Introduction
Cerebral ischemia, particularly during the period of
ischemia/reperfusion, involves complex biochemical mechanisms, and
compared with other organs, brain tissue is more likely to produce
free radicals and lipid peroxides; therefore, oxidative stress is
likely to be the key factor responsible for the irreversible damage
caused during cerebral ischemia/reperfusion (1). It has been confirmed that oxidative
stress can affect glial cells, which are associated with neuronal
death or a decrease in neuronal cell proliferation (2). At present, however, we have not
acquired a satisfactory curative effect with regards to the
application of exogenous antioxidants and anti-apoptotic agents. It
is well known that the endogenous antioxidant defense system is
crucial to the survival of nerve cells (3). Therefore, endogenous antioxidants
and anti-apoptotic agents may provide an effective therapeutic
intervention in ischemic brain injury. In addition, an improved
understanding of the disease mechanisms is crucial for developing
applicable treatment strategies.
Sulfiredoxin 1 (Srxn1), a central endogenous
antioxidant protein belonging to the sulfiredoxin family of
antioxidants, was initially characterized in yeast (4) and subsequently in mammalian cells
(5) before it was reported to
have an important function in neuroprotection (3). Srxn1 functions as one of the
reactive oxygen species (ROS) management systems that counter cell
oxidative stress-induced damage (6). It has been demonstrated that Srxn1
protects brain tissue from damage through the regulation of
glutathionylation/deglutathionylation in Parkinson’s disease (PD)
(7). The induction of Srxn1
expression has been shown to exert neuroprotective effects against
hydrogen peroxide (H2O2)-induced cell death
(8). The reduction of Srxn1
expression has been shown to lead to an increased sensitivity to
oxidative stress, and this sensitivity was reduced by the
overexpression of intracellular Srxn1 (6,9,10).
In addition, it has been reported that the induction of the
expression of Srxn1 contributes to neuroprotective ischemic
preconditioning in response to oxygen-glucose deprivation (OGD)
in vitro and following brief ischemic episodes in
vivo (11). Moreover, Srxn1
can enter the mitochondria to maintain a balance between
mitochondrial H2O2 production and elimination
(12). Furthermore, Srxn1 may be
a novel component in maintaining the balance between
H2O2 production and elimination and
protecting A549 cells or wild-type mouse embryonic fibroblasts
(MEFs) from apoptosis (13). This
may be related to the inhibition of the release of cytochrome
c (Cyt.C) and the activation of caspase-9 and caspase-3
(13). Nevertheless, the specific
function and related regulatory mechanisms of action of Srxn1 in
cerebral ischemia and in damage induced by apoptosis have not been
extensively investigated.
In our previous study, we demonstrated that the
knockdown of Srxn1 resulted in an increased sensitivity to
H2O2 in PC12 cells, indicating that Srxn1 was
essential for antioxidant proteins (1). The aim of this study was to
investigate the function of Srxn1 with respect to apoptosis in rat
cortical astrocytes, as well as the potential protective mechanisms
of action of Srxn1.
Materials and methods
Reagents
Glucose-high Dulbecco’s modified Eagle’s medium/F12
(DMEM/F12), glucose-free DMEM and fetal bovine serum (FBS) were
purchased from Gibco (Grand Island, NY, USA); poly-L-lysine and
H2O2 were from Sigma-Aldrich (Milan, Italy);
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazol-carbocyanine
iodide (JC-1), Fluo-3, AM and Annexin V-FITC/propidium iodide (PI)
were obtained from Molecular Probes, Invitrogen (Milan, Italy), and
MTS was from Promega Corp. (Madison, WI, USA); Hank’s solution and
trypsin were obtained from HyClone (Logan, UT, USA), and Hoechst
33342, phosphate-buffered saline (PBS) and penicillin/streptomycin
(Pen/Strep) were from Beyotime (Shanghai, China). The lactate
dehydrogenase (LDH) assay kit was purchased from Roche Molecular
Biochemicals (Indianapolis, IN, USA).
Primary culture of rat cortical
astrocytes
All procedures were in accordance with the Guide for
the Care and Use of Laboratory Animals adopted by the National
Institutes of Health (Bethesda, MA, USA). Newborn Sprague-Dawley
(SD) rats (day 0–1-old) were obtained from the Laboratory Animal
Center, Chongqing Medical University, Chongqing, China. Cortical
astrocytes from newborn SD rats were obtained as previously
described (14,15). The cells in DMEM/F12 medium
containing 10% FBS and 1% Pen/Strep were plated onto
poly-L-lysine-coated 75 cm2 flasks at a density of
1.5×105 cells/ml at 37°C in a incubator (Thermo 3111;
Thermo Scientific, Waltham, MA, USA) containing 5% CO2
and 95% air. When firmly attached to the bottom of the flask, the
cells were passaged. Tha astrocytes obtained using this procedure
were then sub-cultured twice. Under these conditions, the
astrocytes were contained at >90% as determined by glial
fibrillary acidic protein (GFAP) staining.
Transfection with short hairpin RNA
(shRNA) against Srxn1
A Srxn1 lentivirus (LV2-nonGFP)-encoding shRNA and a
negative control (NC) shRNA were prepared by Shanghai GenePharma
(Shanghai, China), using the rat Srxn1 gene sequence NM_001047858
available in the NCBI database. The effective knockdown fragment
sequence of shRNA against rat Srxn1 was CAT CCA CAC CAG ACT TGC
AGT, and the sequence of the negative control shRNA against rat was
TTC TCC GAA CGT GTC ACG T. The astrocytes were transfected with the
shRNA according to the directions provided by the manufacturer and
as described in the study by Liu et al (16). Briefly, after the culture medium
was changed, 1 μl of lenti-virus per 1×104 cells
was added to the cells, which were 60–80% confluent, for infection
for approximately 24 h. The cells were then washed with PBS, and
the culture medium was changed. Following transfection for
approximately 72 h, the cells were ready for exposure to OGD or
H2O2. The Srxn1 shRNA knockdown efficiency
was confirmed by western blot analysis and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) at
approximately 72 h post-transfection.
Cell groups and exposure to OGD or
H2O2
The astrocytes were divided into the following 9
groups: i) the control group: astrocytes without any treatment; ii)
the negative control (NC) group: astrocytes were transfected with
negative control shRNA; iii) the sh-Srxn1 group: astrocytes were
transfected with Srxn1 shRNA (sh-Srxn1); iv) the control + OGD
group: astrocytes were exposed to OGD; v) the NC + OGD group:
astrocytes were transfected with negative control shRNA followed by
exposure to OGD; vi) the sh-Srxn1 + OGD group: astrocytes were
transfected with Srxn1 shRNA followed by exposure to OGD; vii) the
control + H2O2 group: astrocytes were exposed
to H2O2; viii) the NC +
H2O2 group: astrocytes were transfected with
negative control shRNA followed by exposure to
H2O2; and ix) the
sh-Srxn1+H2O2 group: astrocytes were
transfected with Srxn1 shRNA followed by exposure to
H2O2.
OGD was performed according to a previously
described method (15). Briefly,
the control and the cells transfected with the shRNA for 72 h were
exposed to OGD for 4 h, followed by oxygen-glucose reoxygenation
for 24 h at 37°C. Subsequently, the cells were ready for further
experiments. H2O2 was administered according
to a previously described method (1). The control and the cells transfected
with the shRNA for 72 h were exposed to H2O2
at an optimal concentration of 100 μM for 6 h at 37°C; the
cells were then ready for the following experiments.
