YS, Zhang J, Li Y, Xu JF,
Zhu J, Zhao W, Chu DK and Wiedemann P: Role of PI3K/Akt and MEK/ERK
in mediating hypoxia-induced expression of HIF-1alpha and VEGF in
laser-induced rat choroidal neovascularization. Invest Ophthalmol
Vis Sci. 50:1873–1879. 2009. View Article : Google Scholar
|
|
5
|
Ke Q and Costa M: Hypoxia-inducible
factor-1 (HIF-1). Mol Pharmacol. 70:1469–1480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Banerjee S, Kong D, Wang Z, Bao B, Hillman
GG and Sarkar FH: Attenuation of multi-targeted
proliferation-linked signaling by 3,3′-diindolylmethane (DIM): From
bench to clinic. Mutat Res. 728:47–66. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brignall MS: Prevention and treatment of
cancer with indole-3-carbinol. Altern Med Rev. 6:580–589. 2001.
|
|
8
|
Shertzer HG and Senft AP: The
micronutrient indole-3-carbinol: Implications for disease and
chemoprevention. Drug Metabol Drug Interact. 17:159–188. 2000.
View Article : Google Scholar
|
|
9
|
Lee SH, Kim JS, Yamaguchi K, Eling TE and
Baek SJ: Indole-3-carbinol and 3,3′-diindolylmethane induce
expression of NAG-1 in a p53-independent manner. Biochem Biophys
Res Commun. 328:63–69. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kunimasa K, Kobayashi T, Kaji K and Ohta
T: Antiangiogenic effects of indole-3-carbinol and
3,3′-diindolylmethane are associated with their differential
regulation of ERK1/2 and Akt in tube-forming HUVEC. J Nutr.
140:1–6. 2010. View Article : Google Scholar
|
|
11
|
Yu BC, Lee DS, Bae SM, Jung WK, Chun JH,
Urm SH, Lee DY, Heo SJ, Park SG, Seo SK, et al: The effect of
cilostazol on the expression of matrix metalloproteinase-1 and type
I procollagen in ultraviolet-irradiated human dermal fibroblasts.
Life Sci. 92:282–288. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kiriakidis S, Andreakos E, Monaco C,
Foxwell B, Feldmann M and Paleolog E: VEGF expression in human
macrophages is NF-kappaB-dependent: Studies using adenoviruses
expressing the endogenous NF-kappaB inhibitor IkappaBalpha and a
kinase-defective form of the IkappaB kinase 2. J Cell Sci.
116:665–674. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sodhi A, Montaner S, Miyazaki H and
Gutkind JS: MAPK and Akt act cooperatively but independently on
hypoxia inducible factor-1alpha in rasV12 upregulation of VEGF.
Biochem Biophys Res Commun. 287:292–300. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vadlapatla RK, Vadlapudi AD, Pal D,
Mukherji M and Mitra AK: Ritonavir inhibits HIF-1α-mediated VEGF
expression in retinal pigment epithelial cells in vitro. Eye
(Lond). 28:93–101. 2014. View Article : Google Scholar
|
|
15
|
Guo M, Song LP, Jiang Y, Liu W, Yu Y and
Chen GQ: Hypoxia-mimetic agents desferrioxamine and cobalt chloride
induce leukemic cell apoptosis through different hypoxia-inducible
factor-1α independent mechanisms. Apoptosis. 11:67–77. 2006.
View Article : Google Scholar
|
|
16
|
Ratcliffe PJ, O’Rourke JF, Maxwell PH and
Pugh CW: Oxygen sensing, hypoxia-inducible factor-1 and the
regulation of mammalian gene expression. J Exp Biol. 201:1153–1162.
1998.PubMed/NCBI
|
|
17
|
Ratcliffe PJ, Pugh CW and Maxwell PH:
Targeting tumors through the HIF system. Nat Med. 6:1315–1316.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fay J, Varoga D, Wruck CJ, Kurz B,
Goldring MB and Pufe T: Reactive oxygen species induce expression
of vascular endothelial growth factor in chondrocytes and human
articular cartilage explants. Arthritis Res Ther. 8:R1892006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xia C, Meng Q, Liu LZ, Rojanasakul Y, Wang
XR and Jiang BH: Reactive oxygen species regulate angiogenesis and
tumor growth through vascular endothelial growth factor. Cancer
Res. 67:10823–10830. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cervellati F, Cervellati C, Romani A,
Cremonini E, Sticozzi C, Belmonte G, Pessina F and Valacchi G:
Hypoxia induces cell damage via oxidative stress in retinal
epithelial cells. Free Radic Res. 48:303–312. 2014. View Article : Google Scholar
|
|
21
|
Gupta B, Chiang L, Chae K and Lee DH:
Phenethyl isothiocyanate inhibits hypoxia-induced accumulation of
HIF-1α and VEGF expression in human glioma cells. Food Chem.
141:1841–1846. 2013. View Article : Google Scholar : PubMed/NCBI
|