Introduction
Hepatocellular carcinoma (HCC) is one of the most
aggressive types of cancer in humans and remains the second leading
cause of cancer-associated mortality worldwide. The prognosis of
HCC patients is poor, with a 5-year survival rate of <12%,
predominantly due to late diagnosis, early metastasis and high
resistance to chemotherapy (1).
Despite advances in diagnostic techniques, drug development and
surgical procedures, the incidence of HCC is almost equal to the
mortality rate (1,2). Local surgical ablation therapies can
also prolong survival rates, however, due to locally extended and
metasta-sized disease, only 15% of patients are eligible for
resection, which is often followed by local recurrence (3). In HCC, the rates of incidence and
mortality are higher in males than in females (3). HCC is generally poorly responsive to
cytotoxic chemotherapy. Sorafenib, a Food and Drug Administration
approved agent for HCC, only increases median survival rates from
~8–10 months (4). One of the
major causes of morbidity and mortality associated with the disease
is poor understanding of the molecular signaling pathway, and novel
therapeutic strategies and drugs are desirable for HCC.
The Wnt/β-catenin signaling pathway is important in
embryonic development and adult homeostasis, including
proliferation, migration and differentiation (5,6).
The role of β-catenin was first recognized as a membrane-associated
protein, involved in cell-cell adhesion. Cytoplasmic β-catenin
binds to the carboxyl terminus of E-cadherin at the plasma
membrane, and this complex recruits α-catenin, which further
recruits other structural proteins to form the cell-cell junctions
(7,8). In addition to its role as an
adhesion protein, β-catenin can also act as a transcription
co-activator. β-catenin is a key component of downstream signaling
in the Wnt/Wingless pathway, which is important to embryonic
development. This pathway is also involved in disease development
if misregu-lation of β-catenin occurs (9). In development, Wnt protein binds to
the Frizzled receptor and inhibits the adenomatous polyposis coli
(APC)/glycogen synthase kinase-3β (GSK-3β) complex, which
phosphorylates β-catenin, targeting it to proteasomal degradation.
When β-catenin is protected from degradation, it enters the nucleus
and associates with the T-cell factor and lymphoid enhancer
factor-1 (TCF/LEF-1) family of transcription factors, and this
association activates the transcription of β-catenin target genes,
including regulators of cell growth and proliferation, modulators
of cell death pathways and cell-cell communication (10). Several studies have demonstrated
that the Wnt/β-catenin pathway is activated in HCC, and this
appears to be important in the aggressive nature of this disease,
including its resistance to chemotherapy (11–13). Analyses of different genetic
alterations have led to the identification of several major
oncogenic pathways, which are deregulated in HCC, including the
p53, Rb, E-cadherin and Wnt/β-catenin pathways (14).
In continuation of previous studies on
tetrahydropyridinol derivatives as antibacterial and
antimycobacterial agents (15,16), the present study synthesized three
novel analogs, N-(bromo-acetyl)-3-carboxyethyl-2,6-diphenyl-4-O-
(pentafluorobenzoyl)- Δ3-tetrahydropyridine (5a),
N-(chloroacetyl)-3-carboxy-
ethyl-2,6-diphenyl-4-O-(pentafluorobenzoyl)-Δ3-tetrahydro- pyridine
(5b) and
N-(2-bromopropanoyl)-3-carboxyethyl-2,6-diphenyl-4-O-(penta
fluorobenzoyl)-Δ3-tetrahydropyridine (5c), and investigated their
anticancer effects on human HCC and breast cancer cell lines.
Materials and methods
Synthesis of tetrahydropyridinol
derivatives
All solvents and reagents used in the present study
were of reagent grade, obtained from commercial sources and used
without further purification, unless otherwise stated. The course
of the reactions and purity of the products were assessed by using
TLCon Silica Gel 60 F254 (Sigma-Aldrich, St. Louis, MO, USA)
pre-coated plates. The melting points were read and recorded using
an Electrothermal-9100 (Shimadzu, Kyoto, Japan) instrument. The
nuclear magnetic resonance (NMR) spectra were run on a JNM ECP-400
instrument (JEOL, Ltd., Tokyo, Japan), operating at 400 MHz for 1 h
at 100.6 MHz for complete proton decoupled 13C, using
CDCl3 (Sigma-Aldrich) as a solvent and tetramethylsilane
(TMS; Sigma-Aldrich) as an internal standard. The chemical shift
values were reported in parts/million (ppm) relative to TMS, or
with the solvent reference relative to TMS, used as the internal
standard (CDCl3; δ=7.26 ppm for proton and 77.00 ppm for
carbon NMR). Mass spectra were obtained using a JMS-700 (JEOL,
Ltd.) instrument. Purification of the final compounds was performed
using silica gel (Sigma-Aldrich) (200–400 mesh-60Å). The
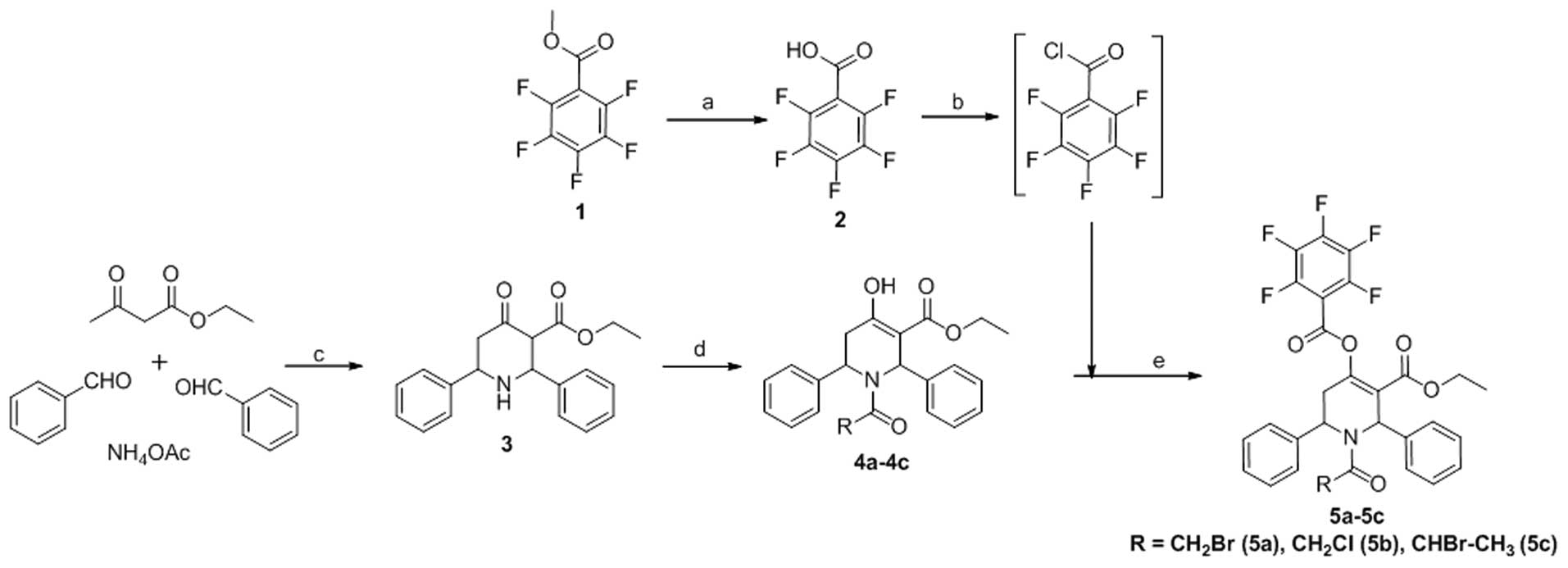
commercially available methylpentafluorobenzoate 1 upon base
hydrolysis furnished the corresponding acid 2 (Fig. 1). Compounds 4a-4c were obtained
using methods described in our previous studies (15,16). The desired esters, 5a-5c, were
synthesized by treating 4a-4c with pentafluorobenzoyl chloride
(Sigma-Aldrich), which was obtained from acid 2 (Fig. 1) via thionyl chloride
(Sigma-Aldrich) treatment in moderate yields (61–81%).
Synthesis of pentafluorobenzoyl
chloride
Pentafluorobenzoic acid (2–4)
was obtained by base hydrolysis of methylpentaf-luorobenzoate
(Sigma-Aldrich) in methanol. To a mixture of pentafluorobenzoic
acid (0.005 mol) in dry toluene (15 ml; Sigma-Aldrich), freshly
distilled thionyl chloride (2 ml) was added and refluxed for ~6 h.
The reaction mixture was cooled and the solvent was evaporated
in vacuo. Another 10 ml of toluene was added to the reaction
mixture and complete evaporation of the contents was performed
again. The residue remaining was dissolved in dry DCM
(Sigma-Aldrich) and was used for esterification with the respective
hydroxyl derivatives (4a–4c) to obtain the desired target compounds
(5a–5c).
Synthesis of 5a-5c
To an ice cooled solution of the respective N-acyl
derivatives 4a–4c (0.005 mol) in dry DCM (20 ml), either DMAP or
NEt3 (0.015 mol each) (both from Sigma-Aldrich) was
added and stirred well. To this, a solution of
pentafluorobenzoylchloride in dry DCM was added drop-wise and
stirred at room temperature over a period of 5 h. Following
completion of the reaction, the solvent was removed in
vacuo, the residue was dissolved in ethyl acetate, and washed
with bicarbonate, water and then brine. The organic layer was dried
over Na2SO4, and the residue obtained by
evaporation was purified in a column (0.5:10–1:10, ethyl
acetate:hexane) to obtain the pure products.