Western blot analysis
The cells were lysed with PRO-PREP™ liquid
(Beyotime) in the presence of a protease inhibitor cocktail (Roche,
Mannheim, Germany), and subsequently centrifuged at 12,000 rpm for
15 min at 4°C. The total cytoplasmic protein concentration was
determined using a bicinchoninic acid (BCA) assay kit (Beyotime).
Equal amounts (20 μg) of total protein were separated by
16-6% sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE) gels and
transferred onto polyvinylidene fluoride (PVDF) membranes (0.22 and
0.45 μm; Millipore, Bedford, MA, USA). The membranes were
blocked with 5% non-fat milk in TBST buffer for 1 h and then
sequentially incubated with rabbit polyclonal anti-Srxn1 (1:400;
bs-8329R; Bioss, Beijing, China), rabbit polyclonal anti-Cyt.C
(1:1,000; ab92509; Abcam Ltd., Cambridge, UK), rabbit polyclonal
anti-caspase-3 (1:500; sc-7148), mouse polyclonal anti-caspase-9
(1:400; sc-8355) (both from Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA), rabbit polyclonal anti-caspase-12 (1:400; bs-1105R;
Bioss), rabbit polyclonal anti-poly(ADP-ribose) polymerase (1:800;
ab6070; Abcam Ltd.), rabbit polyclonal anti-Bax (1:400; sc-6236),
rabbit polyclonal anti-Bcl-2 (1:400; sc-492) (both from Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) and mouse monoclonal
anti-β-actin (1:1,000; AP0060; Bioworld Technology, Inc., St. Louis
Park, MN, USA) as the primary antibodies overnight at 4°C. After
being washed, the bound antibodies were detected by incubation for
1.5 h at room temperature with goat anti-rabbit or goat anti-mouse
HRP-conjugated secondary antibodies (1:2,000; BS10350 and BS12478,
respectively; Bioworld Technology, Inc.). The membranes were
visualized by using the electro-chemiluminescence (ECL) detection
system and quantified using Quantity One image analysis software
(both from Bio-Rad, Hercules, CA, USA).
RNA extraction and RT-qPCR
Total RNA was extracted using RNAiso Plus (Takara
Biotechnology Co., Ltd., Dalian, China) according to the
manufacturer’s instructions. The concentration and purity of the
RNA were measured with a spectrophotometer (U-3010; Hitachi Co.,
Tokyo, Japan). Subsequently, cDNA from the total RNA was obtained
using the PrimeScript™ RT reagent kit (Perfect Real-Time) (Takara
Biotechnology Co., Ltd.). The amplification was carried out using
the Thermal Cycler miniopticon real-time PCR system (Bio-Rad) in a
25 μl reaction mixture containing 2 μl of diluted
cDNA templates, 1 μl of each primer and 12.5 μl of 2X
SYBR® Premix Ex Taq™ (Takara Biotechnology Co., Ltd.).
The PCR conditions were as follows: 95°C for 10 sec, followed by 39
cycles consisting of 95°C for 5 sec, 60°C for 15 sec and 72°C for
15 sec, and melt curve at 72°C 5 sec, followed by 95°C keep. The
sequences of the specific primers used in this study were as
follows: Srxn1 forward, 5′-CCC AAG GCG GTG ACT ACT AC-3′ and
reverse, 5′-GGC AGG AAT GGT CTC TCT CTG TG-3′; β-actin forward,
5′-CAC CCG CGA GTA CAA CCT TC-3′ and reverse, 5′-CCC ATA CCC ACC
ATC ACA CC-3′. β-actin was used to normalize the expression levels
of each sample. The reaction of each sample was performed in
triplicate. Data were calculated using 2−ΔΔCt method, as
previously described (17,18)
with Bio-Rad CFX Manager software (Bio-Rad).
3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-
(4-sulfophenyl)-2H-tetrazolium (MTS) assay for cell viability
The number of viable cells was determined using the
CellTiter 96 AQueous One Solution Cell Proliferation Assay
according to the manufacturer’s instructions (Promega, Fitchburg,
WI, USA). In brief, the treated cells (1×104 cells/well)
were added to 20 μl of CellTiter 96 AQueous One Solution
Reagent. The plate was then incubated at 37°C for 3 h. The
absorbance was measured at 490 nm using a microtiter plate reader
(Bio-Rad).
LDH assay
The release of LDH was measured using the LDH assay
kit according to manufacturer’s instructions (Roche Molecular
Biochemicals). Following treatment, the nutrient solution was
collected and transferred to new 96-well plates, then mixed with
the 100 μl reaction solution provided in the kit. The
optical density was measured at 490 nm 30 min using a microplate
reader (Bio-Rad).
Hoechst staining
Apoptotic morphology was measured using Hoechst
33342 staining as previously described (15). Briefly, the treated cells on
coverslips (approximately 1×105 cells/coverslip) were
fixed with 4% paraformaldehyde for 20 min at 37°C. After washing,
the cells were mounted with Hoechst 33342 at room temperature for
15 min. Fluorescent images were acquired using a fluorescence
microscope (Olympus BX51; Olympus, Tokyo, Japan). Nuclear
shrinkage, chromatin condensation and nuclear fragmentation were
the criteria for identifying an apoptotic cell.
Flow cytometric analysis
Apoptosis analyses were performed as previously
described (19), using Annexin
V-FITC and propidium iodide (PI) double staining. The treated cells
were harvested and diluted in 100 μl of 1X Annexin V binding
buffer per assay and incubated with Annexin V-FITC and PI for 15
min in the dark at room temperature. Subsequently, 400 μl of
1X binding buffer were added. The stained cells were immediately
analyzed by flow cytometry (FCM). During apoptosis,
phosphatidylserine is translocated from the inner to the outer
leaflet of the plasma membrane where it can be detected by Annexin
V conjugates. For each sample, at least 1×105 cells were
analyzed by applying CellQuest™ Pro Analysis software (BD
Biosciences, San Jose, CA, USA). The cytogram of the 4 quadrants
was used to distinguish normal (Annexin
V−/PI−), early apoptotic (Annexin
V+/PI−), late apoptotic (Annexin
V+/PI+) and necrotic cells (Annexin
V−/PI+). The sum of the early and late
apoptotic cells represented the total apoptosis.
Measurement of mitochondrial
transmembrane potential (Δψm)
To measure the Δψm, JC-1, a lipophilic
cation sensitive fluorescent probe for Δψm, was used
according to the manufacturer’s instructions (Molecular Probes) and
as described in the study by Han et al (20). The cells were washed and incubated
with 10 μg/ml JC-1 at 37°C for 30 min. After removing JC-1
and washing, images were captured by Laser Scanning Confocal
Microscopy (Nikon TiE; Nikon, Tokyo, Japan) with both red and green
channels. A total of 6 random, non-adjacent fields in each group
were used for statistical analysis. IPP 6.0 software was used to
measure the average fluorescence intensity of the red and green
fluorescence in each group. The Δψm level is represented
by the JC-1 fluorescence ratio, which was calculated as the ratio
of average fluorescence intensity of red/green.
Measurements of intracellular calcium
([Ca2+]i) levels
To monitor changes in [Ca2+]i levels,
Fluo-3 AM was used according to the manufacturer’s instructions
(Invitrogen). The cells were washed and incubated with 5 μM
of the Ca2+ indicator, Fluo-3/AM, at 37°C for 60 min.
After removing Ca2+ and washing, changes in
[Ca2+]i levels (Fluo-3 fluorescence intensity) were
measured by Laser Scanning Confocal Microscopy (Nikon TiE; Nikon).
