Introduction
Epilepsy is one of the most common diseases of the
central nervous system. In certain female patients, seizures may
cluster in association with the menstrual cycle, and this is known
as catamenial epilepsy (1). The
association between seizures and fluctuations of the sex hormone
levels during the ovarian cycles in certain women with epilepsy was
suggested as early as the 19th century (2). Since then, clinicians have
investigated this observation further and identified that it may be
attributed to the neuroactive properties of steroid hormones and
their cyclic variation in serum levels. However, the association
between estrogens and epileptic seizures remains unclear (3).
Voltage-gated potassium (Kv) channels are important
physiological regulators of membrane potentials, action potential
shape, firing adaptation and neuronal excitability in excitable
tissues (4). The voltage-gated
potassium (Kv) currents are divided into two sections: The
transient current and the sustained current (5,6).
Previous studies have demonstrated that the transient outward
potassium current, also known as the A-type potassium current
(IA), has an important role in controlling the membrane
excitability and it contributes to remodel neuronal excitation
under pathological conditions (7,8).
In the Kv channel family, Kv4 is the major subtype mediating the
A-type current (IA). IA is encoded by
homomultimeric or heteromultimeric complexes of the Kv channel
subunits within identical subfamilies (9,10).
In the hippocampus, dendrites of CA1 pyramidal neurons contain a
high density of transient A-type potassium channels, and this
dampening effect reduces the ability of dendrites to initiate
action potentials, decreases the amplitude of back-propagating
action potentials, and reduces the magnitude of EPSPs (11). Taken together, these reveal that
the A-type potassium current has an inhibitory modulation effect on
neuronal activities in the hippocampus, which suggest that
inhibition of Kv channels would increase the cell excitability.
Indeed, in pilocarpine seizure rat model, by using
electrophysiological recording, the function of A-type potassium
channels were found to be decreased, which most likely accounts for
the increased dendritic excitability and the epileptic seizure
generation (12).
In one of our previous studies, we reported that
phenol red, a weak estrogen receptor agonist, as well as
17-β-estradiol, by activation of estrogen receptors, exhibited a
dose-dependent U-shape-like effect on the spontaneous epileptiform
bursting activities in cultured hippocampal neurons (13); however, the underlying mechanism
remains unknown. Hoffman et al (14) also reported that modulation of
voltage-gated potassium channels by estrogen reduced seizure and
induced the spread and degree of neuronal loss in the kainate acid
(KA) epilepsy model, which was modulated by progesterone (15). Previous studies have shown that
estrogen receptor activation affects the function of various
potassium channels in the brain. 17-β-estradiol has been reported
to inhibit the activity of the outward potassium currents in the
rat parabrachial nucleus (16)
and modulate the small conductance calcium-activated SK3 potassium
channels in the hypothalamus of the female guinea pig (17). In GT1-7 cells, estrogen has been
reported to enhance the Kv4.2 mRNA expression level and transient
outward current (18). Thus,
activation of estrogen receptors is likely to affect the neuronal
excitability and in turn reduces the occurrence of epilepsy, and
potassium channels may be involved in this process. However, the
mechanism of how estrogen receptor activation modulates the
transient outward potassium channel and affects the epileptiform
activities is not fully understood.
Therefore, we hypothesized that estrogen modulation
of epileptiform activity may occur, at least in part, via affecting
the transient outward potassium current. In the present study, the
effect of estrogen on the excitability and outward potassium
currents of cultured hippocampal neurons was investigated. The
results showed that 17-β-estradiol has a suppressive effect on the
epileptiform bursting activities in cultured hippocampal neurons
and an enhancing effect on the transient outward potassium currents
with the similar dose-related U-shape manner. Inhibition of the
transient outward potassium currents reversed the low dose
17-β-estradiol-evoked inhibition on epileptiform bursting
activities, indicating that the transient outward potassium
currents mediated the anti-seizure effect of low dose
17-β-estradiol.