Composition of
N-(bromoacetyl)-3-carboxyethyl-2,6-dipheny l-4-O-(penta
fluorobenzoyl)-Δ3-tetrahydropyridine (5a)
Yield: 65%, a semi solid. 1H NMR (400
MHz; CDCl3) δ ppm: 7.19–7.03 (m, 10H); 6.88 (bs, 1H),
5.51 (bs, 1H), 4.31 (d, J=12.86 Hz, 1H), 4.16 [d (not
resolved well), 1H], 4.05 (q, J=7.05 Hz, 2H), 3.14 (dd,
J=17.84 Hz, J=4.98 Hz, 1H), 2.98 (dd, J=17.84
Hz, J=6.22 Hz, 1H), 0.99 (t, J=7.05 Hz, 3H).
13C NMR (100 MHz; CDCl3) δ ppm: 166.57,
162.42, 157.23, 152.79, 148.09 (m), 145.47 (m), 139.04 (m), 138.15,
138.02, 128.51–126.76 (phenyl carbons), 120.17, 103.13 (t,
JC-F=11.14), 61.18, 55.34, 53.82, 42.33, 33.20,
13.64; MS [electrospray ionisation (ESI), positive] m/z calculated
for C29H21BrF5NO5
(M+H): 637.05; identified 637.3.
Composition of
N-(chloroacetyl)-3-carboxyethyl-2,6-dipheny l-4-O-(penta
fluorobenzoyl)-Δ3-tetrahydropyridine (5b)
Yield: 81%. White solid, mp 142–144°C. 1H
NMR (400 MHz; CDCl3) δ ppm: 7.20–7.02 (m, 10H, phenyl
protons); 6.95 (bs, 1H), 5.51 (bs, 1H),4.31 (d, J=12.86 Hz,
1H), 4.14 [d (not resolved well), 1H], 4.06 (q, J=7.05 Hz,
2H), 3.14 (dd, J=17.84 Hz, J=5.39 Hz, 1H), 2.98 (dd,
J=17.43 Hz, J=6.22 Hz, 1H), 0.99 (t, J=7.05
Hz, 3H); 13C NMR (100 MHz, CDCl3) δ ppm:
166.62, 162.44, 157.24, 152.78, 148.13 (m), 145.40 (m), 139.06 (m),
138.19, 138.03, 128.53–126.72 (phenyl carbons), 120.20, 103.17 (t,
JC-F=11.19), 61.19, 55.27, 53.83, 42.32, 33.23,
13.65; MS (ESI, positive) m/z calculated for
C29H21ClF5NO5 (M+H):
593.1; identified 593.3.
Composition of
N-(2-bromopropanoyl)-3-carboxyethyl-2,6-d iphenyl-4-O-(penta
fluorobenzoyl)-Δ3-tetrahydropyridine (5c)
Yield: 61%, a white solid, mp 140–141°C.
1H NMR (400 MHz, CDCl3) δ ppm: 7.19–7.03 (m,
10H), 6.98 (s, 1H), 5.51 (s, 1H), 4.65 [q (not resolved well), 1H],
4.02 [q (not resolved well), 2H], 3.15 (s, 2H), 1.87 (d,
J=6.22 Hz, 3H), 0.96 (t, J=7.05 Hz, 3H);
13C NMR (100 MHz, CDCl3) δ ppm: 169.13,
162.40, 157.29, 152.24, 148.06 (m), 145.60 (m), 139.08 (m), 138.54,
138.43, 128.64–126.26 (phenyl carbons), 119.97, 103.31 (t,
JC-F=11.15), 61.07, 55.07, 53.11, 39.10, 32.50,
22.01, 13.62; MS (ESI, positive) m/z calculated for
C30H23BrF5NO5 (M+H):
651.07; identified 651.1.
Biology
All the cell lines used in the present study were
purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA). All the cells were treated with the half
maximal inhibitory concentration (IC50) of each compound
for the indicated durations. The IC50 concentrations
were determined using Microsoft excel: 12 µM of 5a and 6
µM of 5b for the Sk-Hep1 cells; 24 µM of 5a and 11
µM of 5b for the Hep3B cells; 48 µM of 5a and 20
µM of 5b for the MDA-MB-231 cells were used and the cells
were treated with the compounds for 24 h at 37°C. An
antiproliferative assay was performed using a previously described
method (17) with
modifications.
Cell culture and antiproliferative
assay
Human HCC Sk-Hep1 and Hep3B cells were maintained in
minimum essential medium with Earle’s balanced salts (MEM/EBSS),
containing 10% fetal bovine serum (FBS) at 37°C in a humidified
atmosphere containing 5% CO2 in air. The human breast
adenocarcinoma MDA-MB-231 cells and non-cancerous human embryonic
kidney HEK293 cells were maintained in Dulbecco’s modified Eagle’s
medium (DMEM) containing 10% FBS at 37°C in a humidified atmosphere
containing 5% CO2. The THLE-3 human normal liver cells
were maintained in bronchial epithelial cell basal medium (BEBM)
containing 10% FBS at 37°C in a CO2 incubator. Stock
solutions (10 mM) of the three compounds were obtained by
dissolving them in 100% dimethylsulfoxide (DMSO) and the stock
solution of the 3 different compounds was diluted with distilled
water to yield a concentration of 1 mM for treating the cells. Each
well of a 96-well tissue culture microtiter plate was inoculated
with 100 µl complete medium containing 1×104
cells. The plates were incubated at 37°C in a humidified 5%
CO2 incubator for 24 h prior to the experiments.
Following removal of the medium, 100 µl fresh medium
containing the compound at different concentrations was added to
each well (4, 8, 12, 16, 20 µM of 5a for the HEK293, THLE-3
and Sk-Hep1 cells; 10, 20, 30, 40, 50 µM of 5a for the Hep3B
and MDA-MB-231 cells; 2, 4, 6, 8, 10 µM of 5b for the
HEK293, THLE-3 and Sk-Hep1 cells; 4, 8, 12, 16, 20 µM of 5b
for the Hep3B and MDA-Mb-231 cells; 20, 40, 60, 80, 100 µM
of 5c for the Sk-Hep1, Hep3b and MDA-MB-231 cells) and incubated at
37°C for 24 h. Following incubation, 10 µl of EZ-Cytox Cell
Viability Assay Solution WST-1® reagents (Daeil Lab
Service, Seoul, Korea) were added to each well followed by further
incubation for 4 h at 37°C. The absorbance at 460 nm was measured
using a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
The IC50 was defined as the compound concentration
required to inhibit cell proliferation by 50%, compared with the
cells treated with DMSO only (deemed 100% viable).
Flow cytometry
The cells were treated with different concentrations
of the compounds (12 µM 5a and 6 µM 5b for the
Sk-Hep1 cells; 24 µM 5a and 11 µM 5b for the Hep3B
cells; 48 µM 5a and 20 µM 5b for the MDA-MB-231
cells) and incubated for 12 h at 37°C. The cells were then
harvested by trypsinization and centrifugation for 3 min at 1,200
rpm at room temperature, washed with phosphate-buffered saline
(PBS), and fixed with ice-cold 70% ethanol for overnight at 4°C.
The fixed cells were collected by centrifugation for 3 min at 1,200
rpm at 4°C, washed with PBS, snd resuspended in 0.5 ml PBS
containing propidium iodide (40 µg/ml) and RNase A (200
µg/ml). The cells were incubated on ice, in the dark, for 30
min. The cell cycle distribution was analyzed using a FACSCalibur
flow cytometer (BD Biosciences).
Deoxycholic acid (DCA) analysis
The cells were divided into 4 groups: i) control;
ii) DCA (5 µM) only; iii) DCA (5 µM) + 5a; and iv)
DCA (5 µM) + 5b. The cells were treated with DCA for 30 min
and were then immediately exposed to 5a and 5b compounds
independently. Following treatment for 12 h, the total proteins
were extracted from the cells and subjected to western blot
analyses with antibodies against phosphorylated forms of β-catenin,
E-cadherin, Axin and GSK-3β.
