Introduction
Non-alcoholic fatty liver disease (NAFLD) is
characterized by fat accumulation in the liver that is not due to
alcohol abuse. Epidemiological data indicate that NAFLD is not
simpl a liver disease, but rather a concentrated reflection of
obesity, hyper-lipidemia and insulin resistance and other metabolic
disorders in the liver (1,2).
Although low-grade NAFLD is considered a benign disease, without
appropriate treatment, it can cause more severe liver injury, and
may develop into non-alcoholic steatohepatitis, fibrosis,
cirrhosis, and in some cases, hepatocellular carcinoma (3,4).
Previous studies have revealed that NAFLD is more prevalent in
obese individuals and patients with type 2 diabetes (5,6),
suggesting an association between metabolic syndrome and insulin
resistance (1,4,7–9).
However, the pathogenesis of NAFLD is not yet fully understood, and
an effective treatment for NAFLD is yet to be developed.
Peroxisome proliferator-activated receptors (PPARs)
are members of the steroid hormone nuclear receptor family
(10). The PPAR family
encompasses PPARα, PPARγ and PPARβ/δ, which are all
ligand-activated transcription factors that mediate the hormonal
control of gene expression. PPARs serve as important modulators in
glucose and fatty acid metabolic pathways, as well as in adipocyte
proliferation, differentiation and apoptosis (11–14). Agonists of PPARs have been shown
to be effective in the treatment of metabolic syndrome, such as
type 2 diabetes mellitus and NAFLD (15,16). Thiazolidinediones (TZDs), a group
of agonists of PPARγ, have been widely studied and used in the
treatment of type 2 diabetes since 1997 (15). Of these, pioglitazone, a selective
agonist to the nuclear receptor PPARγ and to a lesser extent PPARα,
has been shown to improve insulin resistance (17,18). PPARs are considered to have
medical significance (19).
Another PPARγ agonist, rosiglitazone, has been shown to ameliorate
non-alcoholic steatohepatitis (20). PPARγ has also been shown to
regulate genes involved in insulin/insulin-like growth factor
signaling, lipid metabolism and adipocyte-specific development
(21).
PPARδ is ubiquitously expressed in skeletal muscle
cells, as well as in liver cells (22,24). It has been documented that
GW501516, a potent agonist to PPARδ, modulates glyco-metabolism,
fatty acid metabolism and alleviates insulin resistance (22–24). GW501516 increases fatty acid
oxidation by increasing the levels of hepatic mRNAs associated with
fatty acid β-oxidation, such as acyl-CoA oxidase (ACOX), carnitine
palmitoyltransferase-1 (CPT-1) and liver fatty acid binding protein
(L-FABP) (23,25). The increase in fatty droplets
within hepatocytes and the development of steato-hepatitis in mice
fed a methionine- and choline-deficient diet has been shown to be
inhibited by GW501516 (25).
However, the effects of GW501516 on NAFLD remain controversial, and
the underlying mechanisms remain to be elucidated.
In the present study, we explored the effects of
GW501516 in a rat model of NAFLD. It was found that GW501516
effectively alleviated NAFLD. Glucose and fatty acid metabolism, as
well as the related enzymes, were also found to be regulated by
GW501516.
Materials and methods
Animals and treatment
Forty-six 8-week-old male Sprague-Dawley (SD) rats
obtained from the Laboratory Animal Center of Xi'an Jiaotong
University (Xi'an, China) were used in the present study. All the
rats were housed under specific pathogen-free conditions with free
access to water. All the experiments were carried out in adherence
to a protocol approved by the Institutional Animal Care and Use
Committee of Xi'an Jiaotong University. Of the 46 rats, some were
fed a standard chow diet (Laboratory Animal Center of Xi'an
Jiaotong University, Xi'an, China) and were assigned to the normal
control group (n=10), while the others were used to establish a
model of NAFLD. For the induction of NAFLD, the rats were fed a
high-fat diet (HFD; 10% lard, 2% cholesterol, 5% saccharose, 0.5%
hog bile extract and 82.5% standard chow diet) for 12 weeks. The 36
rats fed the HFD were assigned to the HFD control group (n=12), the
GW501516 treatment group (n=12), or the pioglitazone treatment
group (n=12). Therapeutic intervention with GW501516 (Santa Cruz
Biotechnology, Santa Cruz, CA, USA) or pioglitazone (Santa Cruz
Biotechnology) was initiated after 8 weeks on the HFD. GW501516 (10
mg/kg body weight/day, in water) and pioglitazone (10 mg/kg body
weight/day, in water) were administered orally once a day to the
rats in the respective groups from the 8th week until the 12th
week. The rats in the HFD control group received the same amount of
normal saline. Blood samples were collected from the tail vein and
stored at −70°C until biochemical analysis. At the end of each
experiment, the rats were sacrificed by an intravenous
administration of pentobarbital (overdose). The body weight, liver
wet weight and body length of the rats were then measured. The
hepatosomatic index was calculated as follows: hepatosomatic index
= liver wet weight (g)/body weight (g). The body mass index was
calculated as follows: body mass index = body weight (kg)/body
length (m)2. The livers were cut into a number of
sections and stored at −80°C until tissue examination.
Biochemical assays
Serum fasting blood glucose (FBG), triglyceride
(TG), total cholesterol (TC), low-density lipoprotein (LDL),
high-density lipoprotein (HDL), alanine aminotransferase (ALT),
aspartate aminotransferase (AST), gamma-glutamyl transpeptidase
(GGT) and alkaline phosphatase (ALP) levels were measured using an
automatic biochemical analyzer. The serum insulin levels were
measured using an enzyme-linked immunosorbent assay (ELISA) kit
(EIA2048; DRG International, Inc., Springfield, NJ, USA) according
to the manufacturer's instructions. Insulin resistance was
calculated using the homeostasis model assessment of insulin
ressistance (HOMA-IR) as follows: fasting insulin (mIU/l) x fasting
blood glucose (mmol/l)/22.5, as previously described (26). The serum insulin-like growth
factor-1 (IGF-1) levels were measured using an ELISA kit (IB39431)
purchased from IBL (Minneapolis, MN, USA), according to the
manufacturer's instructions.
Histological analyses
Histological analyses were performed according to a
the methods described in a previous study (27). Briefly, the liver tissues were
fixed in 10% buffered formaldehyde and then embedded in paraffin.
The paraffin-embedded tissues were cut into 5-µm-thick
sections and were then stained with hematoxylin and eosin
(H&E). To visualize the accumulation of fat droplets, sections
of frozen liver tissue were stained with Oil Red O, and hematoxylin
was introduced to labelmark the cell nuclei.
Immunohistochemical staining
The 5-µm-thick paraffin-embedded sections of
liver tissue were deparaffinized by changing the xylene twice, and
this procedure was followed by rehydration in an ethanol gradient.
Following antigen retrieval and non-specific binding blocking, the
slides were incubated with primary antibodies diluted at 1:800 for
60 min at 37°C. After washing 3 times, the slides were then
incubated with HRP-conjugated rabbit anti-rat IgG (Cat. no. ab6734;
Abcam, Cambridge, UK) diluted at 1:500 for 60 min at 37°C. Color
development was performed using a DAB Horseradish Peroxidase Color
Development kit (Beyotime, Nantong, China). Hematoxylin was used
for nuclear compartment visualization. Rabbit anti-rat sterol
regulatory element binding protein-1c (SREBP-1c) and glucose
transporter type 2 (GLUT-2) antibodies were purchased from Santa
Cruz Biotechnology. The results were quantified using a color
density assay using iVision software (version 4.0.9; BioVision
Technologies, Exton, PA, USA).