A total of 6 random, non-adjacent fields in each group were used
for statistical analysis.
Statistical analysis
All the experiments were performed at least 3 times
and the data are presented as the means ± SEM. Data were analyzed
by one-way ANOVA followed by a post hoc Tukey’s test. Statistical
analysis was performed using SPSS 17.0 software (SPSS, Inc.,
Chicago, IL, USA) and a value of P<0.05 was considered to
indicate a statistically significant difference.
Results
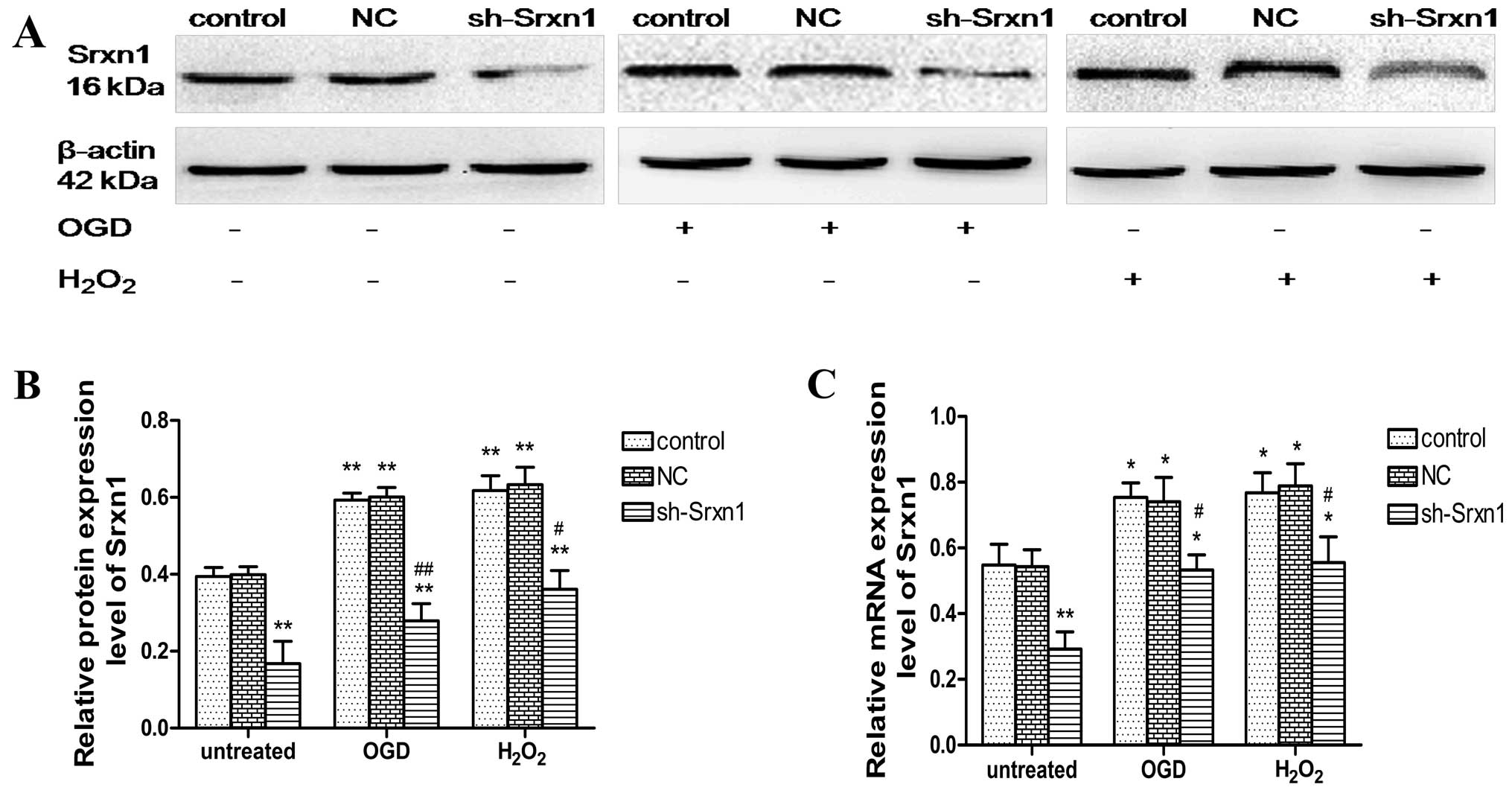
Evaluation of the efficiency of Srxn1
shRNA following exposure to OGD or H2O2
The inhibition of Srxn1 expression in the presence
of OGD or H2O2 was confirmed by western blot
analysis (Fig. 1A and B) and
RT-qPCR (Fig. 1C). Compared with
the control group, Srxn1 knockdown resulted in a ~61% decrease in
protein and a ~47% decrease in Srxn1 mRNA expression (Fig. 1). The exposure of the cells to OGD
(or H2O2) resulted in a significant increase
in Srxn1 protein and mRNA expression, and Srxn1 knockdown prevented
this increase (sh-Srxn1 + OGD group and sh-Srxn1 +
H2O2 group), compared with the control + OGD
(or H2O2) group (P<0.05 and P<0.01).
These results suggest that Srxn1 plays an important role in
protecting astrocytes from damage induced by exposure to OGD or
H2O2.
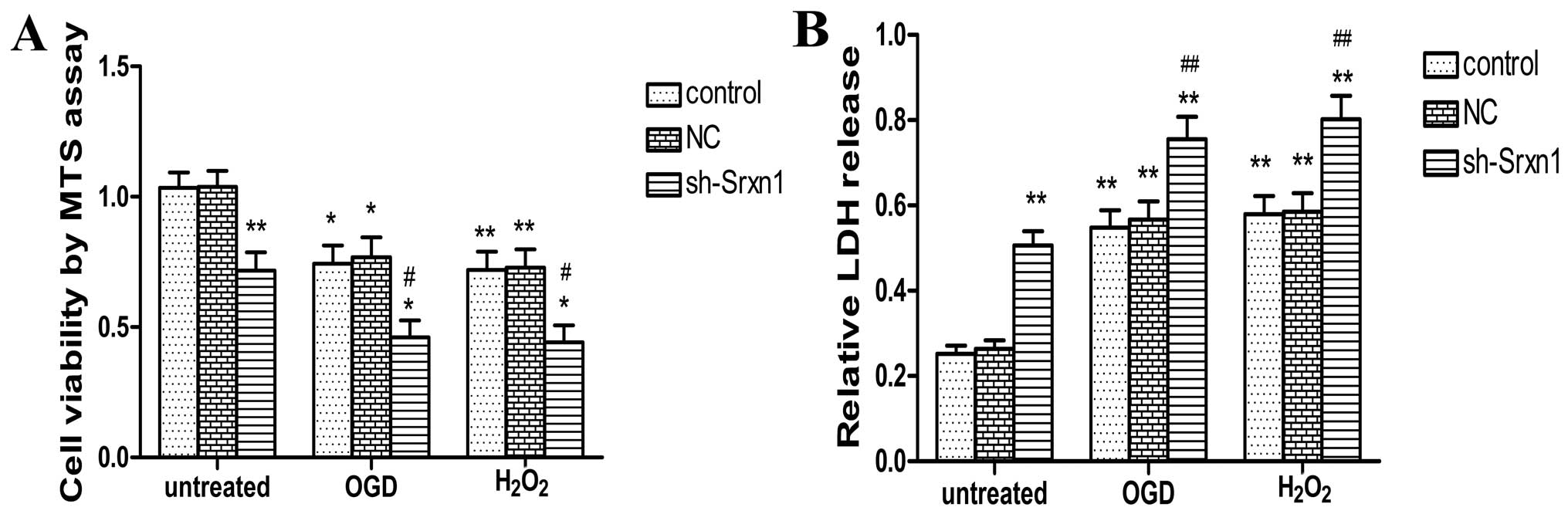
Knockdown of Srxn1 decreases cell
viability and enhances cell damage following exposure to OGD or
H2O2
Following exposure to OGD or
H2O2, cell viability was determined by MTS
assay (Fig. 2A). Compared with
the control group, following exposure to OGD or
H2O2, cell viability was decreased by ~26% in
the control + OGD group and by 27% in the control +
H2O2 group. Following the knockdown of Srxn1,
cell viability decreased more significantly (sh-Srxn1 + OGD group
and sh-Srxn1 + H2O2 group) compared with the
control + OGD (or H2O2) group (P<0.05). We
also determined astrocyte damage by LDH assay (Fig. 2B). Exposure to OGD (or
H2O2) resulted in an increase in the release
of LDH (P<0.05), and following exposure to OGD or
H2O2, the knockdown of Srxn1 resulted in the
release of LDH increasing more significantly compared with the
control + OGD (or H2O2) group (P<0.01;
Fig. 2B). These results indicate
that Srxn1 may protect astrocytes from OGD- or
H2O2-induced damage.