Materials and methods
Ethics statement
All the animal experiments were approved by the
Local Committees of the Use of the Laboratory Animals, Fudan
University (Shanghai, China) and were carried out in accordance
with the National Natural Science Foundation of China animal
research regulation.
Primary hippocampal neuronal culture
Primary hippocampal neurons were prepared from
embryonic day 18 Sprague-Dawley rats, as previously reported
(19). Briefly, the pregnant rat
was anaesthetized with 10% chloral hydrate (intraperitoneal
injection), and the pups were dissected out for tissue preparation.
All the animals were subsequently euthanized with an overdose of
chloral hydrate. Following the dissection of the hippocampus, the
tissue was rinsed in cold Hanks' balanced salt solution and
subsequently digested with 0.05% trypsin-ethylenediaminetetraacetic
acid for ~15 min at 37°C, followed by trituration with pipettes in
the plating media (Dulbecco's modified Eagle's medium with 10%
fetal bovine serum, 10% F12 and 25 µg/ml
penicillin/streptomycin). Subsequent to rinsing twice, cells were
counted and plated onto glass coverslips (12-mm round; Carolina
Biological Supply Co., Burlington, NC, USA) or 35-mm petri dishes
with a 20-mm glass bottom well (Shengyou Biotechnology Co., Ltd.,
Hangzhou, China) precoated with 0.1 mg/ml poly-D-lysine (Sigma
Aldrich, St. Louis, MO, USA). Subsequent to culturing for 1 day,
half of the media was changed into neuronal culture media
(neurobasal media containing 2 mM GlutaMAX™-I
supplement, 2% B27 and 25 µg/ml penicillin/streptomycin)
without phenol red. Ara-C (2 µM; Sigma-Aldrich) was added
6–8 days after plating during the culture medium change, and cells
were fed twice weekly thereafter. All the cells were grown at 37°C
and in 5% CO2.
Drugs and treatment
Hippocampal neurons were cultured with phenol
red-free neurobasal cell culture medium throughout the whole
culture period. Different concentrations of 17-β-estradiol (0, 0.1
and 1 ng/ml) were added into the cell culture medium at 1, 3, 6 and
10 days in vitro (DIV), to maintain the neuron development
and survival until the analysis. For the drug treatment study, the
estrogen receptor antagonist ICI 182,780 (100 nM) (20,21) and selective Kv4.2 and Kv4.3
channel blocker phrixotoxin2 (PaTx2) (100 nM) (22) were added to the culture medium
with 17-β-estradiol at the DIV1 for co-culturing.
Patch-clamp recordings data
acquisition
Whole-cell recordings of pyramidal shaped neurons
were recorded using a conventional patch-clamp technique with
MultiClamp 700B amplifier (Axon Instruments, Foster City, CA, USA).
Patch pipettes were pulled using a Sutter P-97 (Sutter Instrument,
Novato, CA, USA) pipette puller. The bath solution contained 128 mM
NaCl, 5 mM KCl, 25 mM HEPES, 1 mM MgCl2, 30 mM glucose
and 2 mM CaCl2 (pH adjusted to 7.3 with NaOH).
Soft-glass recording pipettes were filled with an internal solution
containing 125 mM potassium gluconate, 10 mM KCl, 10 mM HEPES, 5 mM
EGTA, 10 mM Tris-phosphocreatine, 4 mM MgATP and 0.5 mM
Na2GTP (pH adjusted to 7.2–7.3 with KOH). The pipette
resistance was 3–5MΩ subsequent to filling with internal solution.
The cultured pyramidal shaped neurons selected for
electrophysiological recording exhibited the same morphological
characteristics. All the recordings were performed at room
temperature.
In order to record the action potential, the
membrane potential was held at −70 mV in the current clamp mode. A
large depolarization shift was defined as a membrane potential up
shift for >10 mV, lasting for ≥300 msec. A bursting activity was
defined by ≥5 consecutive action potentials overlaying on the top
of this large depolarization shift. When quantifying the percentage
of neurons showing bursting activity, the criterion is ≥2 bursts
occurring during 10 min of recording, the same as our previous
studies (23,24).