RNA extraction and
quantitative-polymerase chain reaction (RT-qPCR)
Total RNA from the rat livers was extracted using
TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer's instructions. cDNA was synthesized from 5 µg
of the total RNA using a PrimeScript® RT reagent kit
(Takara, Dalian, China) with oligo(dT) primers as per the
manufacturer's instructions. The quantitative PCR (qPCR) reactions
were performed on a Bio-Rad iQ5 real-time thermal cycler using a
SYBR® Premix Ex Taq™ II kit (Takara) in a 25-µl
reaction system. All reactions were performed in triplicate, and
the results are represented as relative mRNA expression data, which
were calculated using the 2−ΔΔCT method, as previously
described (28). The sequences of
the primers used were as follows: SREBP-1c forward, 5′-CGC TAC CGT
TCC TCT ATC AAT GAC-3′ and reverse, 5′-AGT TTC TGG TTG CTG TGC TGT
AAG-3′; GLUT-2 forward, 5′-TCC GCT TGC TCC TCC TCC TAC-3′ and
reverse, 5′-ACA CCGA TGT CAT ATC CGA ACT GG-3′; diacylglycerol
acyltransferase (DGAT) forward, 5′-TCC GCC TCT GGG CAT TC-3′ and
reverse, 5′-GAA TCG GCC CAC AAT CCA-3′; CPT-1 forward, 5′-TAT GTG
AGG ATG CTG CTT CC-3′ and reverse, 5′-CTC GGA GAG CTA AGC TTG
TC-3′; ACOX1 forward, 5′-GTT GAT CAC GCA CAT CTT GGA-3′ and
reverse, 5′-TCG TTC AGA ATC AAG TTC TCA ATT TC-3′; ACOX3 forward,
5′-TGG AGA AGG AAC GAG AAC TGA AC-3′ and reverse, 5′-ACA TGT GGA
GGA CAC ATT TGT TG-3′; and β-actin forward, 5′-CAA CTT GAT GTA TGA
AGG CTT TGG T-3′ and reverse, 5′-ACT TTT ATT GGT CTC AAG TCA GTG
TAC AG-3′. All the primers were synthesized by BGI Tech (Shenzhen,
China).
Statistical analysis
Data are expressed as the means ± SEM from a minimum
of 3 experiments. Differences between groups were analyzed using
the Student's t-test. A value of P<0.05 was considered to
indicate a statistically significant difference.
Results
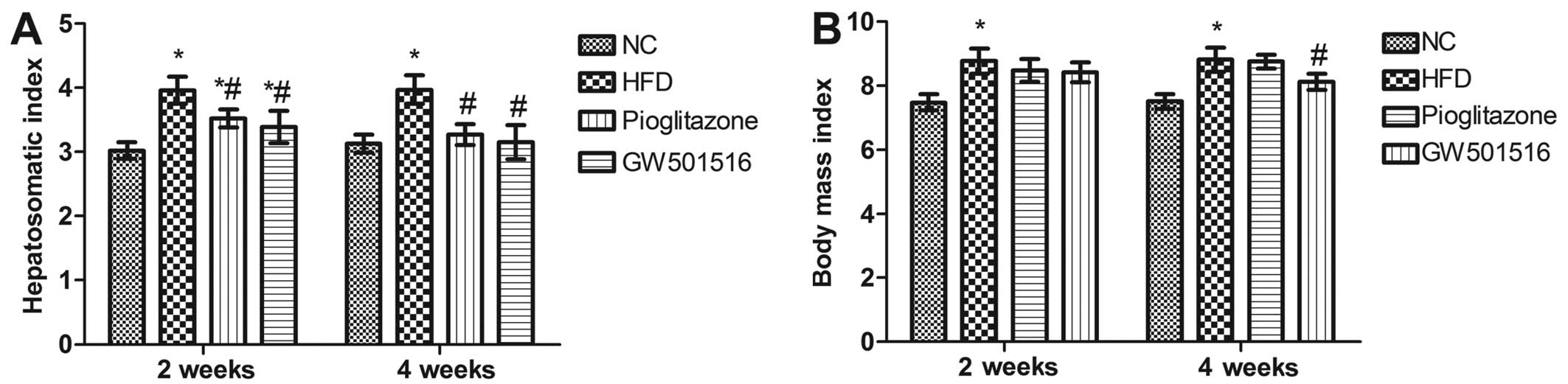
Hepatosomatic index and alterations in
body mass index
Pioglitazone, a prescription drug of the class TZD,
which is used in the treatment of diabetes, was introduced in the
present study as a positive control. After 12 weeks of being fed
the HFD, the hepatosomatic index and body mass index of the rats
were both significantly elevated (P<0.05) compared to the rats
in the normal control group (Fig.
1). However, 2 weeks of treatment with GW501516 or pioglitazone
significantly reduced the hepatosomatic index (P<0.05) compared
to the HFD group, although the hepatosomatic index was still
significantly higher than that of the normal control group
(P<0.05; Fig. 1A). After 4
weeks of treatment with GW501516 or pioglitazone, the hepatosomatic
index was almost reduced to the level of the normal control group
(Fig. 1A). Treatment with
pioglitazone did not reduce the body mass index (Fig. 1B). However, a significant decrease
in body mass index was observed in the rats that were treated with
GW501516 for 4 weeks (Fig.
1B).
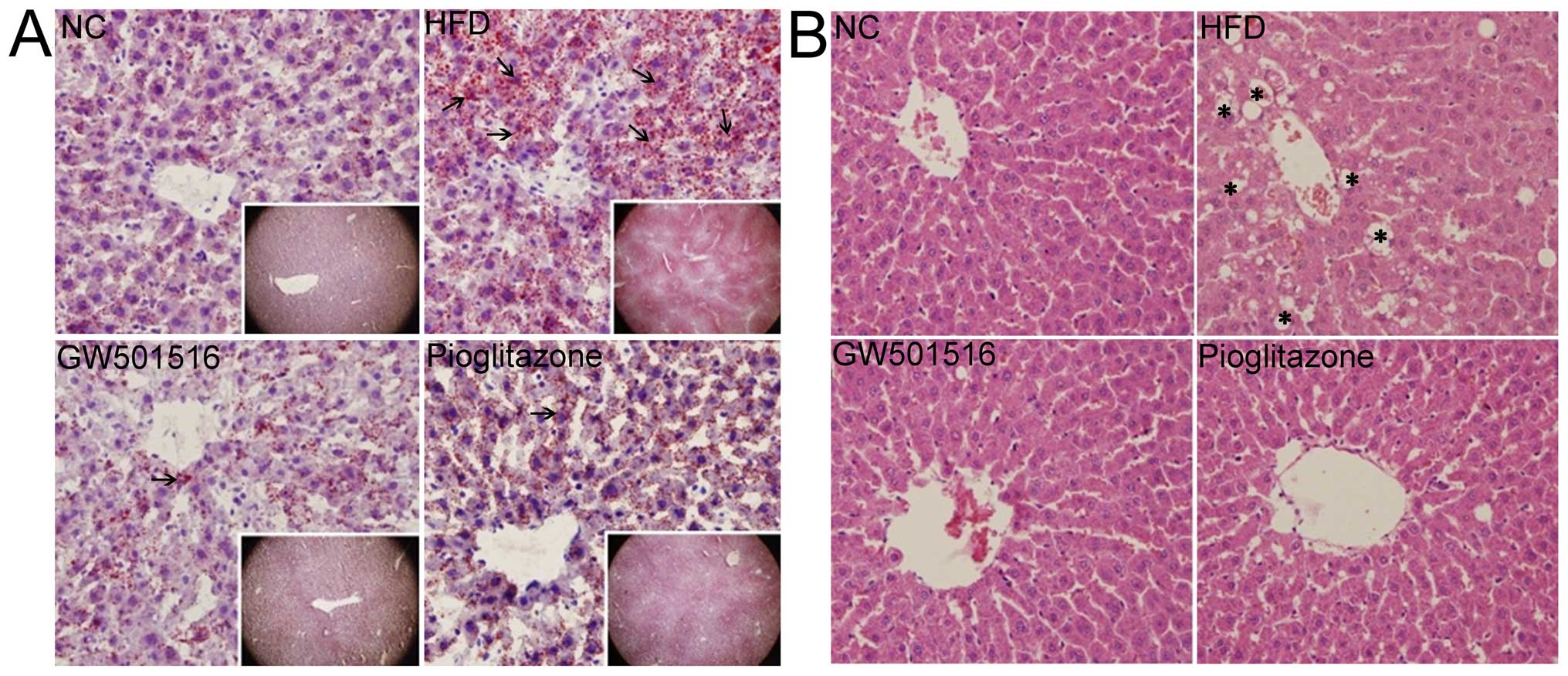
Alleviation of pathological changes in
the liver following treatment with GW501516
After 4 weeks of treatment with normal saline,
GW501516 or pioglitazone, the livers of the rats in all the groups
were collected and analyzed using Oil Red O (Fig. 2A) and H&E (Fig. 2B) staining. The histological
examination of the livers of the rats from the normal control group
exhibited a normal structure and architecture. No obvious lipid
droplets or lobular inflammatory cell infiltration were observed
(Fig. 2). However, typical
vesicular steatosis, hepatocellular ballooning, and lipid droplets
were observed in the HFD group (Fig.