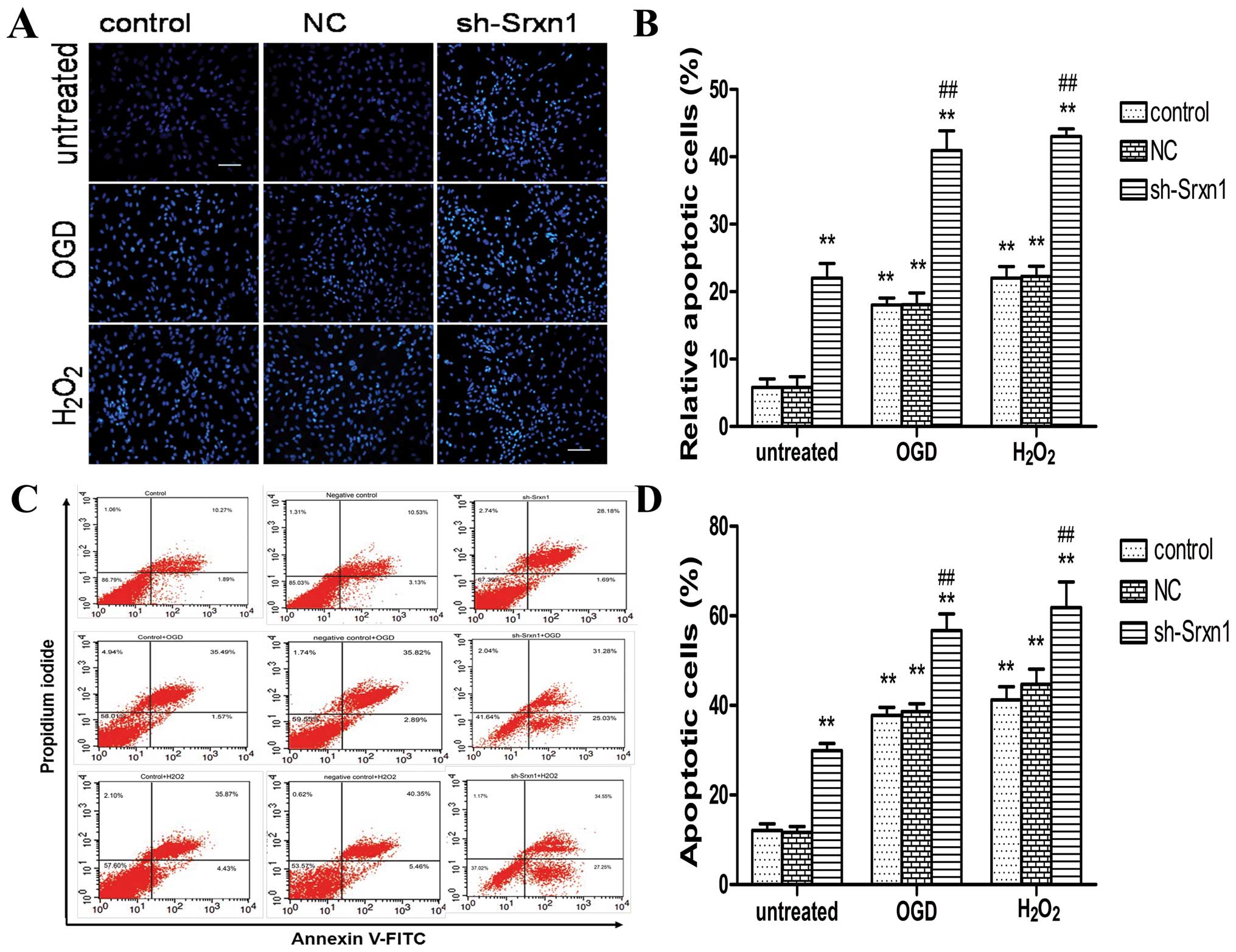
Knockdown of Srxn1 increases apoptosis
following exposure to OGD or H2O2
Apoptosis was detected by combining microscopic
analysis with Hoechst 33342 staining (Fig. 3A and B) and FCM (Fig. 3C and D). Compared with the control
group, exposure to OGD (or H2O2) induced
apoptosis (control + OGD group and control +
H2O2 group) (P<0.01), and following the
knockdown of Srxn1, the cell apoptotic rate increased more
significantly (sh-Srxn1 + OGD group and sh-Srxn1 +
H2O2 group) compared with the control + OGD
(or H2O2) group (P<0.01; Fig. 3A and B). Annexin V-FITC/PI double
staining revealed a significant increase in apoptotis by ~29% in
the control + OGD group and by 31% in the control +
H2O2 group, compared with the control group
(P<0.01; Fig. 3C and D). In
addition, following exposure to OGD or H2O2,
the knockdown of Srxn1 resulted in an even more significant
increase in apoptotis (P<0.01), compared with the control + OGD
(or H2O2) group. Taken together, these
results indicate that Srxn1 plays a vital role in protecting
astrocytes from apoptosis induced by exposure to OGD (or
H2O2).