Protein extraction and western blot
analysis
The cells were incubated in the presence of the
compounds, as described, and, were collected at different time
intervals, centrifuged at 1,200 rpm for 3 min at room temperature
and washed twice with ice-cold PBS. The pellets were then
resuspended in lysis buffer, containing 50 mM Tris-Cl (pH 7.5), 150
mM NaCl, 1 mM DTT, 0.5% NP-40, 1% Triton X-100, 1% deoxycholate,
0.1% sodium dodecyl sulfate (SDS) and a cocktail of protease
inhibitors (Intron Biotechnology, Inc., Seoul, Korea). Following
lysis of the cells on ice for 1 h, the lysates were centrifuged at
14,000 rpm at 4°C for 20 min and the protein lysates were
collected. Equal quantities of 40 µg protein were resolved
using 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred onto a nitrocellulose membrane (Pall Life Sciences,
Port Washington, NY, USA). The membranes were blocked with PBS
buffer with 0.5% Tween-20, containing 5% skim milk overnight at
4°C. The membranes were then incubated with primary antibodies
overnight at 4°C: p53 (1:1,000, monoclonal, rabbit anti-human,
#2527), p16 (1:1,000, polyclonal, rabbit anti-human, #4824),
β-catenin (1:1,000, monoclonal, rabbit anti-human, #8480),
phospho-β-catenin (Ser33/37) (1:1,000, polyclonal, rabbit
anti-human, #2009), E-cadherin (1:1,000, monoclonal, rabbit
anti-human, #3195), Axin (1:1,000, monoclonal, rabbit anti-human,
#2087), GSK-3β (1:1,000, monoclonal, mouse anti-human, #9832),
c-myc (1:1,000, polyclonal, rabbit anti-human, #9402), Cdk2
(1:1,000, monoclonal, rabbit anti-human, #2546), p21 (1:1,000,
monoclonal, rabbit anti-human, #2947), p27 (1:1,000, polyclonal,
rabbit anti-human, #2552), MMP-2 (1:1,000, polyclonal, rabbit
anti-human, #4022) and GAPDH (1:1,000, monoclonal, rabbit
anti-human, #5174) (all purchased from Cell Signaling Technology
Inc., Beverly, MA, USA); Cyclin D1 (1:1,000, monoclonal, mouse
anti-human, ab101430) and phospho-β-catenin (Tyr142) (1:1,000,
polyclonal, rabbit anti-human, ab27798) were purchased from Abcam
(Cambridge, UK). The membranes were subsequently incubated with
horseradish peroxidase-conjugated anti-rabbit IgG secondary
antibodies (1:2,000) or anti-mouse IgG second antibodies (1:2,000)
for 1 h at room temperature. Both secondary antibodies were
purchased from Cell Signaling Technology Inc. All membranes were
visualized using WestSave™ Gold ECL (AbFrontier, Inc., Seoul,
Korea) and exposed to Hyperfilm (GE Healthcare, Little Chalfont,
UK). GAPDH was used as an internal loading control.
Matrigel invasion assay
The invasion of tumor cells was assessed using
Matrigel coated Transwell chambers with a 6.5 mm
polyvinyl/pyrrolidone-free polycarbonate filter of 8 µm pore
size (Corning Life Sciences, Tewksbury, MA, USA), as described
previously (18). The cells
(5×104 each of the Sk-Hep1, Hep3B and MDA-MB-231 cells)
and different concentrations of the compounds were suspended in 100
µl serum-free media, placed in the upper Transwell chamber
and incubated for 24 h at 37°C. The cells on the upper surface of
the filter were removed completely using a cotton swab, and the
lower surface of the filter was fixed with 4% formaldehyde and
stained with crystal violet (Sigma-Aldrich). Following staining,
the lower surface cells were lysed with 2% SDS for 1 h and the
lysate was measured using a microplate reader at 570 nm.
Statistical analysis
The statistical significance of differences between
the values of the compound treated and untreated groups was
determined using SigmaPlot 11 software (trial version). The results
are expressed as the mean ± standard deviation of three independent
experiments. Data were analyzed using one-way analysis of variance
followed by Tukey’s analysis for multiple comparisons. A P-value
<0.001 was considered to indicate a statistically significant
difference.
Results and Discussion
In vitro antiproliferative
activities
The cytotoxic effects of 5a, 5b and 5c were
evaluated in human embryonic kidney HEK293 cells, human normal
liver THLE-3 cells, human Sk-Hep1 and Hep3B cells and human breast
adenocarcinoma MDA-MB-231 cells. The synthesis and structure of the
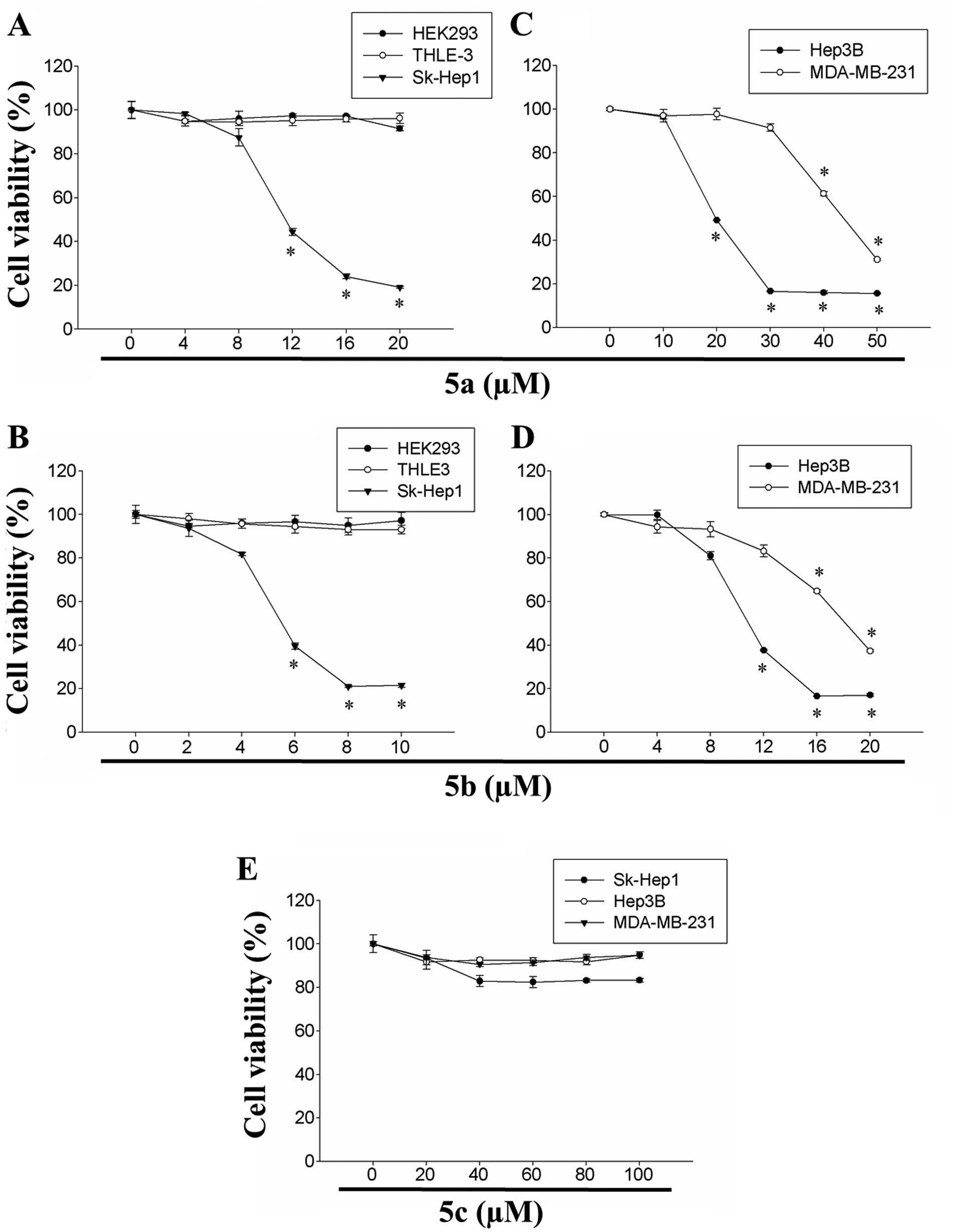
tetrahydropyridinol derivatives are illustrated in Fig. 1. The effects of 5a and 5b on the
viability of HEK293, THLE-3 and Sk-Hep1 cells are shown in Fig. 2A and B. Although the two compounds
exhibited cytotoxicity in a dose-dependent manner, 5b exhibited
significantly higher cytotoxicity, compared with 5a in the Sk-Hep1
cells. By contrast, the two compounds exhibited lower levels of
cytotoxicity towards the non-cancerous HEK293 and THLE-3 cells
(Fig. 2). The IC50 of
compounds 5a and 5b were 12 and 6 µM in the Sk-Hep1 cells,
respectively. The effectiveness of 5a on the viability of Hep3B and
MDA-MB-231 cells was subsequently investigated, and the results
revealed that 5a inhibited the cell proliferation in the two cell
lines in a dose-dependent manner (Fig. 2). The IC50 of 5a in
Hep3B and MDA-MB-231 cells was 24 and 48 µM, respectively.
Similarly, 5b dose-dependently inhibited the growth of the Hep3B
and MDA-MB-231 cells (Fig. 2).
The IC50 of 5b in the Hep3B and MDA-MB-231 cells was 11
and 20 µM, respectively. These results suggested that 5a and
5b were potent in inhibiting the proliferation of human HCC and
breast adenocarcinoma cells.
The present studyy also assessed the cytotoxicity of
5c in human HCC (Sk-Hep1 and Hep3B) and breast carcinoma
(MDA-MB-231) cells. However, no significant cytotoxicity in was
observed in any of the three cell lines was observed at
concentrations up to 100 µM (Fig. 2). These results indicated that the
cytotoxic effects of 5a and 5b compounds were more divergent on
human HCC and breast carcinoma cells. Taken together, the
investigations of cellular viability revealed that 5a and 5b were
significantly effective in inducing cytotoxicity towards Sk-Hep1,
Hep3B and MDA-MB-231 cancer cell lines, and had differential
inhibitory effects. Among these three cell lines, Sk-Hep1 cells
were more sensitive to the two compounds, compared with the other
cell lines, and the inhibitory concentrations were considerably
less, compared with the Hep3B and MDA-MB-231 cells. However, the 5a
and 5b compounds were less toxic towards the non-cancerous human
embryonic kidney HEK293 cells and human liver THLE-3 cells.