2). After 4 weeks of treatment with GW501516 or pioglitazone,
these pathological changes were significantly alleviated. In
addition, it was clear that GW501516 had a more potent effect than
pioglitazone, as lipid droplets, hepatocellular ballooning and
inflammatory cell infiltration were scarcely observed in the group
treated with GW501516 (Fig.
2).
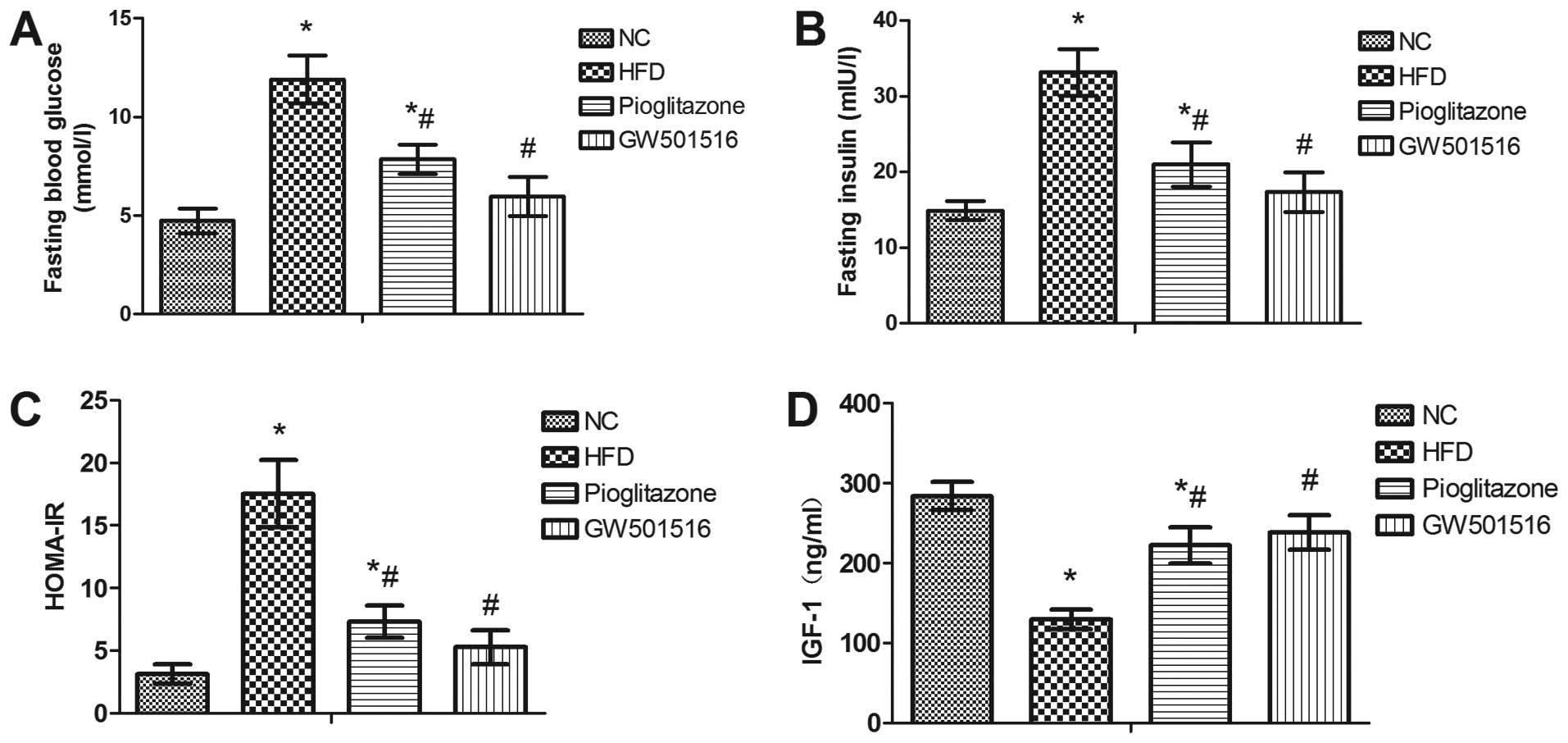
Effect of GW501516 on glucose
metabolism
In order to investigate whether treatment with
GW501516 affects glucose metabolism, serum was collected from the
rats at the end of each treatment for the determination of the
levels of fasting blood glucose, fasting insulin and IGF-1. The
HOMA-IR index was calculated based on the levels of fasting blood
glucose and fasting insulin. As expected, a significant increase in
the serum levels of fasting blood glucose and fasting insulin, as
well as in the HOMA-IR index was observed in the rats in the HFD
group compared to the rats of the normal control group. In
addition, a significant decrease in the levels of IGF-1 was
observed in the HFD group compared to the normal control group
(Fig. 3). However, 4 weeks of
treatment with pioglitazone significantly decreased the levels of
fasting blood glucose and fasting insulin, and the HOMA-IR index,
and increased the levels of IGF-1 compared to the HFD group
(Fig. 3). Nonetheless, these
levels and indexes still differed significantly from those of the
normal control group (Fig. 3).
However, 4 weeks of treatment with GW501516 almost restored these
indexes to normal levels (Fig.
3). This suggested that glucose metabolism is regulated by
GW501516.