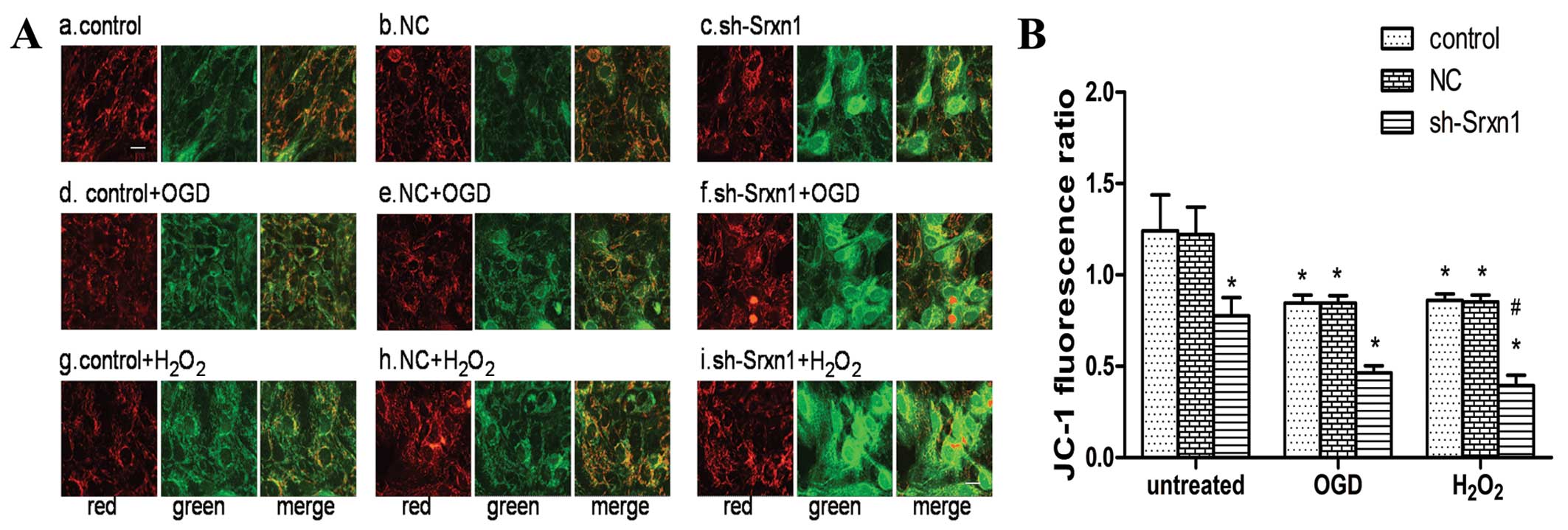
Knockdown of Srxn1 activates the
mitochondrial apoptotic pathway following exposure to OGD or
H2O2
To evaluate whether the mitochondrial apoptotic
pathway was activated, we determined Δψm using JC-1
staining (Fig. 4), as well as the
expression levels of some mitochondrial apoptotic pathway-related
proteins by western blot analysis (Fig. 5). At a high membrane potential,
JC-1 enters the mitochondria and forms aggregates that have a
fluorescence of bright red, whereas JC-1 exists as a green
fluorescence at a low potential (21). As shown in Fig. 4A, the untreated cell mitochondria
exhibited a high Δψm, as indicated by a bright, red
fluorescence and a very low green fluorescence (Fig. 4A panels a and b). The knockdown of
Srxn1 decreased Δψm, and thus the green fluorescence
increased significantly compared with the red fluorescence
(Fig. 4A panel c). In addition,
following exposure to OGD or H2O2, the green
fluorescence was stronger than the red fluorescence [control + OGD
(or H2O2) group], compared with the untreated
control group (Fig. 4A panels d,
e, g and h). Following exposure to OGD or
H2O2, Srxn1 knockdown resulted in a further
decrease in Δψm, and thus the green fluorescence became
much stronger and the red fluorescence became much weaker (Fig. 4A panels f and i). To quantify the
level of Δψm, the JC-1 fluorescence ratio was calculated
using the average optical density ratio of red/green. The JC-1
ratio decreased by ~38% in the sh-Srxn1 group, by 32% in the
control + OGD group and by 31% in the control +
H2O2 group, compared with the untreated
control group (P<0.05; Fig.
4B). Moreover, following exposure to OGD or
H2O2, Srxn1 knockdown markedly decreased the
JC-1 ratio in the sh-Srxn1 + OGD (or H2O2)
group compared with the control + OGD (or
H2O2) group (P<0.05), representing a
dissipation of Δψm. These data indicated that Srxn1
knockdown induced the dissipation of Δψm in the
astrocytes following exposure to OGD or
H2O2.
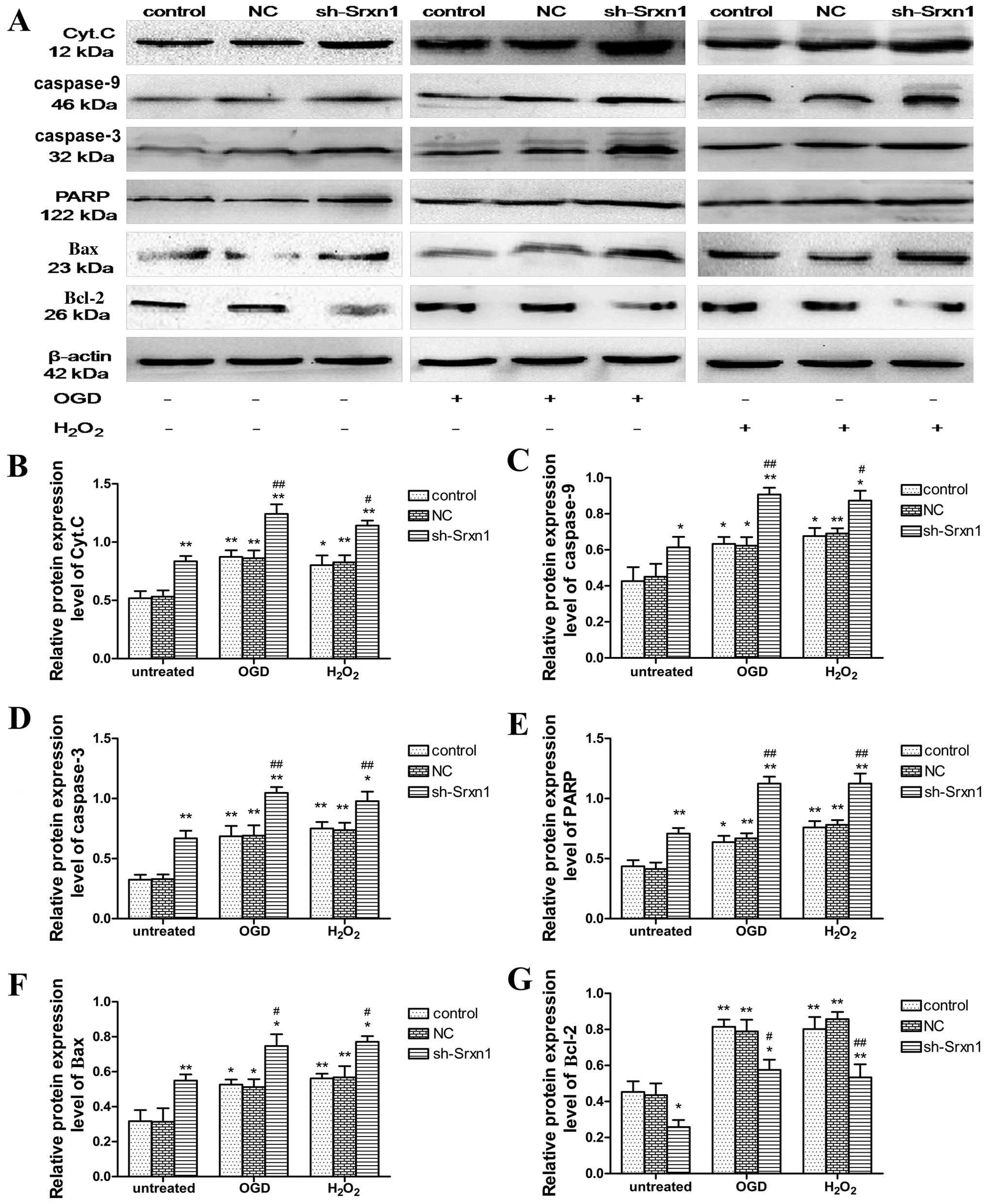
Compared with the control group, exposure to OGD or
H2O2 increased the cytoplasmic release of
Cyt.C, as well as the cytoplasmic levels of caspase-9, caspase-3,
PARP, Bax and Bcl-2 proteins (P<0.05; Fig. 5). Following exposure to OGD or
H2O2, Srxn1 knockdown promoted this increase
in protein expression apart from the expression of Bcl-2; the
expression level of Bcl-2 decreased (sh-Srxn1 + OGD group and
sh-Srxn1 + H2O2 group) compared with the
control + OGD (or H2O2) group (P<0.01 or
P<0.05). These observations indicate that Srxn1 has an
anti-apoptotic function in astrocytes following exposure to OGD or
H2O2, which is related to the mitochondrial
apoptotic pathway.