The significant cytotoxicity profiles of the
chloroacetyl derivative, compared with the corresponding bromo
counterpart and 2-bromopropionyl bromide suggested that the size
and electronegativity of the halogens are important, indicating
that, compared to bromine, chlorine atoms are smaller in size with
increased electronegativity, which may affect its improved cell
permeability and favorable interaction with the binding site of its
biological target. Furthermore, depending on the nature of
substituent at the heterocyclic nitrogen, each of the molecules
preferentially exhibit an energetically favorable non-chair
conformation, which may also be vital with regard to their
individual potency. The poor activity observed with bromofluoro may
be due to the increased size from the incorporation of lipophilic
methyl group at the N-acetyl functionality.
Compounds 5a and 5b differentially
targets β-catenin signaling in human HCC and breast cancer
cells
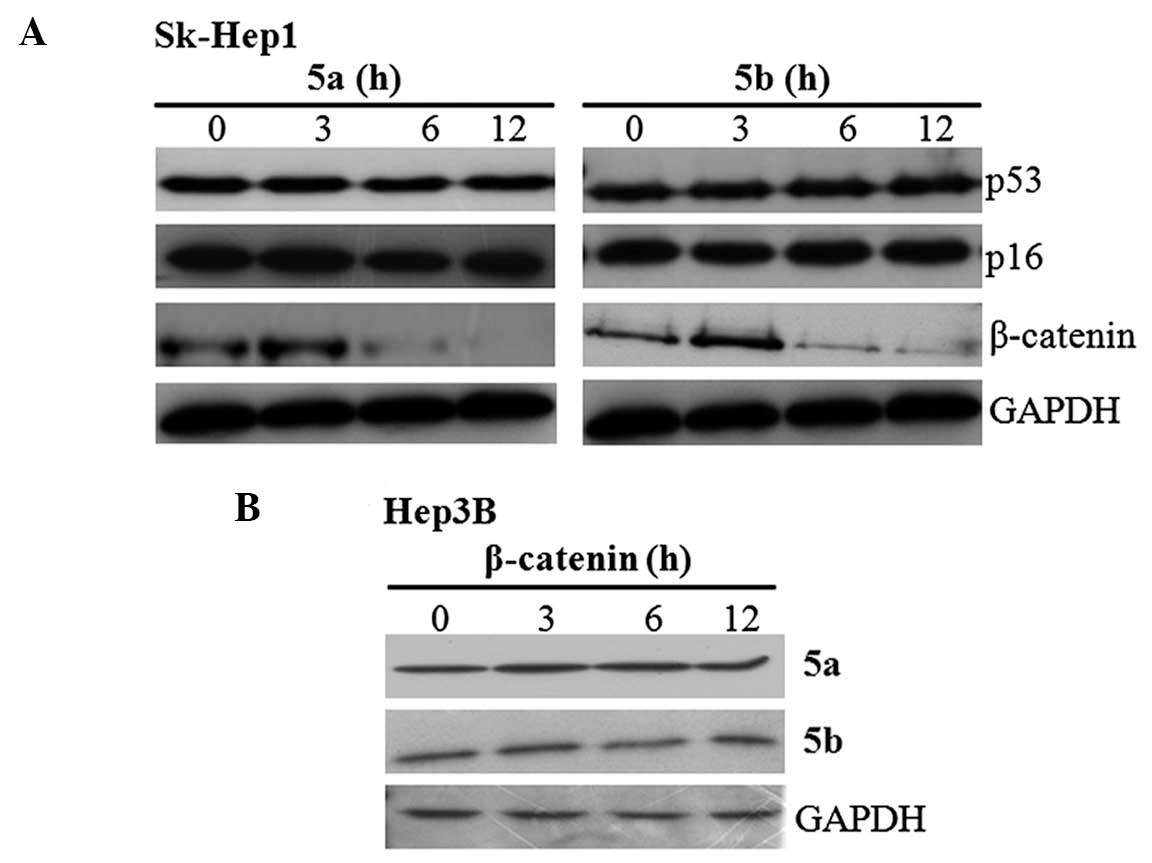
The present study subsequently examined whether the
inhibition of cell proliferation was due to upregulation of tumor
suppressor proteins or downregulation of oncogenes. To clarify
this, Sk-Hep1 cells were treated with 5a and 5b independently for
different time intervals, and the expression of HCC deregulated
proteins, p53, p16 and β-catenin, were examined using western
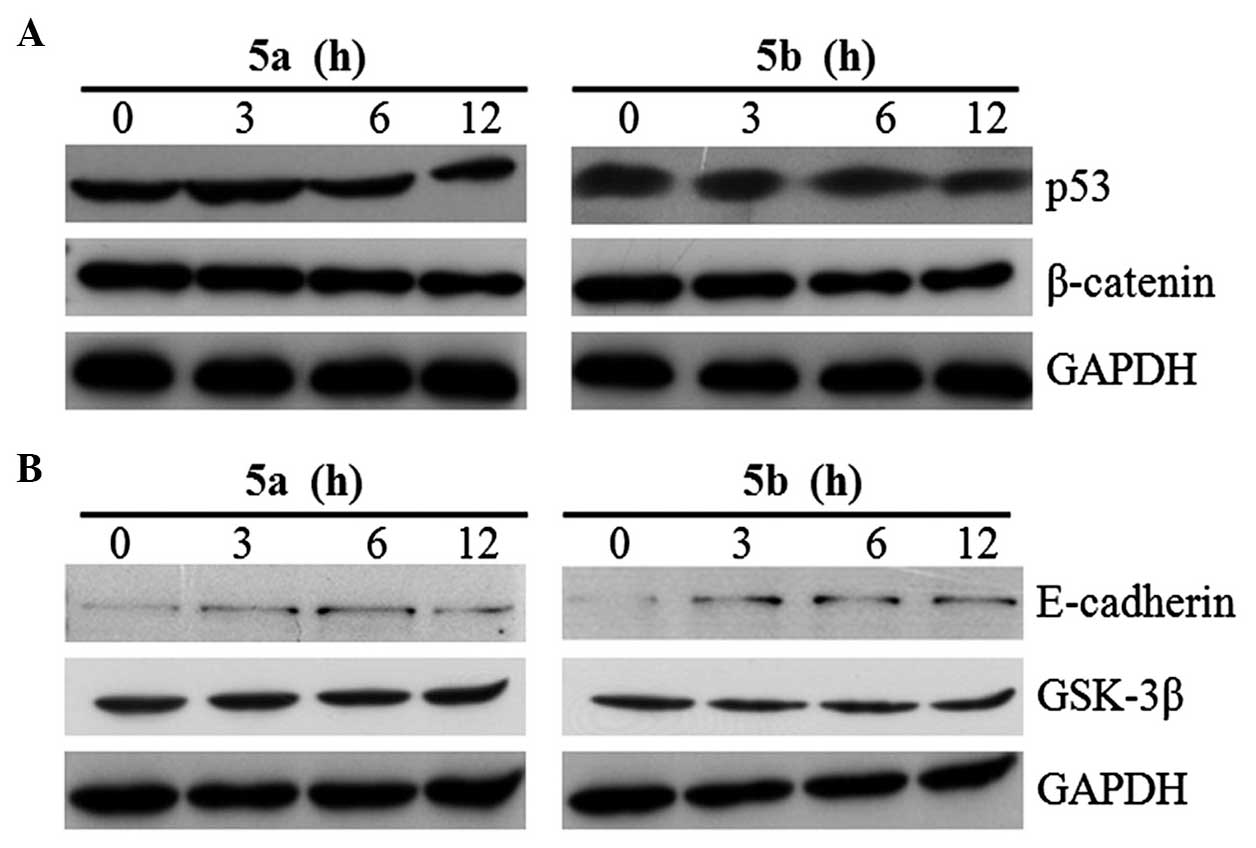
blotting. The blots revealed that the expression of β-catenin was
downregulated in a time-dependent manner by the two compounds
(Fig. 3A), whereas no change in
the expression of the p53 and p16 tumor suppressor proteins was
observed (Fig. 3A). Following
treatment for 12 h, the expression level of β-catenin was
completely inhibited by 5a and 5b in the Sk-Hep1 cells. The
inhibitory effects of 5a and 5b were also analyzed in Hep3B cells.
The results demonstrated that the expression of β-catenin was not
altered, and its expression levels were maintained throughout
treatment (Fig. 3B). These
results raises the question of whether these compounds specifically
inhibit either the expression or activity of β-catenin in Sk-Hep1
cells.