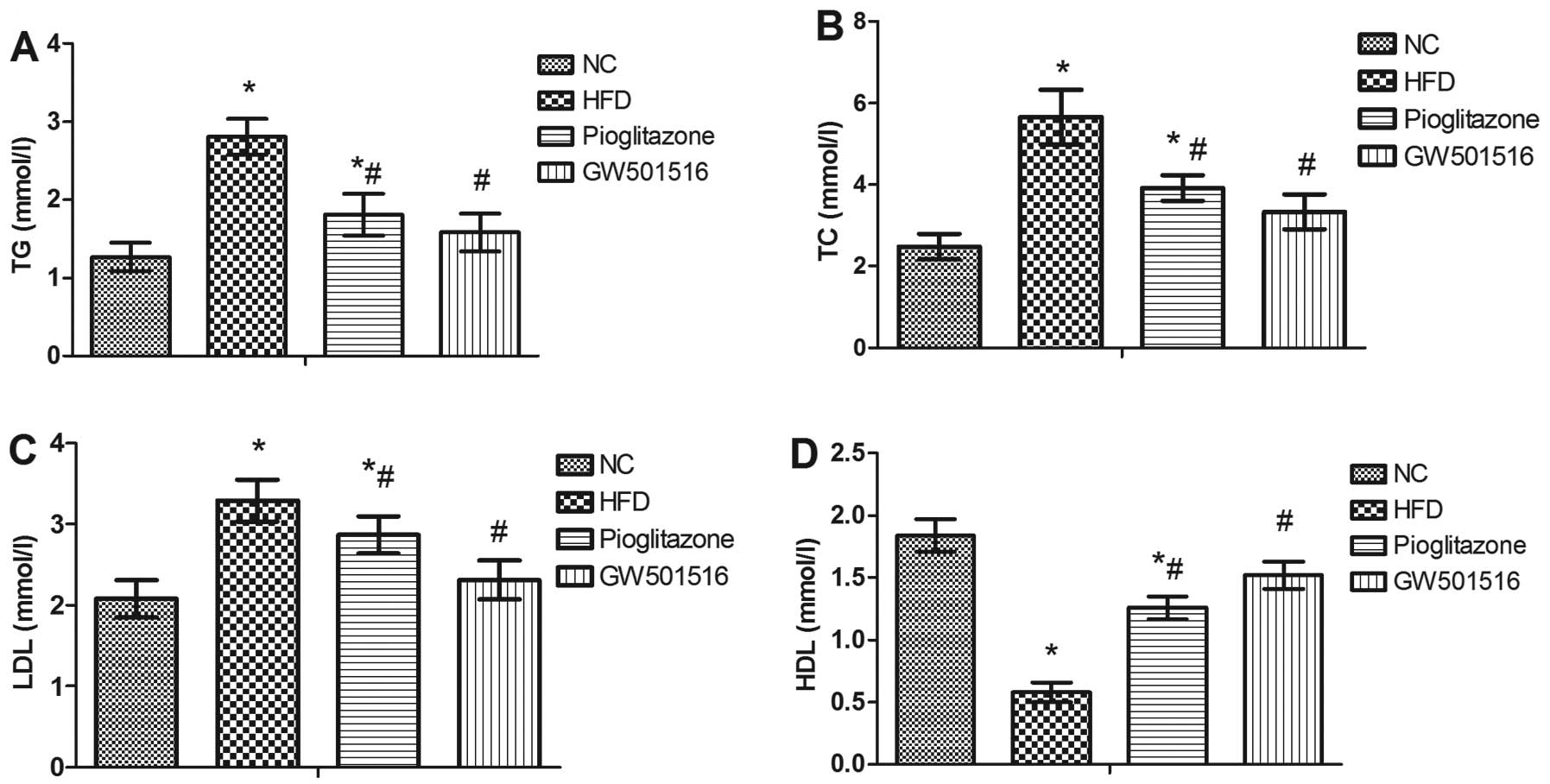
Effect of GW501516 on lipid
metabolism
To determine the effect of GW501516 on lipid
metabolism, the levels of TG, TC, HDL and LDL in the serum were
measured. In contrast to the normal control group, the levels of
TG, TC and LDL were all significantly increased, and the levels of
HDL were significantly decreased in the HFD group (Fig. 4). However, 4 weeks of treatment
with pioglitazone significantly decreased the levels of TG, TC and
LDL, and increased the levels of HDL in comparison to the HFD group
(Fig. 4). Treatment with GW501516
almost restored the TG, TC, LDL and HDL levels to normal levels
(Fig. 4). This suggested that
lipid metabolism may be regulated by GW501516.
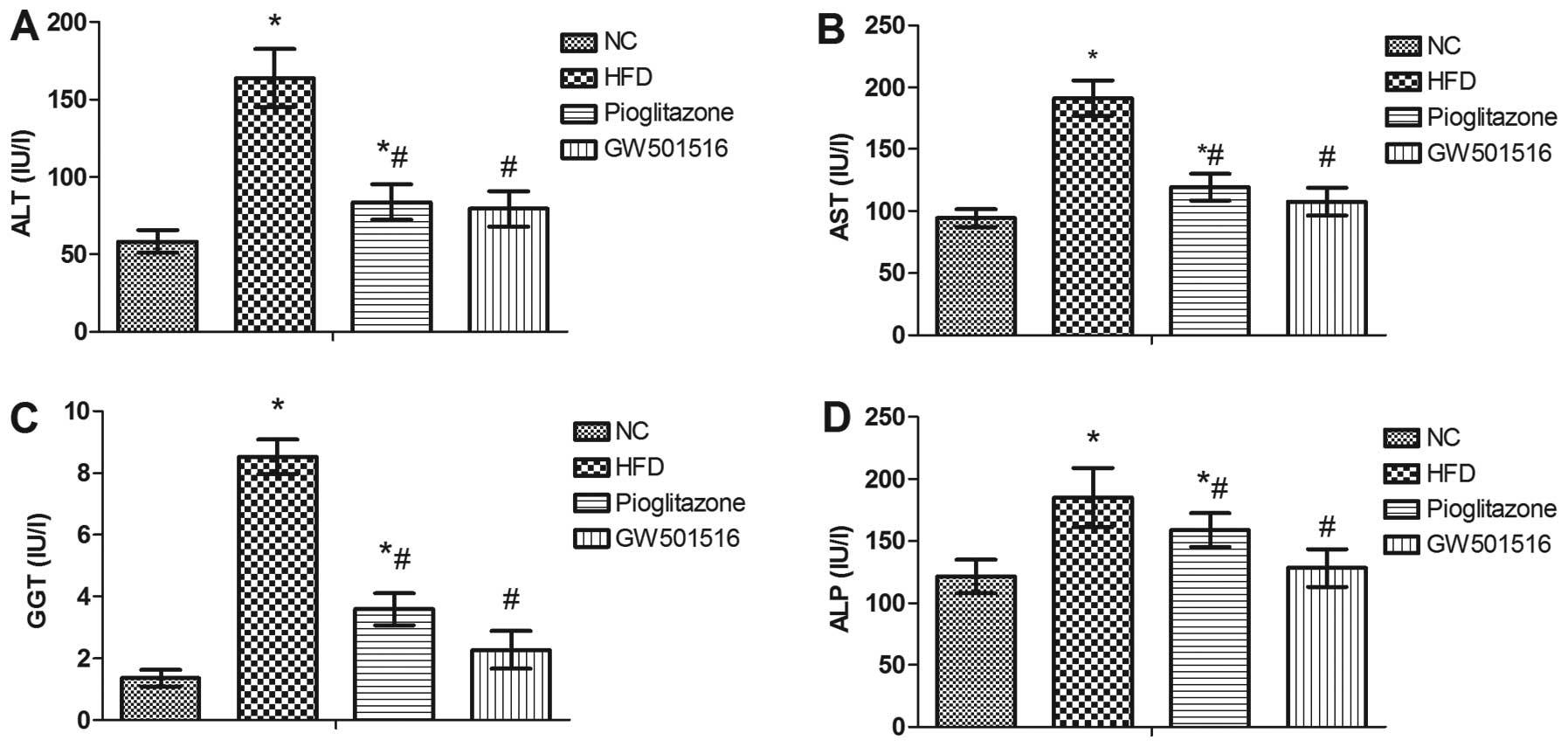
Effect of GW501516 on the levels of liver
enzymes
To evaluate liver function, the serum levels of ALT,
AST, GGT and ALP were determined. As shown in Fig. 5, the levels of ALT, AST, GGT and
ALP were all significantly elevated in the HFD group compared to
those of the normal control group, and these were all markedly
reduced after 4 weeks of treatment with GW501516 compared to the
HFD group (Fig. 5). These data
indicate that GW501516 effectively alleviates NAFLD and that it is
more effective than pioglitazone, since treatment with pioglitazone
did not reduce the ALT, AST, GGT or ALP levels to the same
extent.