IA recordings were obtained in the
voltage clamp mode at −100 mV. To record the fast transient outward
potassium currents, the step command potentials were applied
between −70 and +40 mV, with 10-mV steps and a width of 200 msec.
In order to trigger the A-type potassium current, a prepulse (−100
mV, 200 msec) was applied to the cells immediately prior to the
step commands (18). To isolate
IA, tetraethylamonium (5 mM), tetrodotoxin (1 µM)
and CdCl2 (100 µM) were added to the
extracellular solution to block the calcium channels, delayed
rectifier potassium channels and voltage-gated sodium channels.
All the electrophysiological data were recorded
using a MultiClamp 700B amplifier (Axon Instruments). Data
acquisition and analysis were performed with pClamp 10.2 software
(Axon Instruments).
Equation for curve fitting of the
steady-state activation of IA currents
In the activation experiments, membrane potential
was first held at −100 mV. The total voltage-dependent potassium
currents were evoked by a 200 msec depolarization pulse from −70 to
+40 mV in 10-mV steps at 10-sec intervals. The IA
current value was the difference between the instant peak current
and the steady-state current. Data were analyzed by calculation of
the equation G = I/(Vm−Vrev), for the
membrane potassium conductance. Vm represents the
membrane potential, whereas Vrev is the reversal
potential of K+ (25).
Subsequent to normalizing each current amplitude to the maximal
current amplitude obtained from the depolarization to +40 mV, the
function G/Gmax = 1/{1 + exp[− (Vm −
V1/2)/k]} was used to fit the data. From this equation,
an activation curve of IA was obtained and the
V1/2, the voltage at which the IA current was
half-activated, was calculated.
Data analysis
All the electrophysiological results were analyzed
using two-way analysis of variance (ANOVA) for comparisons between
multiple groups, and post hoc analysis was performed with Fisher's