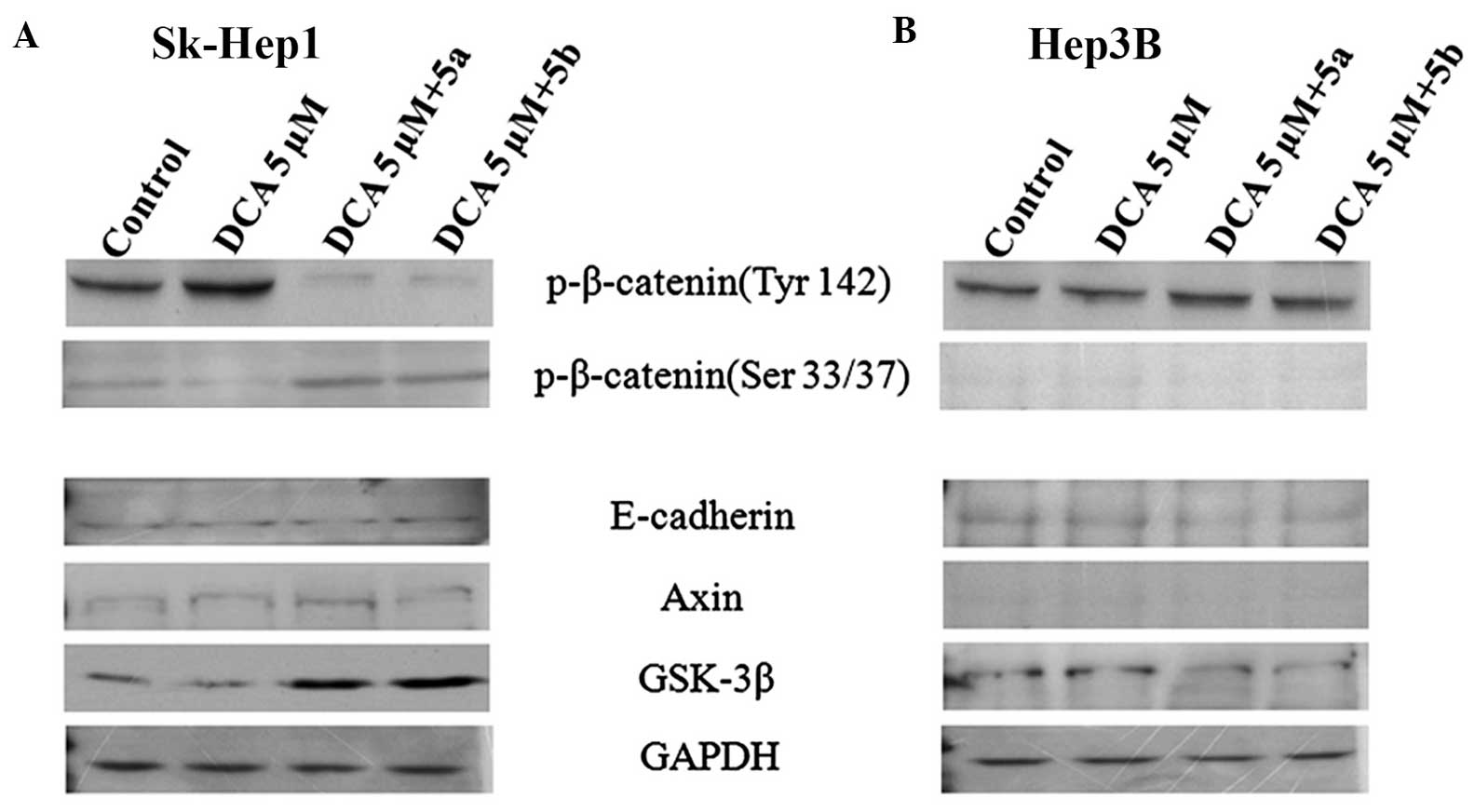
In order to address this question, DCA, which
specifically activates the phosphorylation of β-catenin (19) was used. For this analysis, the
cells were divided into four groups: i) control; ii) DCA (5
µM) only;iii) DCA (5 µM) + 5a; and iv) DCA
(5µM) + 5b. DCA was treated for 30 min and then immediately
exposed the cells to 5a and 5b compounds independently. Following
treatment for 12 h, the proteins were extracted from the cells and
subjected to western blot analyses with antibodies against the
phosphorylated forms of β-catenin. The results of the western
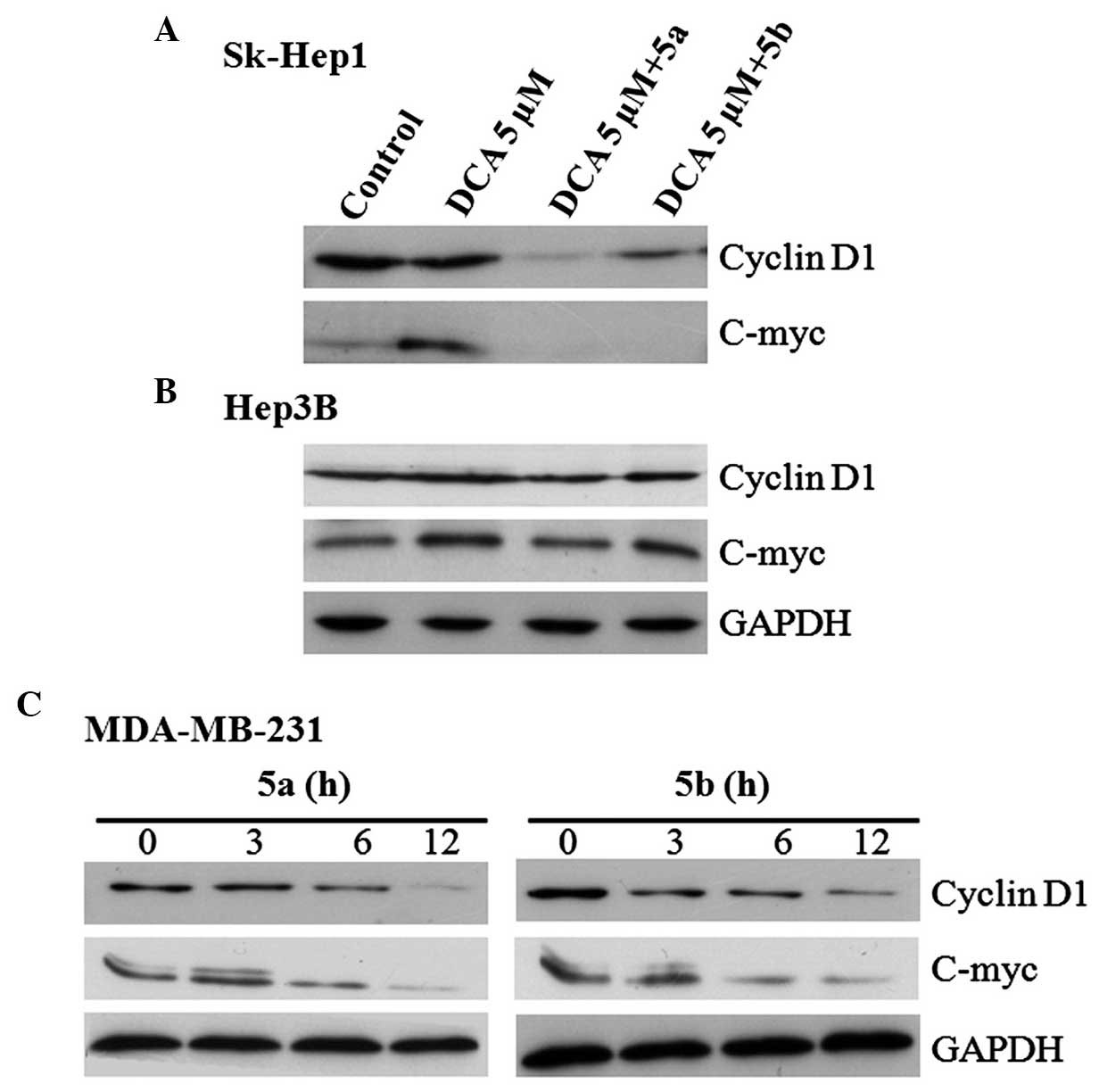
blotting revealed that, in the Sk-Hep1 cells, the expression of
phospho-β-catenin (Tyr142) was increased in the DCA only group and
decreased in the DCA+5a and DCA+5b groups (Fig. 4A), whereas the expression of
phospho-β-catenin (Ser33/37) was not significantly different to
those in the DCA+5a and DCA+5b compound groups (Fig. 4A). However, this type of
expression pattern was not observed in Hep3B cells (Fig. 4B). The phosphorylation of
β-catenin at Ser33/37 by GSK-3β and the association of β-catenin
with other tumor suppressor proteins, Axin and APC, lead to
ubiquitinization of β-catenin and subsequently targets degradation
by the ubiquitin-proteosome complex (20). By contrast, the phosphorylation of
β-catenin at Tyr142 inhibits the incorporation of β-catenin with
the Axin-APC complex, which leads to accumulation of β-catenin in
the cytoplasm and subsequent export to the nucleus. In the nucleus,
β-catenin, in association with its nuclear binding partners, TCF
and LEF, initiates transcription of the β-catenin-targeted genes,
cyclin D1 and c-myc (20–22). The c-myc and cyclin D1 promoters
contain TCF-binding sites, which mediate transcriptional activation
via the TCF/β-catenin complex. The results of the present study
suggeste that compounds 5a and 5b inhibited active β-catenin
expression in highly invasive Sk-Hep1 cells only, but not in the
Hep3B cells (Fig. 4B).
The present study also investigated whether these
compounds inhibit only β-catenin, or affect any other proteins in
the β-catenin signaling pathway. Using western blot analysis, the
expression of E-cadherin, a metastasis-suppressor protein, Axin1, a
tumor-suppressor protein, and GSK-3β were examined. The resulting
data demonstrated that, in the Sk-Hep1 cells, the expression levels
of E-cadherin and Axin1 were low in the control group, and their
levels of expression were not altered by these compounds (Fig. 4A). By contrast, these compounds
upregulated the expression of GSK-3β (Fig. 4A), which resulted in the
phosphorylation of β-catenin at the Ser33/37 residue and
facilitated its degradation. In the Hep3B cells, this upregulation
was not observed, with the proteins expressed at a low level and
remaining unchanged (Fig. 4B).