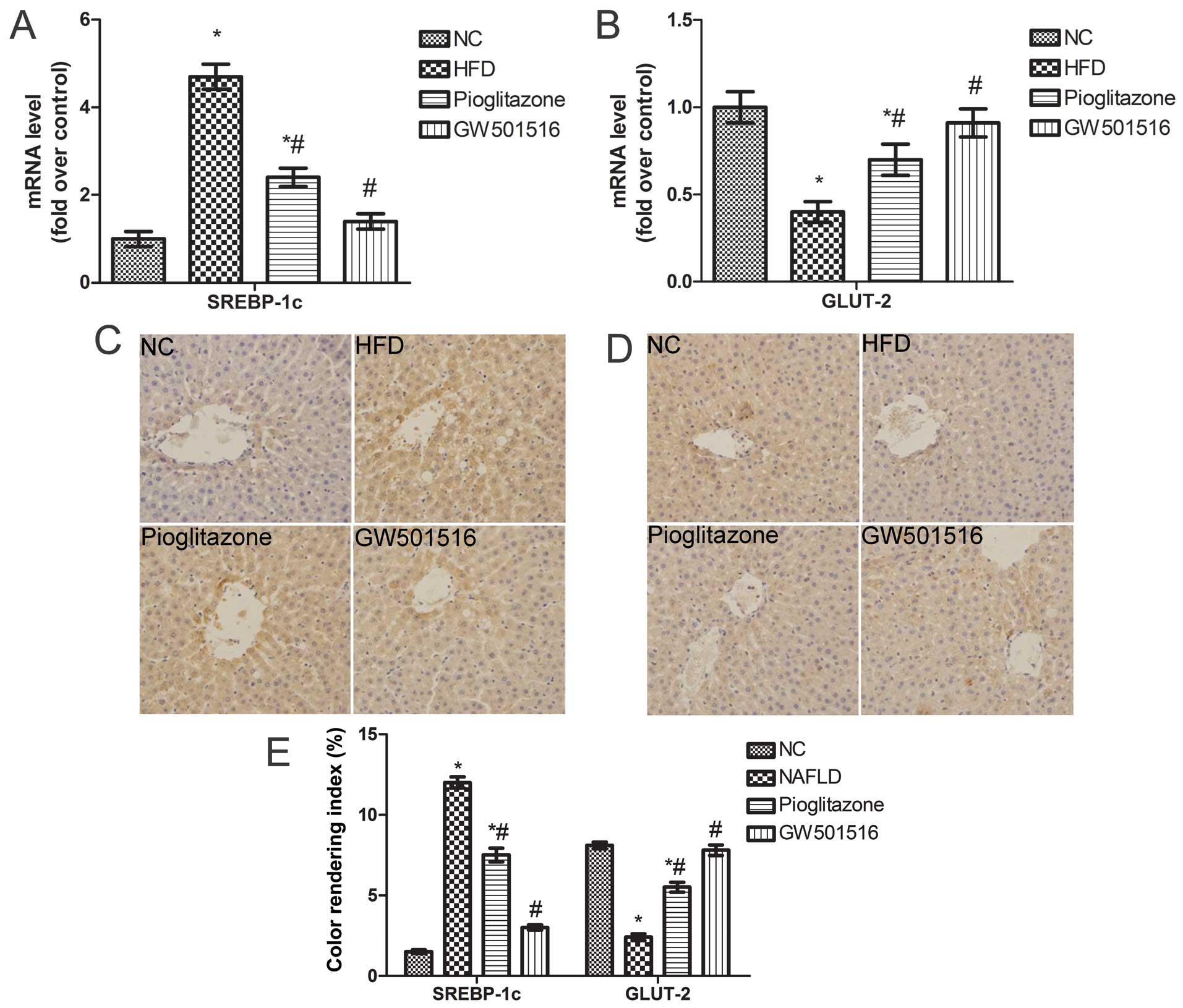
Expression of SREBP-1c and GLUT-2 is
restored following treatment with GW501516
The above-mentioned findings indicate that treatment
with GW501516 regulates glucose and lipid metabolism. To elucidate
the underlying mechanisms responsible for the effects of GW501516
on glucose and lipid metabolism, the expression levels of SREBP-1c,
a key lipogenic transcription factor involved in lipogenesis
(29,30), and GLUT-2 were determined. As
expected, the mRNA levels of SREBP-1c were significantly
upregulated and those of GLUT-2 were significantly downregulated in
the livers of the rats in the HFD group compared to the normal
control group (Fig. 6A and B).
However, these levels were both restored to almost normal levels
after 4weeks of treatment with GW501516 (Fig. 6A and B). A similar trend was
observed in relation to the protein expression of SREBP-1c and
GLUT-2, as was determined by immunohistochemical staining (Fig. 6C and D). These data suggest that
GW501516 modulates glucose and lipid metabolism by regulating
SREBP-1c and GLUT-2 expression.