least significant difference, χ2 test and the Student's
t-test for direct comparison between the two groups. Data were all
presented as mean ± standard error of the mean and analyzed by SPSS
13.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
U-shape dose-dependent effect of
17-β-estradiol on epileptiform bursting activities in cultured
hippocampal neurons
In a previous study, we reported that phenol red, a
weak estrogen receptor agonist, at the concentration of a
supplement ingredient in the commercially available culture medium,
has a suppressive action on epileptiform bursting activities in
cultured neurons by activation of the estrogen receptors (13). Thus, in the present study, all the
experiments were performed in phenol red-free medium cultured
hippocampal neurons. In the phenol red-free neurobasal culture
medium, DIV14 hippocampal neurons were cultured with alcohol (as
the vehicle control for 17-β-estradiol) or 17-β-estradiol at doses
of 0.1 and 1 ng/ml, and were patch-clamp recorded in the current
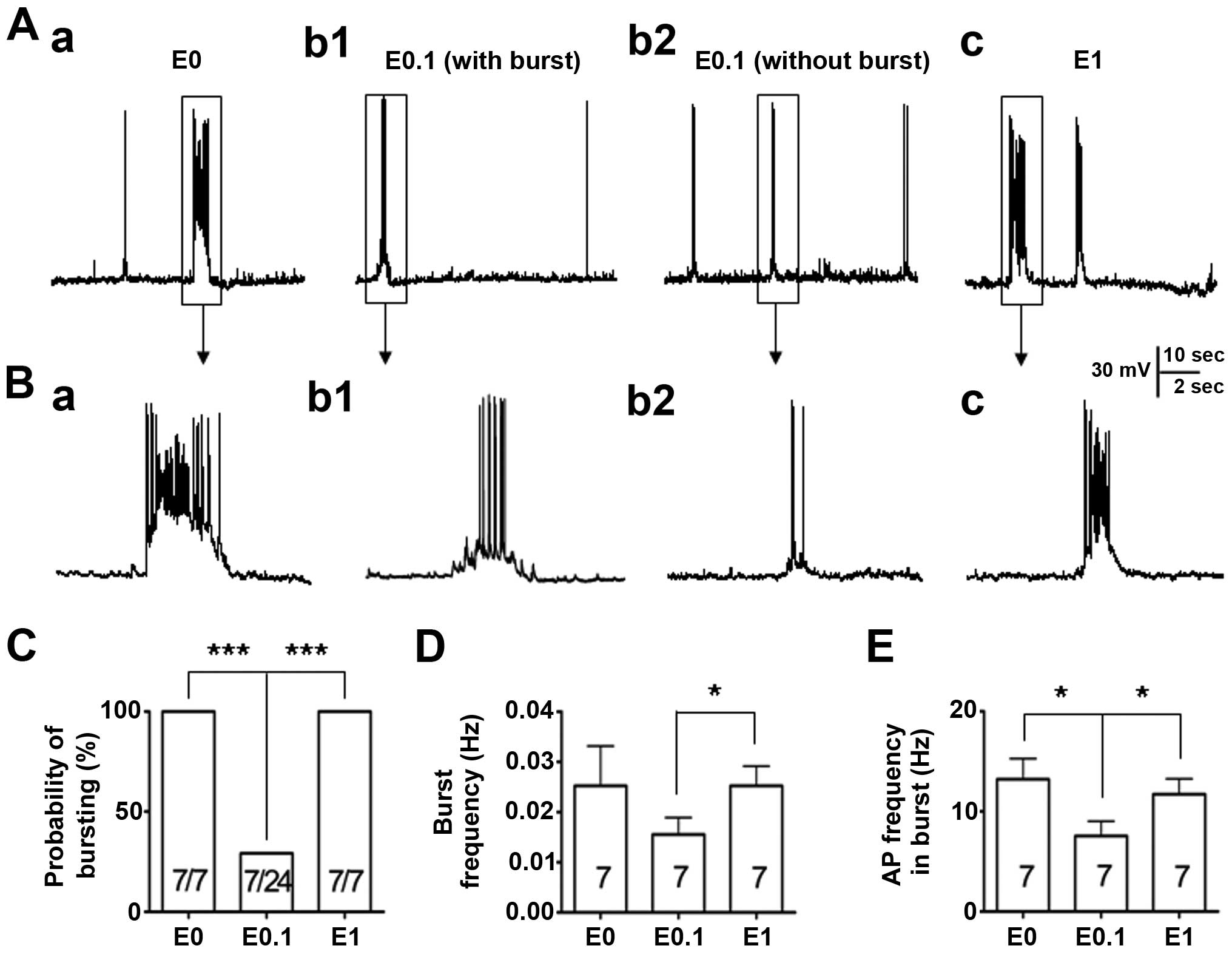
mode to record their firing properties (Fig. 1). Similar to the previous study
(13), all the neurons (7/7,
100%) were showing epileptiform bursting activities while the
neurons were cultured in phenol red-free medium with no added
17-β-estradiol (0 ng/ml). However, when the neurons were cultured
in low dose 17-β-estradiol (0.1 ng/ml)-added medium, the percentage
of neurons showing abnormal epileptiform bursting discharges (7/24,
29%) was significantly reduced (P<0.001) when compared to the
vehicle control. By contrast, when the neurons were cultured in the
high dose of 17-β-estradiol (1 ng/ml), the percentage of neurons
showing epileptiform bursting discharges (7/7, 100%) was not
different to that of the vehicle control group, but significantly
more than that of the low dose 17-β-estradiol (0.1 ng/ml) group
(P<0.001) (Fig. 1A–C).