The levels of β-catenin in the cells is tightly controlled by its
degradation complexm composed of Axin, APC, GSK-3β and β-catenin,
in which GSK-3β phosphorylates β-catenin and thus triggers its
ubiquitination and subsequent proteosomal degradation (20). Similarly, the downregulation of
E-cadherin is more frequently achieved by promoter hypermethylation
in tumor cells, particularly in HCC (23). The overexpression of β-catenin and
downregulation of E-cadherin in poorly differentiated, highly
invasive cancers implicates the involvement of E-cadherin and
β-catenin in the progression of cancer (24). The inhibition of β-catenin, and
the upregulation of GSK-3β and E-cadherin may be crucial for
restraining the progression of cancer. Overexpression of Axin1 in
mammalian cells results in a decrease in levels of β-catenin and
suppression of TCF-dependent gene transcription (25). Together, the results suggested
that, particularly in highly invasive Sk-Hep1 cells, 5a and 5b
inhibited the elevated expression of phospho-β-catenin (Tyr142),
and the upregulation of GSK-3β by these compounds phosphorylated
free cytoplasmic β-catenin at Ser33/37 residues, which, tagged with
the ubiquitin, lead to the degradation of β-catenin.
In order to further confirm the above-mentioned
results, the highly invasive breast cancer, MDA-MB-231, cell line
was examined. The MDA-MB-231 cells were exposed to 5a and 5b
independently, and western blot analysis against β-catenin and p53
was performed. The results demonstrated no changes in the
expression levels of β-catenin or p53 (Fig. 5A). Therefore, the expression of
β-catenin binding partner, E-cadherin and GSK-3β, were examined.
The two compounds upregulated the expression of E-cadherin,
however, the expression of GSK-3β was not altered (Fig. 5B). These results demonstrated that
5a and 5b stimulated the expression of E-cadherin, suggesting
significant attachment may occur between E-cadherin and β-catenin.
Several studies have described a partial or complete loss of
E-cadherin during cancer progression (26–28). The role of E-cadherin as a tumor
suppressor protein and the dual role of its binding partner,
β-catenin, in cell adhesion and Wnt signaling may indicate a
function for E-cadherin in the Wnt pathway. In the absence of an
appropriate Wnt signal, E-cadherin may sequester β-catenin at the
cell membrane, thereby preventing the formation of TCF-β-catenin
complexes in the nucleus. Mutation of E-cadherin in cancer cells
may disrupt the interaction with β-catenin. This disruption
inhibits the formation of destruction complex of β-catenin with
APC, Axin and GSK-3β (29). In
the presnt study, the compounds differentially targetEd the
Wnt/β-catenin signaling by inhibiting the expression of β-catenin
in the Sk-Hep1 cells and upregulating the expression of GSK-3β and
E-cadherin in the Sk-Hep1 and MDA-MB-231 cells, respectively,
specifically modulating the activity of Wnt/β-catenin signaling in
highly invasive cancer cells.
Compounds 5a and 5b induce G1 phase
arrest
In the nucleus, β-catenin, in association with
TCF/LEF, initiates transcription of its downstream target genes,
particularly c-myc and cyclin D1 (20–22). To determine whether these
compounds modulate the expression of β-catenins target genes,
cyclin D1 and c-myc, western blot analysis was performed using the
proteins of the Sk-Hep1, Hep3B and MDA-MB-231 cells. The blots
revealed that the expression levels of cyclin D1 and c-myc were
inhibited in the Sk-Hep1 and MDA-MB-231 cells by the two compounds
(Fig. 6). However, the expression
levels of these β-catenin target proteins were not inhibited and
remained unchanged in the Hep3B cells (Fig. 6).
The present study subsequently examined whether 5a
and 5b induced the downregulation of cyclin D1 in either Sk-Hep1 or
MDA-MB-231 cells, and where growth inhibition of Hep3B cells was
caused by cell cycle arrest. To address this question, the cells
were treated independently with 5a and 5b at IC50
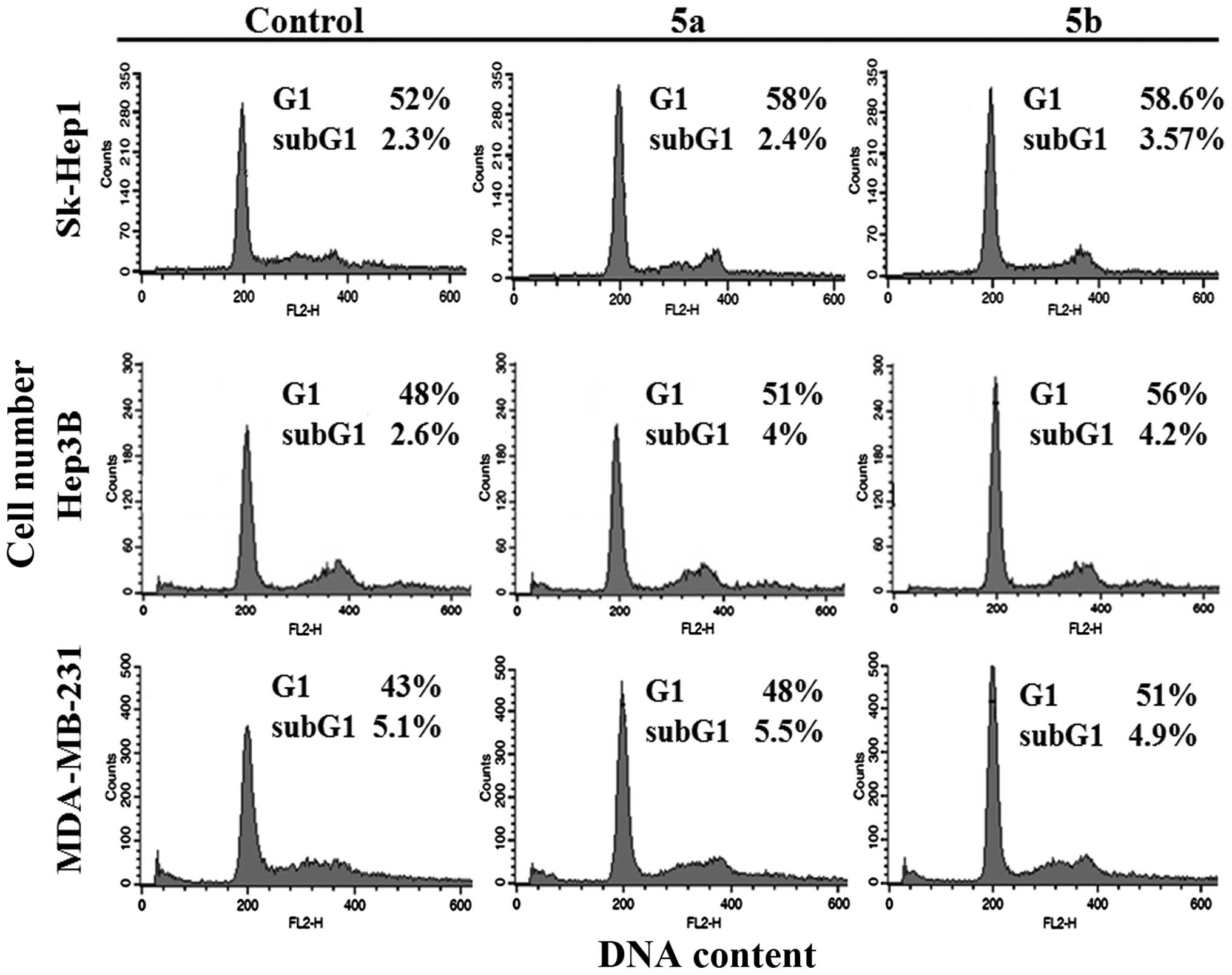
concentrations for 12 h, fixed, and cell cycle populations were
determined using flow cytometry. The results demonstrated that the
percentage of the cell population in the G1 phase was significantly
higher in all three cell lines, compared with the untreated control
group (Fig. 7). These results
confirmed that the growth of Hep3B cells was inhibited by cell
cycle arrest, whereas the growth inhibition in Sk-Hep1 and
MDA-MB-231 cells was mediated by Wnt/β-catenin signaling. The
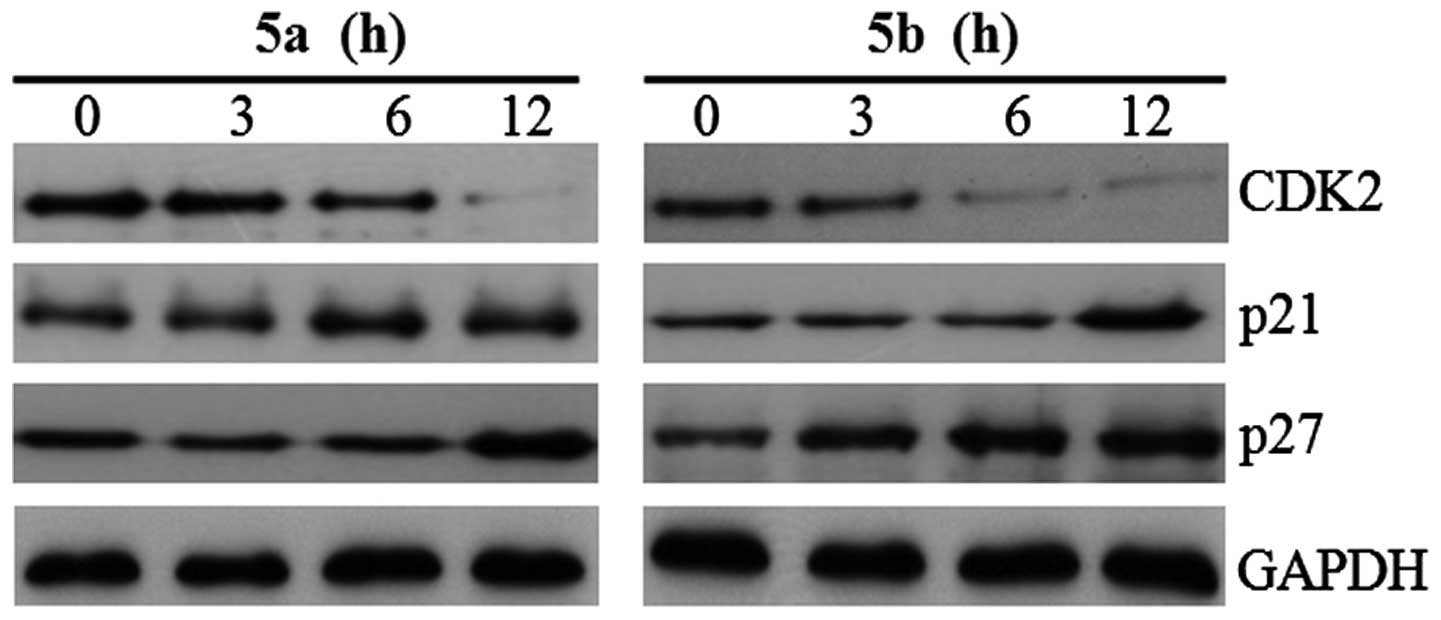
expression levels of cell cycle regulatory proteins, p21, p27 and
CDK2, were also examined in the Hep3B cells. The expression levels
of p21 and p27 were significantly upregulated in the Hep3B cells
following treatment with either of the two compounds, and CDK2 was
significantly decreased following treatment (Fig. 8). In mammalian cells, cyclin D, E
and A are synthesized sequentially during the G1 phase of the cell
cycle. The major catalytic partners of these cyclins are CDK2 and
CDK4, which are negatively regulated by CDK-inhibitors, including
p21, p27 and p53 (30,31). The p53 tumor suppressor is
required for the transcriptional activation of p21 (32). Previously, indole-3-carbinol
tetrameric derivative has been observed to induce G1 cell cycle
arrest in breast cancer cells by upregulating p27kip1 (33). In the present study, as expected,
p21 and p27 were induced by 5a and 5b via a p53-independent pathway
in the Hep3B cells (p53 null type). These results suggested that
downregulation of the expression of cyclin D1 and CDK2, and the
induction of p21 and p27, had a suppressive effect on the growth of
the Sk-Hep1, Hep3B and MDA-MB-231 cells. Taken together, compounds
5a and 5b inhibited the progression of cell cycle differentially
through Wnt/β-catenin mediation in the Sk-Hep1 and MDA-MB-231
cells, and via p53-independent cell cycle arrest in Hep3B
cells.