Knockdown of Srxn1 does not affect
endoplasmic reticulum stress pathway-mediated apoptosis following
exposure to OGD or H2O2
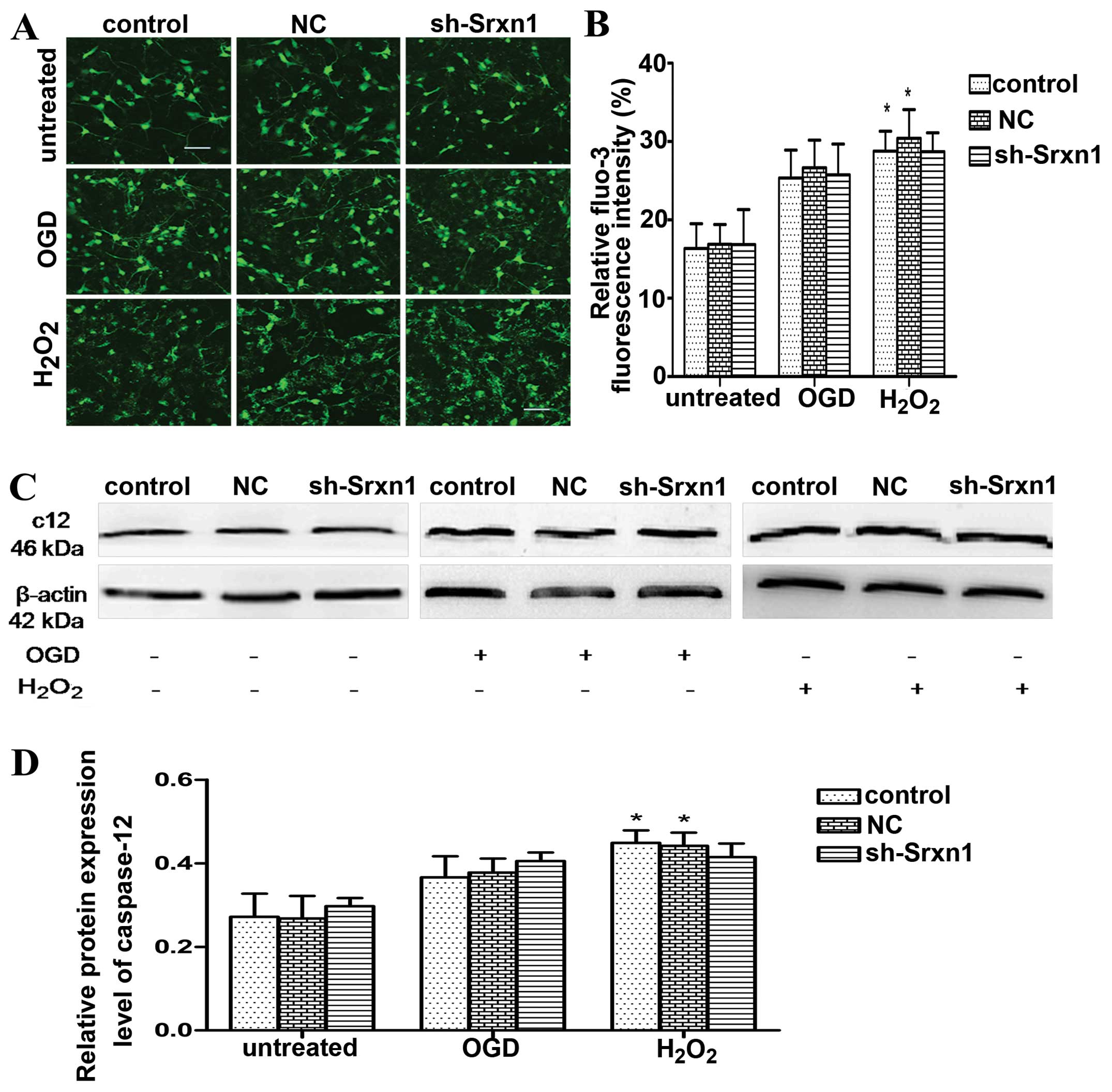
In order to determine whether endoplasmic reticulum
stress pathway-mediated apoptosis had occurred, we detected
[Ca2+]i and caspase-12 levels based on Fluo-3 AM
staining (Fig. 6A and B) and
western blot analysis (Fig. 6C and
D). Of note, we found that the levels of caspase-12 and
[Ca2+]i increased following exposure to OGD or
H2O2 compared with the control group;
however, after the knockdown of Srxn1, these levels did not differ
from those of the control + OGD (or H2O2)
group. Hence, we deduced that the neuroprotective effects of Srxn1
are not related to endoplasmic reticulum stress pathway-mediated
apoptosis.
Discussion
In the present study, we provide evidence that Srxn1
exerts a protective effect against oxidative stress-induced brain
injury and also provide insight into its mechanisms of action. Rat
corital astrocytes were exposed to OGD and
H2O2 in order to mimic oxidative
stress-induced injury in cerebral ischemia and used shRNAs to
knockdown Srxn1. We found that following exposure to OGD or
H2O2, the knockdown of Srxn1 significantly
increased cell death as determined by MTS and LDH assays, increased
cell apoptosis as measured by Hoechst 33342 and Annexin V-FITC/PI
staining. Following exposure to OGD or H2O2,
the knockdown of Srxn1 promoted the activation of the mitochondrial
apoptotic pathway, decreasing Δψm and the expression of
the anti-apoptotic Bcl-2 protein in the cytoplasm, and increasing
the cytoplasmic levels of Cyt.C, caspase-3, caspase-9, PARP and
pro-apoptotic Bax proteins. We also demonstrated that the levels of
[Ca2+]i and caspase-12, which are tightly linked with
endoplasmic reticulum stress pathway-mediated apoptosis, were not
altered after the knockdown of Srxn1. Taken together, these results
indicate that Srxn1 plays a vital role in protecting astrocytes
from apoptosis induced by exposure to OGD or
H2O2. Srxn1 was found play a neuroprotective
role partly by exerting anti-apoptosis effects associated with the
mitochondrial apoptotic pathway.
Cerebral ischemia, particularly during the period
of ischemia/reperfusion, involves complex pathophysiological and
biochemical mechanisms, such as the generation of ROS,
glutamate-mediated excitotoxicity, inflammation, calcium-activated
proteolysis and apoptosis (22,23). It has been widely reported that
apoptosis is the major cell death pathway following
hypoxia-ischemia in developing brains (24–26). Within the central nervous system
(CNS), astrocytes are the most abundant cells and play a role in
synaptic transmission and plasticity, they transport nutrients and
participate in neurotransmission (27–29). It has been demonstrated that
astrocytes in primary culture undergo apoptosis following an
ischemic insult (30,31). Furthermore, studies have indicated
that oxidative stress and glial-derived ROS are critical for the
apoptosis-induced selective loss of neurons (32,33). Moreover, astrocyte apoptosis may
contribute to the pathogenesis of a number of acute and chronic
neurodegenerative disorders, such as cerebral ischemia, Alzheimer’s
disease and PD (34).