In addition, the burst frequency and the action
potential frequency were analyzed within the burst among those
bursting neurons recorded. The data revealed that when neurons were
cultured with 0.1 ng/ml 17-β-estradiol and had burst activities
(n=7), the burst frequency was not different to that of the neurons
in the vehicle control group (P>0.05) (Fig. 1D); however, the action potential
frequency within the burst was significantly lower (P<0.05) than
that in the neurons in the vehicle control group (Fig. 1E). The burst frequency and the
action potential frequency were further compared within the burst
between the bursting neurons from neurons cultured with either low
(0.1 ng/ml) or high (1 ng/ml) 17-β-estradiol medium. The results
showed that the burst frequency and the action potential frequency
within the burst among those bursting neurons were significantly
lower (P<0.05) in neurons treated with 0.1 ng/ml 17-β-estradiol
compared with those in 1 ng/ml 17-β-estradiol group (Fig. 1D and E).
These results indicate that the effect of
17-β-estradiol on the epileptiform bursting discharges is U-shape
like, and 17-β-estradiol, in a certain low dose range, has a
suppressing effect, similar to the effect that we previously
reported (13).
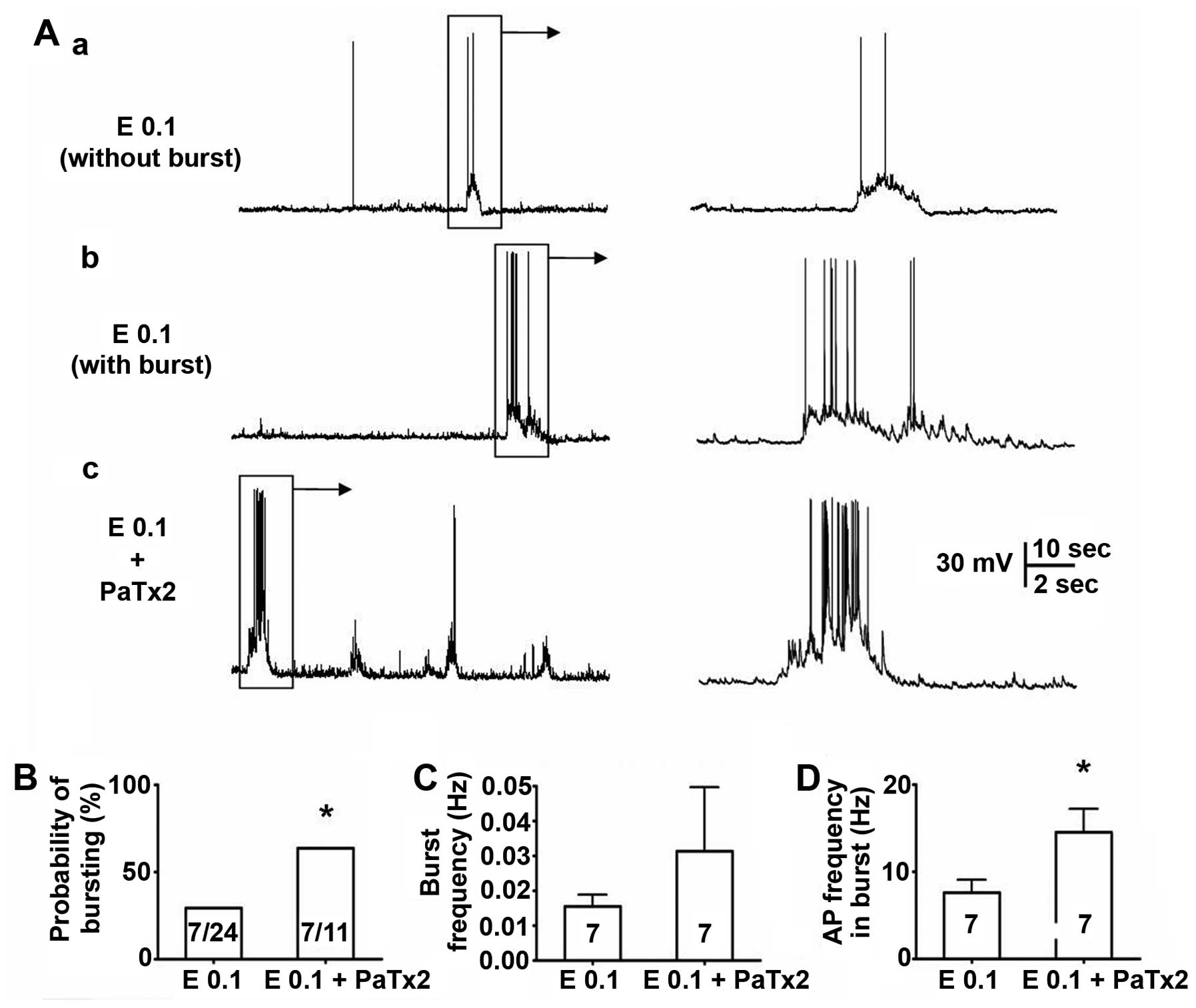
Effect of PaTx2 on epileptiform bursting
activities
Previous studies have shown that 17-β-estradiol has