Effects of 5a and 5b on invasion of
Sk-Hep1 and MDA-MB-231 cells
Non-phosphorylated β-catenin accumulates in the
cytoplasm and, when activated, it enters the nucleus and interacts
with TCF/LEF to control various target genes, which are involved in
cellular proliferation and metastasis (10). Since metastasis is the leading
cause of mortality in human cancer (10), the present study assessed the
chemotherapeutic effects of 5a and 5b on the invasive potential of
human HCC and breast carcinoma cells. For this purpose, a Matrigel
invasion assay was performed using the Sk-Hep1, Hep3B and
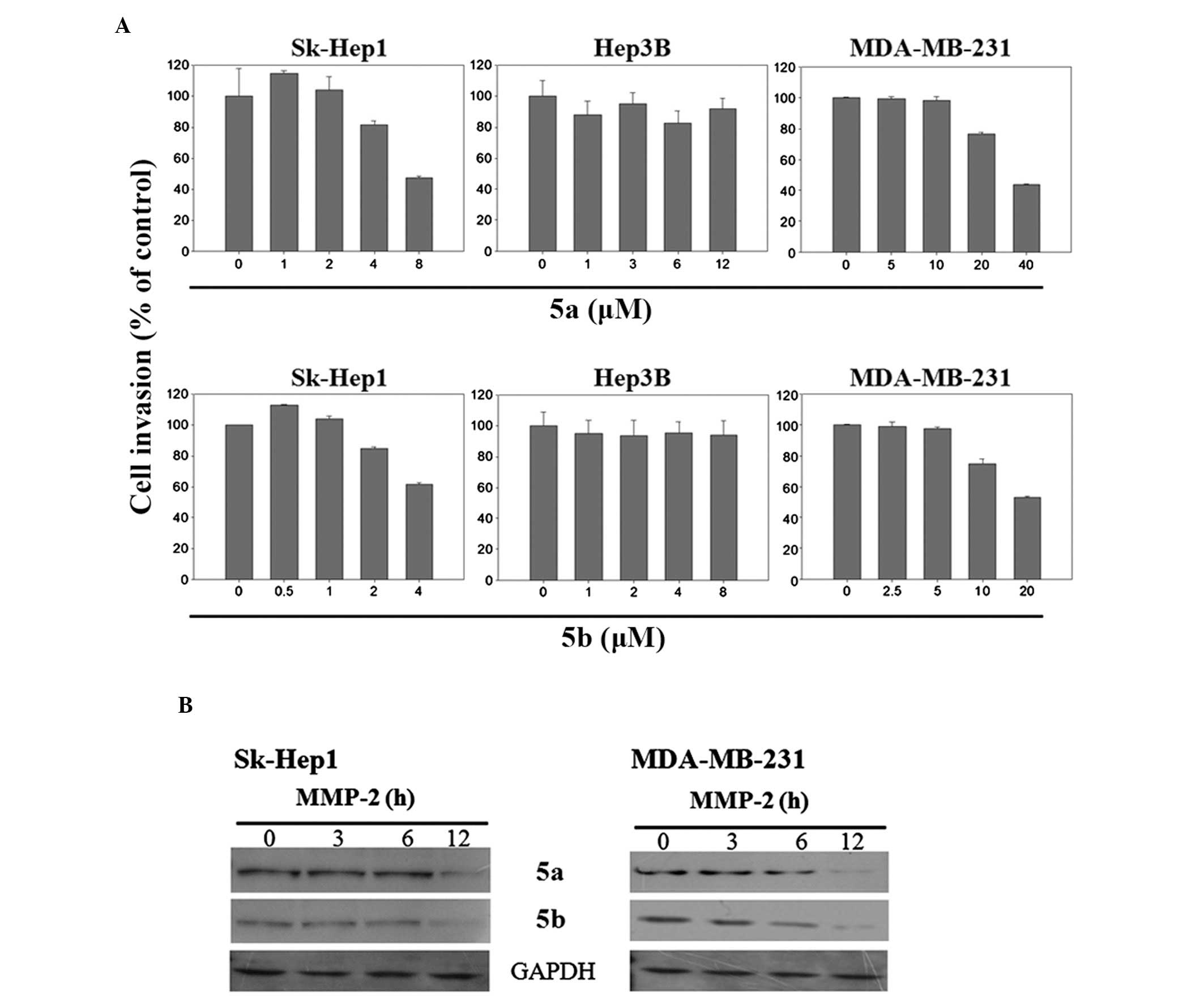
MDA-MB-231 cells. The results of the invasion assay reveled that
the invasive potential of highly metastatic Sk-Hep1 and MDA-MB-231
cells was decreased dose-dependently by 5a and 5b compounds
(Fig. 9A), whereas, in
non-metastatic Hep3B cells, these compounds were not functional
(Fig. 9A). The the expression of
MMP-2 was then determined in the Sk-Hep1 and MDA-MB-231 cells, as
MMP-2 is involved in the process of invasion and its inhibition may
be crucial for the suppression of cancer metastasis. The results
demonstrated that the expression of MMP-2 was reduced after 12 h
treatment with 5a and 5b in the Sk-Hep1 and MDA-MB-231 cells
(Fig. 9B). The expression of
MMPs, including MMP-2 has been observed to be important in the
degradation of the basement membrane in cancer invasion and is
associated with tumor metastasis (34). The expression of cell adhesion
molecule, including E-cadherin, is also associated with the
invasiveness of tumor cells. High expression levels of E-cadherin
are reported to correlate with a low invasive potential of cells,
whereas low levels of the expression are associated with increased
invasiveness (35,36). Hispoplan and lycopene inhibits
metastasis in Sk-Hep1 cells by downregulating the expression levels
of MMP-2 and MMP-9 (37,38). It has been reported that
suppression of breast cancer invasion and migration by
indole-3-carbinol is associated with the upregualtion of
E-cadherin/catenin complex (39).
The results of the present study suggested that the downregulation
of β-catenin, as well as the inhibition of MMP-2 may facilitate the
inhibition of invasion potential of Sk-Hep1 and MDA-MB-231 cells.
As the level of β-catenin was not downregulated in the Hep3B cells,
the inhibition of invasion was not observed, and further confirmed
that the inhibitory effect was associated with the β-catenin
signaling pathway.
In conclusion, accumulating evidence has suggested
that several natural compounds may be used alone or in combination
with traditional chemotherapeutic agents to prevent the occurrence
of cancer and its metastatic spread. In addition, several
non-steroid inflammatory drugs may target directly or indirectly to
β-catenin signaling. The findings of the present study suggested
that compounds 5a and 5b have the ability to interfere β-catenin
signaling by downregulating β-catenin and its target genes in
Sk-Hep1 cells, upregulate the expression of E-cadherin in
MDA-MB-231 cells and also inhibit the invasive potential of Sk-Hep1
and MDA-MB-231 cells. In Hep3B cells, however, these compounds did
not affect the β-catenin pathway, and affected only the cell cycle
progression by modulating the cell cycle regulatory proteins. These
results suggested that 5a and 5b are specific for highly invasive
cells and inhibit cell proliferation and invasion by targeting the
β-catenin signaling pathway. Thus, the antiproliferative and
anti-invasive activitievs of 5a and 5b could serve as a basis for
chemopreventative therapy in human HCC and breast carcinoma.