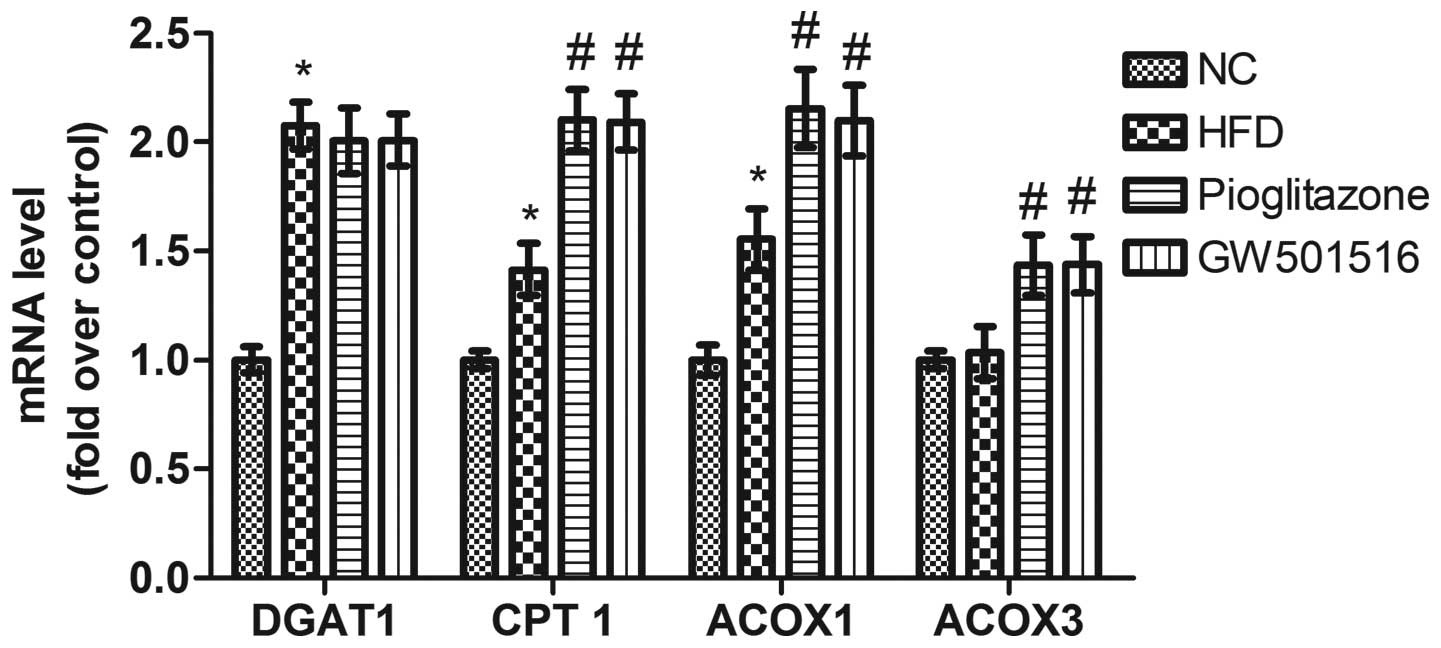
Expression of genes related to fatty acid
synthesis is regulated by GW501516
Previous studies have demonstrated that the
expression of genes related to lipid metabolism is modulated by
PPARs (31–35), and it has been shown that the
expression of SREBP-1c, a key lipogenic transcription factor, is
regulated by GW501516 (36). As
DGAT, CPT-1, ACOX1 and ACOX3 are important rate-limiting enzymes
which are related to lipid metabolism, we determined the
transcriptional levels of these genes in order to evaluate the
effects of GW501516 on lipid metabolism. As shown in Fig. 7, the transcriptional levels of
DGAT1, CPT-l and ACOX1 were all significantly upregulated; however,
those of ACOX3 were only slightly altered by the HFD. Four weeks of
treatment with GW501516 did not greatly alter the transcription
levels of DGAT1, in comparison to those of the HFD group. This
suggests that triglyceride synthesis is not significantly affected
by GW501516, as DGAT1 is the rate-limiting enzyme for the synthesis
of TGs from diacylg-lycerols (37). However, the transcriptional levels
of CPT-l, ACOX1 and ACOX3 were significantly upregulated following
tretment with GW501516 compared to the levels of the HFD group
(Fig. 7). These data suggest that
the β-oxidation of fatty acids in the mitochondria and peroxisomes
is improved by treatment with GW501516, as CPT-l is the
rate-limiting enzyme for β-oxidation in the mitochondria (38), and ACOX1/ACOX3 are the
rate-limiting enzymes for β-oxidation in peroxisomes (39).
Discussion
NAFLD is an increasingly common condition,
characterized by the increased accumulation of lipids in the liver,
which may lead to the development of progressive hepatic fibrosis,
cirrhosis and hepatic failure. The increase in fat deposits in the
liver can be caused by an increase in the uptake of free fatty
acids into the liver, impaired fatty acid β-oxidation, or the
increased incidence of de novo lipogenesis (40). NAFLD is closely related to
obesity, hyperlipidemia and insulin resistance, as well as other
other metabolic disorders in the liver (1,2). A
recent study emphasized the important role that PPARs play in
certain metabolic disorders and NAFLD (41). PPARs, which are known to be
important regulators of energy metabolism and homeostasis, play a
role in modulating the accumulation of TGs in the liver, which is a
hallmark of the development of NAFLD (32,41,42).
GW501516, as a potent agonist of PPARδ, regulates
glycometabolism, fatty acid metabolism and insulin resistance
(22–24). GW501516 has been shown to enhance
the β-oxidation of liver fatty acids by increasing the expression
of PPARα-target genes involved in fatty acid oxidation (23). Several studies have highlighted
that insulin resistance is a main characteristic of NAFLD (1,7,8).
In skeletal muscle cells, treatment with GW501516 has been shown to
prevent fatty acid-induced inflammation and insulin resistance in
by preventing the phosphorylation of insulin receptor substrate-1
at Ser307 and the inhibition of insulin-stimulated Akt
phosphorylation, which is caused by exposure to the saturated fatty
acid, palmitate (22). The
expression of two well-known PPARδ target genes involved in fatty
acid oxidation, CPT-1 and pyruvate dehydrogenase kinase 4, has also
been shown to be elevated following treatment with GW501516
(22). In human liver cells,
treatment with GW501516 has been shown to increase the adenosine
monophosphate (AMP)/adenosine triphosphate (ATP) ratio and decrease
the ATP/adenosine diphosphate (ADP) ratio, thus contributing to the
prevention of insulin resistance (24). GW501516 can also prevent
endoplasmic reticulum stress-associated inflammation and insulin
resistance in skeletal muscle cells by activating AMPK (43,44). The Akt and ERK signaling pathways
have also been shown to be involved in the effects of GW501516
(45). In a previous study, in
INS-1E cells treated with GW501516, the expression of v-maf avian
musculoaponeurotic fibrosarcoma oncogene homolog A (MafA), GLUT-2,
and the upregulation of insulin, indicated that the c-Jun
N-terminal kinase (JNK)-MafA-GLUT-2 pathway was involved in the
enhanced effects of PPARδ on insulin secretion (46). GW501516 also possesses the ability
to reduce the proliferative potential of hepatoma cells (47). All these studies suggested that
GW501516 alleviates NAFLD, as GW501516 is able to increase fatty
acid oxidation, reduce insulin resistance, prevent endoplasmic
reticulum stress and hepatocyte proliferation and induce insulin
secretion. However, GW501516 cannot diminish the progression of
liver fibrosis. The induction of hepatic stellate cell (HSC)
apoptosis is a potent strategy used to diminish the progression of
liver fibrosis. However, GW501516-activated PPARβ/δ promotes liver
fibrosis through the p38-JNK MAPK-induced HSC proliferation
(48), and decreases the soluble
egg antigen (SEA)-induced HSC apoptosis (49). However, KD3010, another agonist of
PPARδ, has been shown to have hepatoprotective and antifibrotic
effects in hepatocytes and in a model of cholestasis-induced liver
injury and fibrosis (50).
Therefore, the effects of GW501516 on NAFLD are still
debatable.
In the present study, we demonstrated that treatment
with GW501516 significantly alleviated the pathological changes in
the livers of rats in our model of HFD-induced NAFLD. GW501516
significantly decreased the HOMA-IR index, and increased the levels
of IGF-1 in the rats with HFD-induced NAFLD, indicating that
insulin resistance was reduced by GW501516. This finding was
consistent with that of previous studies (22,24,43). We also demonstrated that the
protective effects of GW501516 were more potent than those of
pioglitazone. The decreased expression of GLUT-2 in the livers of
the rats in our NAFLD model was significantly increased following
treatment with GW501516, almost to normal levels (Fig. 6B). Cao et al (46) demonstrated that GW501516
upregulated the expression of GLUT-2 through the JNK-MafA-GLUT-2
pathway. These results suggest that glucose metabolism is modulated
by GW501516. In our study, the expression of SREBP-1c, a key
lipogenic transcription factor involved in lipogenesis, was also
reduced to normal levels following treatment with GW501516
(Fig. 6A). We also demonstrated
that the mRNA levels of DGAT1 were not significantly altered, but
those of CPT-1, ACOX1 and ACOX3 were all significantly upregulated
following treatment with GW501516 (Fig. 7). DGAT1 catalyzes the formation of
TGs from diacylglycerol and Acyl-CoA (37). This is considered the terminal and
only committed step in TG synthesis. CPT-1 is the rate-limiting
enzyme involved in the β-oxidation of long-chain fatty acids
(38). ACOX1 catalyzes the first,
rate-limiting step in peroxisome β-oxidation, of medium to very
long straight-chain fatty acids, and ACOX3 is involved in the
desaturation of branched fatty acids in peroxisomes (39). Our observations indicated that TG
synthesis was not significantly affected by GW501516 treatment, but
that the β-oxidation of fatty acids was significantly enhanced.