a modulation effect on the transient (mediated by Kv4.2) and
sustained outward potassium current (major mediated by Kv2 family)
(18), and Kv4.2 is associated
with the seizure occurrence (12). Thus, whether the suppressive
effect of the low dose (0.1 ng/ml) 17-β-estradiol on spontaneous
epileptiform bursting activities in cultured neurons is due to
modulation of the transient outward potassium current mediated by
Kv4.2 was investigated using the selective Kv4.2/Kv4.3 channel
blocker PaTx2. After 14 days co-culturing in low dose
17-β-estradiol (0.1 ng/ml) with PaTx2 (100 nM), neurons at DIV14
were patch-clamp studied of their spontaneous activities (Fig. 2). As expected, PaTx2 significantly
blocked the low dose 17-β-estradiol-induced suppressive effect on
the epileptiform activities (Fig.
2A). The percentage of neurons showing epileptiform activity in
the PaTx2-treated group was 64% (7 in 11), which was significantly
higher than those cultured with low dose 17-β-estradiol alone (29%,
7 in 24; P<0.05) (Fig. 2B). In
addition, PaTx2 also significantly inhibited low dose
17-β-estradiol-induced reduction of the average action potential
frequency within the burst (P<0.05) (Fig. 2D). It should be noted that we did
not compare the effect of PaTx2 and high dose 17-β-estradiol on
bursting activity in this study, but aim to do so in the
future.
This result indicates that the potassium channel