Further investigations are required to fully elucidate the
signaling pathways in other invasive and non-invasive cancer cell
lines, and in vivo.
Acknowledgments
This study was supported by a grant from the Next
Generation Bio Green 21 Program, Rural Development Administration,
Republic of Korea (grant. no. SSAC-PJ01106901 to W.-Y.K. and
SSAC-PJ01137901 to S.Y.L.).
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
EMEM
|
Eagle’s minimal essential medium
|
|
BEBM
|
bronchial epithelial cell basal
medium
|
|
FBS
|
fetal bovine serum
|
|
WST-1®
|
2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium,
monosodium salt
|
|
PBS
|
phosphate-buffered saline
|
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ries L, Melbert D, Krapcho M, Stinchcomb
D, Howlader N, Horner M, Mariotto A, Miller B, Feuer E and
Altekruse S: SEER Cancer Statistics Review. Based on November 2007
SEER data submission, posted to the SEER web site. National Cancer
Institute; Bethesda, MD: 2008
|
|
3
|
Roayaie S, Blume IN, Thung SN, Guido M,
Fiel MI, Hiotis S, Labow DM, Llovet JM and Schwartz ME: A system of
classifying microvascular invasion to predict outcome after
resection in patients with hepatocellular carcinoma.
Gastroenterology. 137:850–855. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lang L: FDA approves sorafenib for
patients with inoperable liver cancer. Gastroenterology.
134:3792008.PubMed/NCBI
|
|
5
|
Clevers H: Wnt/beta-catenin signaling in
development and disease. Cell. 127:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cadigan KM: Wnt-β-catenin signaling. Curr
Biol. 18:R943–R947. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kemler R: From cadherins to catenins:
cytoplasmic protein interactions and regulation of cell adhesion.
Trends Genet. 9:317–321. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jamora C and Fuchs E: Intercellular
adhesion, signalling and the cytoskeleton. Nat Cell Biol.
4:E101–E108. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Clevers H: Wnt/β-catenin signaling in
development and disease. Cell. 127:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Polakis P: The many ways of Wnt in cancer.
Curr Opin Genet Dev. 17:45–51. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zucman-Rossi J, Benhamouche S, Godard C,
Boyault S, Grimber G, Balabaud C, Cunha A, Bioulac-Sage P and
Perret C: Differential effects of inactivated Axin1 and activated
β-catenin mutations in human hepatocellular carcinomas. Oncogene.
26:774–780. 2007. View Article : Google Scholar
|
|
12
|
Fujie H, Moriya K, Shintani Y, Tsutsumi T,
Takayama T, Makuuchi M, Kimura S and Koike K: Frequent beta-catenin
aberration in human hepatocellular carcinoma. Hepatol Res.
20:39–51. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim YD, Park CH, Kim HS, Choi SK, Rew JS,
Kim DY, Koh YS, Jeung KW, Lee KH and Lee JS: Genetic alterations of
Wnt signaling pathway-associated genes in hepatocellular carcinoma.
J Gastroenterol Hepatol. 23:110–118. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Laurent-Puig P and Zucman-Rossi J:
Genetics of hepatocellular tumors. Oncogene. 25:3778–3786. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Aridoss G, Amirthaganesan S, Kumar NA, Kim
JT, Lim KT, Kabilan S and Jeong YT: A facile synthesis,
antibacterial, and antitubercular studies of some piperidin-4-one
and tetrahydro-pyridine derivatives. Bioorg Med Chem Lett.
18:6542–6548. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Aridoss G, Amirthaganesan S and Jeong YT:
Synthesis, crystal and antibacterial studies of diversely
functionalized tetrahy-dropyridin-4-ol. Bioorg Med Chem Lett.
20:2242–2249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Awasthi N, Schwarz MA, Verma V, Cappiello
C and Schwarz RE: Endothelial monocyte activating polypeptide II
interferes with VEGF-induced proangiogenic signaling. Lab Invest.
89:38–46. 2009. View Article : Google Scholar
|
|
18
|
Hung WC and Chang HC: Indole-3-carbinol
inhibits Sp1-induced matrix metalloproteinase-2 expression to
attenuate migration and invasion of breast cancer cells. J Agric
Food Chem. 57:76–82. 2009. View Article : Google Scholar
|
|
19
|
Pai R, Tarnawski AS and Tran T:
Deoxycholic acid activates β-catenin signaling pathway and
increases colon cell cancer growth and invasiveness. Mol Biol Cell.
15:2156–2163. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
MacDonald BT, Tamai K and He X:
Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev
Cell. 17:9–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tetsu O and McCormick F: β-catenin
regulates expression of cyclin D1 in colon carcinoma cells. Nature.
398:422–426. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
He TC, Sparks AB, Rago C, Hermeking H,
Zawel L, da Costa LT, Morin PJ, Vogelstein B and Kinzler KW:
Identification of c-MYC as a target of the APC pathway. Science.
281:1509–1512. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wei Y, Van Nhieu JT, Prigent S,
Srivatanakul P, Tiollais P and Buendia MA: Altered expression of
E-cadherin in hepa-tocellular carcinoma: correlations with genetic
alterations, β-catenin expression, and clinical features.
Hepatology. 36:692–701. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Matsumura T, Makino R and Mitamura K:
Frequent down-regulation of E-cadherin by genetic and epigenetic
changes in the malignant progression of hepatocellular carcinomas.
Clin Cancer Res. 7:594–599. 2001.PubMed/NCBI
|
|
25
|
Sakanaka C, Weiss JB and Williams LT:
Bridging of β-catenin and glycogen synthase kinase-3β by axin and
inhibition of β-catenin-mediated transcription. Proc Natl Acad Sci
USA. 95:3020–3023. 1998. View Article : Google Scholar
|
|
26
|
Acs G, Lawton TJ, Rebbeck TR, LiVolsi VA
and Zhang PJ: Differential expression of E-cadherin in lobular and
ductal neoplasms of the breast and its biologic and diagnostic
implications. Am J Clin Pathol. 115:85–98. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rakha EA, Patel A, Powe DG, et al:
Clinical and biological significance of E-cadherin protein
expression in invasive lobular carcinoma of the breast. Am J Surg
Pathol. 34:1472–1479. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gould Rothberg BE and Bracken MB:
E-cadherin immunohisto-chemical expression as a prognostic factor
in infiltrating ductal carcinoma of the breast: a systematic review
and meta-analysis. Breast Cancer Res Thr. 100:139–148. 2006.
View Article : Google Scholar
|
|
29
|
Orsulic S, Huber O, Aberle H, Arnold S and
Kemler R: E-cadherin binding prevents beta-catenin nuclear
localization and beta-catenin/LEF-1-mediated transactivation. J
Cell Sci. 112:1237–1245. 1999.PubMed/NCBI
|
|
30
|
Harper JW and Elledge SJ: Cdk inhibitors
in development and cancer. Curr Opin Genet Dev. 6:56–64. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yew PR: Ubiquitin-mediated proteolysis of
vertebrate G1- and S-phase regulators. J Cell Physiol. 187:1–10.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zeng YX and el-Deiry WS: Regulation of
p21WAF1/CIP1 expression by p53-independent pathways. Oncogene.
12:1557–1564. 1996.PubMed/NCBI
|
|
33
|
Brandi G, Paiardini M, Cervasi B, Fiorucci
C, Filippone P, De Marco C, Zaffaroni N and Magnani M: A new
indole-3-carbinol tetrameric derivative inhibits cyclin-dependent
kinase 6 expression, and induces G1 cell cycle arrest in both
estrogen-dependent and estrogen-independent breast cancer cell
lines. Cancer Res. 63:4028–4036. 2003.PubMed/NCBI
|
|
34
|
Nakajima M, Welch DR, Belloni PN and
Nicolson GL: Degradation of basement membrane type IV collagen and
lung subendothelial matrix by rat mammary adenocarcinoma cell
clones of differing metastatic potentials. Cancer Res.
47:4869–4876. 1987.PubMed/NCBI
|
|
35
|
Mareel MM, Van Roy FM and De Baetselier P:
The invasive phenotypes. Cancer Metastasis Rev. 9:45–62. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Christofori G and Semb H: The role of the
cell-adhesion molecule E-cadherin as a tumour-suppressor gene.
Trends Biochem Sci. 24:73–76. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Huang GJ, Yang CM, Chang YS, Amagaya S,
Wang HC, Hou WC, Huang SS and Hu ML: Hispolon suppresses SK-Hep1
human hepatoma cell metastasis by inhibiting matrix
metal-loproteinase-2/9 and urokinase-plasminogen activator through
the PI3K/Akt and ERK signaling pathways. J Agric Food Chem.
58:9468–9475. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hwang ES and Lee HJ: Inhibitory effects of
lycopene on the adhesion, invasion, and migration of SK-Hep1 human
hepatoma cells. Exp Biol Med. 231:322–327. 2006.
|
|
39
|
Meng Q, Qi M, Chen DZ, Yuan R, Goldberg
ID, Rosen E, Auborn K and Fan S: Suppression of breast cancer
invasion and migration by indole-3-carbinol: associated with
up-regulation of BRCA1 and E-cadherin/catenin complexes. J Mol Med.
78:155–165. 2000. View Article : Google Scholar : PubMed/NCBI
|