This observation was in line with that of a previous study, in
which fatty acid oxidation was increased by the activation of PPARδ
in human myotubes (51).
Although we demonstrated that treatment with
GW501516 alleviated NAFLD by modulating glucose and lipid
metabolism, the contribution of the activation of PPARδ to this
process remains to be clarified, as PPARα and PPARγ may also be
activated by GW501516. However, it has been suggested that PPARδ
plays a major role in the effects of GW501516 on NAFLD since
GW501516 displays a high affinity (Ki = 1 nM) and potency
(EC50 = 1 nM) for PPARδ that is 1,000-fold greater than
that for PPARα and PPARγ (52).
In conclusion, in the present study, we demonstrated
that GW501516 prevented the progression of NAFLD in our rat model
of HFD-induced NAFLD. GW501516 improved glucose and fatty acid
metabolism by modulating the levels of glucose and fatty acid
metabolic enzymes. Our results thus suggest that treatment with
GW501516 may prove to be a potential therapeutic strategy for
NAFLD.
Acknowledgments
The present study was supported by a grant from the
National Natural Science Foundation of China (no. 81070328).
References
|
1
|
Sanyal AJ, Campbell-Sargent C, Mirshahi F,
Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML and Clore
JN: Nonalcoholic steatohepatitis: association of insulin resistance
and mitochondrial abnormalities. Gastroenterology. 120:1183–1192.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sanyal AJ: American Gastroenterological
Association: AGA technical review on nonalcoholic fatty liver
disease. Gastroenterology. 123:1705–1725. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Day CP: Pathogenesis of steatohepatitis.
Best Pract Res Clin Gastroenterol. 16:663–678. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Browning JD, Szczepaniak LS, Dobbins R,
Nuremberg P, Horton JD, Cohen JC, Grundy SM and Hobbs HH:
Prevalence of hepatic steatosis in an urban population in the
United States: impact of ethnicity. Hepatology. 40:1387–1395. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gaggini M, Morelli M, Buzzigoli E,
DeFronzo RA, Bugianesi E and Gastaldelli A: Non-alcoholic fatty
liver disease (NAFLD) and its connection with insulin resistance,
dyslipidemia, atherosclerosis and coronary heart disease.
Nutrients. 5:1544–1560. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Williams CD, Stengel J, Asike MI, Torres
DM, Shaw J, Contreras M, Landt CL and Harrison SA: Prevalence of
nonalcoholic fatty liver disease and nonalcoholic steatohepatitis
among a largely middle-aged population utilizing ultrasound and
liver biopsy: a prospective study. Gastroenterology. 140:124–131.
2011. View Article : Google Scholar
|
|
7
|
Yki-Jarvinen H: Liver fat in the
pathogenesis of insulin resistance and type 2 diabetes. Dig Dis.
28:203–209. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fabbrini E, Magkos F, Mohammed BS, Pietka
T, Abumrad NA, Patterson BW, Okunade A and Klein S: Intrahepatic
fat, not visceral fat, is linked with metabolic complications of
obesity. Proc Natl Acad Sci USA. 106:15430–15435. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yki-Jarvinen H: Fat in the liver and
insulin resistance. Ann Med. 37:347–356. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Desvergne B and Wahli W: Peroxisome
proliferator-activated receptors: nuclear control of metabolism.
Endocr Rev. 20:649–688. 1999.PubMed/NCBI
|
|
11
|
Aoyama T, Peters JM, Iritani N, Nakajima
T, Furihata K, Hashimoto T and Gonzalez FJ: Altered constitutive
expression of fatty acid-metabolizing enzymes in mice lacking the
peroxisome proliferator-activated receptor alpha (PPARalpha). J
Biol Chem. 273:5678–5684. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fan CY, Pan J, Chu R, Lee D, Kluckman KD,
Usuda N, Singh I, Yeldandi AV, Rao MS, Maeda N and Reddy JK:
Hepatocellular and hepatic peroxisomal alterations in mice with a
disrupted peroxisomal fatty acyl-coenzyme A oxidase gene. J Biol
Chem. 271:24698–24710. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Leone TC, Weinheimer CJ and Kelly DP: A
critical role for the peroxisome proliferator-activated receptor
alpha (PPARalpha) in the cellular fasting response: the
PPARalpha-null mouse as a model of fatty acid oxidation disorders.
Proc Natl Acad Sci USA. 96:7473–7478. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tontonoz P, Hu E and Spiegelman BM:
Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a
lipid-activated transcription factor. Cell. 79:1147–1156. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Larsen TM, Toubro S and Astrup A:
PPARgamma agonists in the treatment of type II diabetes: is
increased fatness commensurate with long-term efficacy? Int J Obes
Relat Metab Disord. 27:147–161. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Seo YS, Kim JH, Jo NY, Choi KM, Baik SH,
Park JJ, Kim JS, Byun KS, Bak YT, Lee CH, et al: PPAR agonists
treatment is effective in a nonalcoholic fatty liver disease animal
model by modulating fatty-acid metabolic enzymes. J Gastroenterol
Hepatol. 23:102–109. 2008.PubMed/NCBI
|
|
17
|
Gillies PS and Dunn CJ: Pioglitazone.
Drugs. 60:333–345. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Smith U: Pioglitazone: mechanism of
action. Int J Clin Pract. (Suppl): 13–18. 2001.
|
|
19
|
Vamecq J and Latruffe N: Medical
significance of peroxisome proliferator-activated receptors.
Lancet. 354:141–148. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Neuschwander-Tetri BA, Brunt EM, Wehmeier
KR, Oliver D and Bacon BR: Improved nonalcoholic steatohepatitis
after 48 weeks of treatment with the PPAR-gamma ligand
rosiglitazone. Hepatology. 38:1008–1017. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oger F, Dubois-Chevalier J, Gheeraert C,
Avner S, Durand E, Froguel P, Salbert G, Staels B, Lefebvre P and
Eeckhoute J: Peroxisome proliferator-activated receptor gamma
regulates genes involved in insulin/insulin-like growth factor
signaling and lipid metabolism during adipogenesis through
functionally distinct enhancer classes. J Biol Chem. 289:708–722.
2014. View Article : Google Scholar :
|
|
22
|
Coll T, Alvarez-Guardia D, Barroso E,
Gómez-Foix AM, Palomer X, Laguna JC and Vázquez-Carrera M:
Activation of peroxisome proliferator-activated receptor-{delta} by
GW501516 prevents fatty acid-induced nuclear factor-{kappa}B
activation and insulin resistance in skeletal muscle cells.
Endocrinology. 151:1560–1569. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barroso E, Rodriguez-Calvo R,
Serrano-Marco L, Astudillo AM, Balsinde J, Palomer X and
Vázquez-Carrera M: The PPARbeta/delta activator GW501516 prevents
the down-regulation of AMPK caused by a high-fat diet in liver and
amplifies the PGC-1alpha-Lipin 1-PPARalpha pathway leading to
increased fatty acid oxidation. Endocrinology. 152:1848–1859. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Serrano-Marco L, Barroso E, El Kochairi I,
Palomer X, Michalik L, Wahli W and Vázquez-Carrera M: The
peroxi-some proliferator-activated receptor (PPAR) beta/delta
agonist GW501516 inhibits IL-6-induced signal transducer and
activator of transcription 3 (STAT3) activation and insulin
resistance in human liver cells. Diabetologia. 55:743–751. 2012.
View Article : Google Scholar
|
|
25
|
Nagasawa T, Inada Y, Nakano S, Tamura T,
Takahashi T, Maruyama K, Yamazaki Y, Kuroda J and Shibata N:
Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARdelta
agonist, on development of steatohepatitis in mice fed a
methionine- and choline-deficient diet. Eur J Pharmacol.