There are three main apoptotic signaling pathways
that have been discovered: i) the mitochondrial pathway (intrinsic
pathway) (35,36); ii) the death receptor-associated
pathway (extrinsic pathway) (37); and iii) the endoplasmic
reticulum-associated pathway (38,39). Currently, the apoptosis of the
mitochondria and the endoplasmic reticulum pathway, which are
endogenous organelles, are hotspot research topics. A variety of
conditions can disturb the functions of the mitochondria or the
endoplasmic reticulum and lead to mitochondrial or endoplasmic
reticulum stress. These conditions include ischemia, hypoxia,
exposure to free radicals, as well as others. When mitochondrial or
endoplasmic reticulum stress conditions persist, the initiation of
the apoptotic processes is promoted.
Under different pathological conditions, cell death
is associated with or initiated by mitochondrial function
impairment and dissipation of Δψm (40). Δψm, which can modulate
the release of Cyt.C by controlling membrane permeability, plays a
crucial role in the terminal step of apoptosis (41,42). The depolarization of mitochondrial
membrane potential induced by hypoxic-ischemic injury implicates
the loss of Δψm in the brain. The loss of Δψm
leads to mitochondrial membrane permeabilization, and thus the
release of Cyt.C from the mitochondria into the cytoplasm, which
binds and activates caspase-9, thereby resulting in the activation
of other downstream caspases and, ultimately, caspase-3 (43), triggering apoptosis (41,42). Caspase-3 has been identified as a
main executioner of the apoptotic response inside cells. Finally,
activated caspase-3 cleaved effector proteins, including PARP and
the induction of DNA fragmentation in the nucleus eventually lead
to cell death (44,45). Bcl-2 family members are the
arbiters of the mitochondrial apoptotic pathway and decide whether
a cell lives or dies (46). This
family includes anti-apoptotic (e.g., Bcl-2) and pro-apoptotic
genes (e.g., Bax) (47,48). Bcl-2, an inhibitor of apoptosis,
prevents neuronal apoptosis by blocking the destruction of the
mitochondria and the subsequent release of Cyt.C and caspase
activation (49). In this study,
our results demonstrated that OGD or H2O2
increased cell death, including apoptosis, decreased Δψm
and increased the subsequent cytosolic translocation of Cyt.C,
thereby promoting downstream caspase activation, resulting in
increased levels of caspase-9, caspase-3, PARP (the substrate of
caspase-3), anti-apoptotic protein Bcl-2 and pro-apoptotic protein
Bax in the cytoplasm. Following exposure to OGD or
H2O2, Srxn1 knockdown promoted cell death,
apoptosis, dissipation of Δψm, and promoted an increase
in protein expression apart from Bcl-2 expression (the expression
of Bcl-2 decreased). These data indicated that the knockdown of
Srxn1 promoted the activation of the mitochondrial apoptotic
pathway following exposure to OGD or H2O2.
Thus, the protective effects of Srxn1 are associated with the
inhibition of the mitochondrial apoptotic pathway.
It has been reported that caspase-12-mediated
apoptosis is a specific apoptotic pathway of the endoplasmic
reticulum (39,50). Caspase-12 appears to be necessary
for apoptosis induced by a variety of endoplasmic
reticulum-directed pro-apoptotic signals (39). The cytoplasmic calcium
[Ca2+]c signal has been shown to mediate the
calpain/caspase-12-dependent apoptotic pathway primarily in cancer
cells, and it has been shown that alterations in [Ca2+]c
levels may potentially induce apoptotic cell death (51). In this study, exposure to OGD or
H2O2 resulted in endoplasmic reticulum stress
and thus incresaed the expression of caspase-12 and
[Ca2+]i. However, after the knockdown of Srxn1, these
levels were not altered. This suggests that the knockdown of Srxn1
does not affect endoplasmic reticulum stress pathway-mediated
apoptosis.
In conclusion, the data from the present study
demonstrate that Srxn1 exerts a protective effect against the
oxidative stress-induced brain injury and that this effect is
partly mediated through its anti-apoptosis effects, which are
associated with the inhibition of the mitochondrial apoptotic
pathway. The findings of this study suggest that Srxn1 may be a
novel target for neuroprotective intervention in neurodegenerative
diseases. We aim to continue this area of investigation in future
in vivo studies using the chemical synthesis of RNA
interference and the overexpression of Srxn1 to verify the
association between Srxn1 and apoptosis.
Acknowledgments
This study was supported by grants from the
National Natural Science Foundation of China (nos. 81271460,
81171090 and 81301125), and the Natural Science Foundation of
Chongqing Education Committee, China (no. KJ110313).
References
|
1
|
Li Q, Yu S, Wu J, Zou Y and Zhao Y:
Sulfiredoxin-1 protects PC12 cells against oxidative stress induced
by hydrogen peroxide. J Neurosci Res. 91:861–870. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kinsner A, Pilotto V, Deininger S, et al:
Inflammatory neurodegeneration induced by lipoteichoic acid from
Staphylococcus aureus is mediated by glia activation, nitrosative
and oxidative stress, and caspase activation. J Neurochem.
95:1132–1143. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Papadia S, Soriano FX, Leveille F, et al:
Synaptic NMDA receptor activity boosts intrinsic antioxidant
defenses. Nat Neurosci. 11:476–487. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Biteau B, Labarre J and Toledano MB:
ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae
sulphiredoxin. Nature. 425:980–984. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rhee SG, Jeong W, Chang TS and Woo HA:
Sulfiredoxin, the cysteine sulfinic acid reductase specific to
2-Cys peroxiredoxin: its discovery, mechanism of action, and
biological significance. Kidney Int Suppl. 72:S3–S8. 2007.
View Article : Google Scholar
|
|
6
|
Vivancos AP, Castillo EA, Biteau B, et al:
A cysteine-sulfinic acid in peroxiredoxin regulates
H2O2-sensing by the antioxidant Pap1 pathway.
Proc Natl Acad Sci USA. 102:8875–8880. 2005. View Article : Google Scholar
|
|
7
|
Findlay VJ, Tapiero H and Townsend DM:
Sulfiredoxin: a potential therapeutic agent? Biomed Pharmacother.
59:374–379. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Soriano FX, Leveille F, Papadia S, et al:
Induction of sulfiredoxin expression and reduction of peroxiredoxin
hyperoxidation by the neuroprotective Nrf2 activator
3H-1,2-dithiole-3-thione. J Neurochem. 107:533–543. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kang KW, Lee SJ and Kim SG: Molecular
mechanism of nrf2 activation by oxidative stress. Antioxid Redox
Signal. 7:1664–1673. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Singh A, Ling G, Suhasini AN, et al:
Nrf2-dependent sulfiredoxin-1 expression protects against cigarette
smoke-induced oxidative stress in lungs. Free Radic Biol Med.