536:182–191. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Matthews DR, Hosker JP, Rudenski AS,
Naylor BA, Treacher DF and Turner RC: Homeostasis model assessment:
insulin resistance and beta-cell function from fasting plasma
glucose and insulin concentrations in man. Diabetologia.
28:412–419. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang HN, Wang YR, Liu GQ, Liu Z, Wu PX,
Wei XL and Hong TP: Inhibition of hepatic interleukin-18 production
by rosiglitazone in a rat model of nonalcoholic fatty liver
disease. World J Gastroenterol. 14:7240–7246. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
29
|
Ferre P and Foufelle F: Hepatic steatosis:
a role for de novo lipo-genesis and the transcription factor
SREBP-1c. Diabetes Obes Metab. 12(Suppl 2): S83–S92. 2010.
View Article : Google Scholar
|
|
30
|
Shimano H, Yahagi N, Amemiya-Kudo M, Hasty
AH, Osuga J, Tamura Y, Shionoiri F, Iizuka Y, Ohashi K, Harada K,
et al: Sterol regulatory element-binding protein-1 as a key
transcription factor for nutritional induction of lipogenic enzyme
genes. J Biol Chem. 274:35832–35839. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Keller H, Dreyer C, Medin J, Mahfoudi A,
Ozato K and Wahli W: Fatty acids and retinoids control lipid
metabolism through activation of peroxisome proliferator-activated
receptor-retinoid X receptor heterodimers. Proc Natl Acad Sci USA.
90:2160–2164. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Keller H, Mahfoudi A, Dreyer C, Hihi AK,
Medin J, Ozato K and Wahli W: Peroxisome proliferator-activated
receptors and lipid metabolism. Ann NY Acad Sci. 684:157–173. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Keller H and Wahli W: Peroxisome
proliferator-activated receptors A link between endocrinology and
nutrition? Trends Endocrinol Metab. 4:291–296. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kliewer SA, Sundseth SS, Jones SA, Brown
PJ, Wisely GB, Koble CS, Devchand P, Wahli W, Willson TM, Lenhard
JM and Lehmann JM: Fatty acids and eicosanoids regulate gene
expression through direct interactions with peroxisome
prolif-erator-activated receptors alpha and gamma. Proc Natl Acad
Sci USA. 94:4318–4323. 1997. View Article : Google Scholar
|
|
35
|
Forman BM, Chen J and Evans RM:
Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids
are ligands for peroxisome proliferator-activated receptors alpha
and delta. Proc Natl Acad Sci USA. 94:4312–4317. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lee CH, Olson P, Hevener A, Mehl I, Chong
LW, Olefsky JM, Gonzalez FJ, Ham J, Kang H, Peters JM and Evans RM:
PPARdelta regulates glucose metabolism and insulin sensitivity.
Proc Natl Acad Sci USA. 103:3444–3449. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cases S, Smith SJ, Zheng YW, Myers HM,
Lear SR, Sande E, Novak S, Collins C, Welch CB, Lusis AJ, et al:
Identification of a gene encoding an acyl CoA: diacylglycerol
acyltransferase, a key enzyme in triacylglycerol synthesis. Proc
Natl Acad Sci USA. 95:13018–13023. 1998. View Article : Google Scholar
|
|
38
|
Eaton S: Control of mitochondrial
beta-oxidation flux. Prog Lipid Res. 41:197–239. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Poirier Y, Antonenkov VD, Glumoff T and
Hiltunen JK: Peroxisomal beta-oxidation - a metabolic pathway with
multiple functions. Biochim Biophys Acta. 1763:1413–1426. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Koo SH: Nonalcoholic fatty liver disease:
molecular mechanisms for the hepatic steatosis. Clin Mol Hepatol.
19:210–215. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tailleux A, Wouters K and Staels B: Roles
of PPARs in NAFLD: potential therapeutic targets. Biochim Biophys
Acta. 1821:809–818. 2012. View Article : Google Scholar
|
|
42
|
Wahli W and Michalik L: PPARs at the
crossroads of lipid signaling and inflammation. Trends Endocrinol
Metab. 23:351–363. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Salvado L, Barroso E, Gomez-Foix AM,
Palomer X, Michalik L, Wahli W and Vázquez-Carrera M:
PPARbeta/delta prevents endoplasmic reticulum stress-associated
inflammation and insulin resistance in skeletal muscle cells
through an AMPK-dependent mechanism. Diabetologia. 57:2126–2135.
2014. View Article : Google Scholar
|
|
44
|
Palomer X, Capdevila-Busquets E, Botteri
G, Salvadó L, Barroso E, Davidson MM, Michalik L, Wahli W and
Vázquez-Carrera M: PPARbeta/delta attenuates palmitate-induced
endoplasmic reticulum stress and induces autophagic markers in
human cardiac cells. Int J Cardiol. 174:110–118. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ding H, Zhang Y, Liu L, Yuan H, Qu J and
Shen R: Activation of peroxisome proliferator activator receptor
delta in mouse impacts lipid composition and placental development
at early stage of gestation. Biol Reprod. 91:572014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cao M, Long Y, Tong Y, Wan J and Tong N:
Activation of PPARdelta up-regulates the expression of insulin gene
transcription factor MafA and ameliorates glucose-induced insulin
secretion impaired by palmitate. Mol Cell Biochem. 366:183–189.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Vacca M, D'Amore S, Graziano G, D'Orazio
A, Cariello M, Massafra V, Salvatore L, Martelli N, Murzilli S, Lo
Sasso G, et al: Clustering nuclear receptors in liver regeneration
identifies candidate modulators of hepatocyte proliferation and
hepatocarcinoma. PLoS One. 9:e1044492014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kostadinova R, Montagner A, Gouranton E,
Fleury S, Guillou H, Dombrowicz D, Desreumaux P and Wahli W:
GW501516-activated PPARbeta/delta promotes liver fibrosis via
p38-JNK MAPK-induced hepatic stellate cell proliferation. Cell
Biosci. 2:342012. View Article : Google Scholar
|
|
49
|
Wang J, Xu F, Zhu D, Duan Y, Chen J, Sun
X, He X, Li P, Sun W and Feng J: Schistosoma japonicum soluble egg
antigens facilitate hepatic stellate cell apoptosis by
downregulating Akt expression and upregulating p53 and DR5
expression. PLoS Negl Trop Dis. 8:e31062014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Iwaisako K, Haimerl M, Paik YH, Taura K,
Kodama Y, Sirlin C, Yu E, Yu RT, Downes M, Evans RM, et al:
Protection from liver fibrosis by a peroxisome
proliferator-activated receptor delta agonist. Proc Natl Acad Sci
USA. 109:E1369–E1376. 2012. View Article : Google Scholar
|
|
51
|
Feng YZ, Nikolic N, Bakke SS, Boekschoten
MV, Kersten S, Kase ET, Rustan AC and Thoresen GH: PPARdelta
activation in human myotubes increases mitochondrial fatty acid
oxidative capacity and reduces glucose utilization by a switch in
substrate preference. Arch Physiol Biochem. 120:12–21. 2014.
View Article : Google Scholar
|
|
52
|
Oliver WR Jr, Shenk JL, Snaith MR, Russell
CS, Plunket KD, Bodkin NL, Lewis MC, Winegar DA, Sznaidman ML,
Lambert MH, et al: A selective peroxisome proliferator-activated
receptor delta agonist promotes reverse cholesterol transport. Proc
Natl Acad Sci USA. 98:5306–5311. 2001. View Article : Google Scholar : PubMed/NCBI
|