46:376–386. 2009. View Article : Google Scholar
|
|
11
|
Bell KF, Al-Mubarak B, Fowler JH, et al:
Mild oxidative stress activates Nrf2 in astrocytes, which
contributes to neuroprotective ischemic preconditioning. Proc Natl
Acad Sci USA. 108:E1–E2. 2011. View Article : Google Scholar :
|
|
12
|
Noh YH, Baek JY, Jeong W, Rhee SG and
Chang TS: Sulfiredoxin translocation into mitochondria plays a
crucial role in reducing hyperoxidized peroxiredoxin III. J Biol
Chem. 284:8470–8477. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Baek JY, Han SH, Sung SH, et al:
Sulfiredoxin protein is critical for redox balance and survival of
cells exposed to low steady-state levels of
H2O2. J Biol Chem. 287:81–89. 2012.
View Article : Google Scholar :
|
|
14
|
Wu J, Li Q, Wang X, et al: Neuroprotection
by curcumin in ischemic brain injury involves the Akt/Nrf2 pathway.
PLoS One. 8:e598432013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu X, Zhao J, Yu S, Chen Y, Wu J and Zhao
Y: Sulforaphane protects primary cultures of cortical neurons
against injury induced by oxygen-glucose deprivation/reoxygenation
via anti-apoptosis. Neurosci Bull. 28:509–516. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu SB, Zhang N, Guo YY, et al:
G-protein-coupled receptor 30 mediates rapid neuroprotective
effects of estrogen via depression of NR2B-containing NMDA
receptors. J Neurosci. 32:4887–4900. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Song S, Zhou J, He S, et al: Expression
levels of microRNA-375 in pancreatic cancer. Biomed Rep. 1:393–398.
2013.
|
|
19
|
Lin CJ, Chen TH, Yang LY and Shih CM:
Resveratrol protects astrocytes against traumatic brain injury
through inhibiting apoptotic and autophagic cell death. Cell Death
Dis. 5:e11472014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Han F, Tao RR, Zhang GS, et al: Melatonin
ameliorates ischemic-like injury-evoked nitrosative stress:
Involvement of HtrA2/PED pathways in endothelial cells. J Pineal
Res. 50:281–291. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sung DK, Chang YS, Kang S, Song HY, Park
WS and Lee BH: Comparative evaluation of hypoxic-ischemic brain
injury by flow cytometric analysis of mitochondrial membrane
potential with JC-1 in neonatal rats. J Neurosci Methods.
193:232–238. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao J, Kobori N, Aronowski J and Dash PK:
Sulforaphane reduces infarct volume following focal cerebral
ischemia in rodents. Neurosci Lett. 393:108–112. 2006. View Article : Google Scholar
|
|
23
|
Chan PH: Reactive oxygen radicals in
signaling and damage in the ischemic brain. J Cereb Blood Flow
Metab. 21:2–14. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hossain MA: Hypoxic-ischemic injury in
neonatal brain: involvement of a novel neuronal molecule in
neuronal cell death and potential target for neuroprotection. Int J
Dev Neurosci. 26:93–101. 2008. View Article : Google Scholar
|
|
25
|
Kawamura M, Nakajima W, Ishida A, Ohmura
A, Miura S and Takada G: Calpain inhibitor MDL 28170 protects
hypoxic-ischemic brain injury in neonatal rats by inhibition of
both apoptosis and necrosis. Brain Res. 1037:59–69. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Northington FJ, Graham EM and Martin LJ:
Apoptosis in perinatal hypoxic-ischemic brain injury: how important
is it and should it be inhibited? Brain Res Brain Res Rev.
50:244–257. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Stellwagen D and Malenka RC: Synaptic
scaling mediated by glial TNF-alpha. Nature. 440:1054–1059. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Perea G, Yang A, Boyden ES and Sur M:
Optogenetic astrocyte activation modulates response selectivity of
visual cortex neurons in vivo. Nat Commun. 5:32622014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lopez-Hidalgo M and Schummers J: Cortical
maps: a role for astrocytes? Curr Opin Neurobiol. 24:176–189. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Giffard RG and Swanson RA:
Ischemia-induced programmed cell death in astrocytes. Glia.
50:299–306. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yu AC, Wong HK, Yung HW and Lau LT:
Ischemia-induced apoptosis in primary cultures of astrocytes. Glia.
35:121–130. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang J, Deng X, Zhang F, Chen D and Ding
W: ZnO nanoparticle-induced oxidative stress triggers apoptosis by
activating JNK signaling pathway in cultured primary astrocytes.
Nanoscale Res Lett. 9:1172014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xie Z, Smith CJ and Van Eldik LJ:
Activated glia induce neuron death via MAP kinase signaling
pathways involving JNK and p38. Glia. 45:170–179. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Takuma K, Baba A and Matsuda T: Astrocyte
apoptosis: implications for neuroprotection. Prog Neurobiol.
72:111–127. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Desagher S and Martinou JC: Mitochondria
as the central control point of apoptosis. Trends Cell Biol.
10:369–377. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang X: The expanding role of mitochondria
in apoptosis. Genes Dev. 15:2922–2933. 2001.PubMed/NCBI
|
|
37
|
Sun XM, MacFarlane M, Zhuang J, Wolf BB,
Green DR and Cohen GM: Distinct caspase cascades are initiated in
receptor-mediated and chemical-induced apoptosis. J Biol Chem.
274:5053–5060. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ferri KF and Kroemer G: Organelle-specific
initiation of cell death pathways. Nat Cell Biol. 3:E255–E263.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nakagawa T, Zhu H, Morishima N, et al:
Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and
cytotoxicity by amyloid-beta. Nature. 403:98–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Leist M and Nicotera P: Apoptosis,
excitotoxicity, and neuropathology. Exp Cell Res. 239:183–201.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Iijima T, Mishima T, Akagawa K and Iwao Y:
Mitochondrial hyperpolarization after transient oxygen-glucose
deprivation and subsequent apoptosis in cultured rat hippocampal
neurons. Brain Res. 993:140–145. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ye R, Li N, Han J, et al: Neuroprotective
effects of ginsenoside Rd against oxygen-glucose deprivation in
cultured hippocampal neurons. Neurosci Res. 64:306–310. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Acehan D, Jiang X, Morgan DG, Heuser JE,
Wang X and Akey CW: Three-dimensional structure of the apoptosome:
implications for assembly, procaspase-9 binding, and activation.
Mol Cell. 9:423–432. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ji YB, Qu ZY and Zou X: Juglone-induced
apoptosis in human gastric cancer SGC-7901 cells via the
mitochondrial pathway. Exp Toxicol Pathol. 63:69–78. 2011.
View Article : Google Scholar
|
|
45
|
Khan M, Ding C, Rasul A, et al:
Isoalantolactone induces reactive oxygen species mediated apoptosis
in pancreatic carcinoma PANC-1 cells. Int J Biol Sci. 8:533–547.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wong WW and Puthalakath H: Bcl-2 family
proteins: the sentinels of the mitochondrial apoptosis pathway.
IUBMB Life. 60:390–397. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
47
|
Moldoveanu T, Follis AV, Kriwacki RW and
Green DR: Many players in BCL-2 family affairs. Trends Biochem Sci.
39:101–111. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Martinou JC and Youle RJ: Mitochondria in
apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev
Cell. 21:92–101. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Shacka JJ and Roth KA: Regulation of
neuronal cell death and neurodegeneration by members of the Bcl-2
family: therapeutic implications. Curr Drug Targets CNS Neurol
Disord. 4:25–39. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Szegezdi E, Fitzgerald U and Samali A:
Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann
NY Acad Sci. 1010:186–194. 2003. View Article : Google Scholar
|
|
51
|
Sergeev IN: Calcium signaling in cancer
and vitamin D. J Steroid Biochem Mol Biol. 97:145–151. 2005.
View Article : Google Scholar : PubMed/NCBI
|