Kv4.2 is possibly involved in the low dose of
17-β-estradiol-induced suppressive effect on epileptiform
activities.
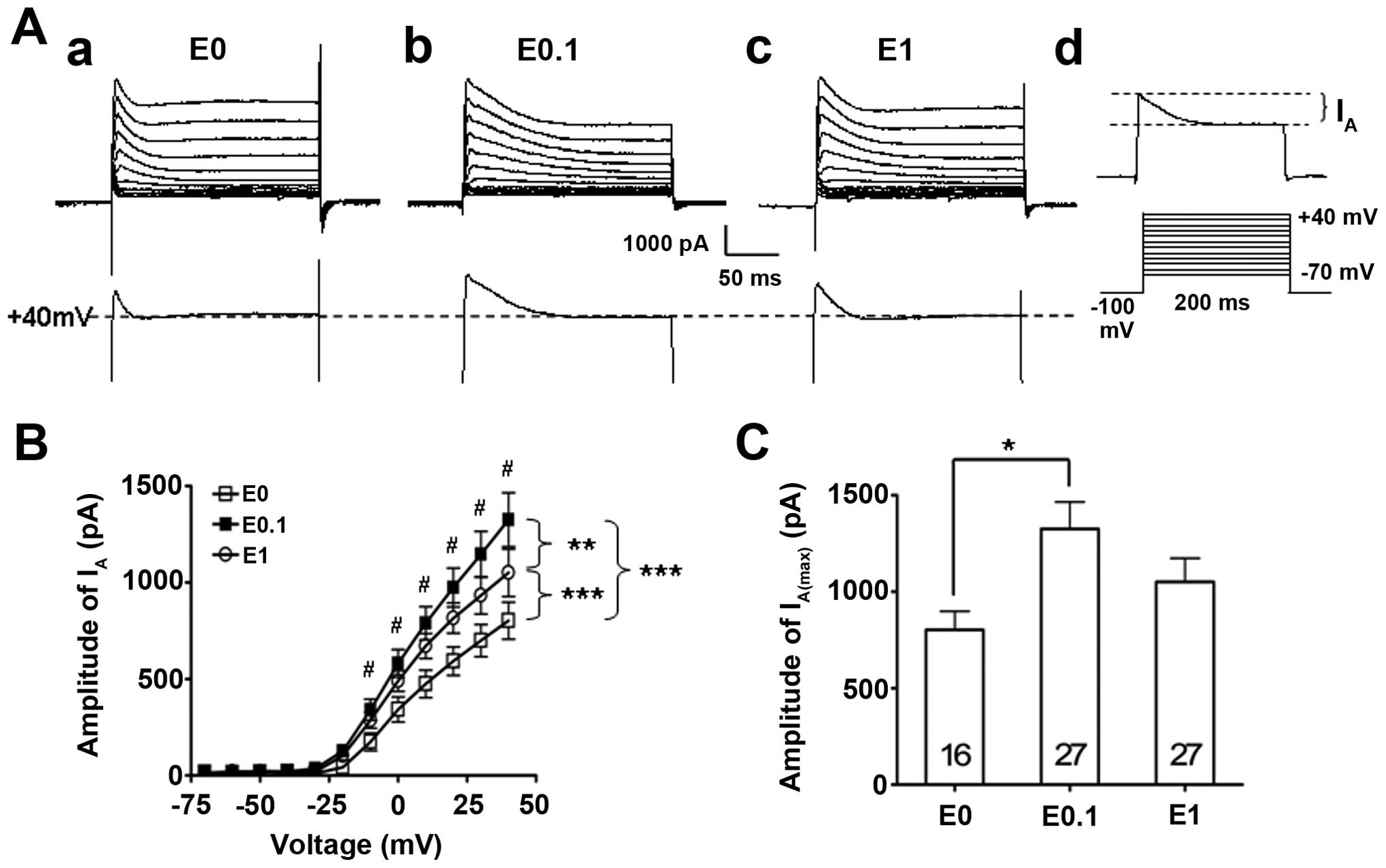
Dose-dependent effect of 17-β-estradiol
on fast transient outward potassium currents in cultured
hippocampal neurons
In order to study the mechanism of how
17-β-estradiol modulates epileptiform activity of hippocampal
neurons through the potassium Kv4.2 channel, the fast transient
outward potassium currents (IA), which are mostly
mediated by Kv4.2 channel, were further tested by the patch-clamp
technique in different 17-β-estradiol-treated neuron groups.
Similar to the effect of 17-β-estradiol on epileptiform bursting
activities in cultured hippocampal neurons, a dose-related 'bell'
shape-like effect of 17-β-estradiol on fast transient outward
potassium currents was identified (Fig. 3). Statistical analysis of the I–V
curves of the IA revealed that low (0.1 ng/ml, n=27) and
high dose (1 ng/ml, n=27) 17-β-estradiol significantly enhanced
IA compared to the 17-β-estradiol-free vehicle control
group (P<0.001, two-way ANOVA) (Fig. 3A and B). However, further
statistical analyses between the low and high dose of the
17-β-estradiol-treated groups data demonstrated that the low dose
of 17-β-estradiol (0.1 ng/ml) had the largest enhancement effect on
the IA, while further increasing the 17-β-estradiol
concentration to a higher dose (1 ng/ml) caused a reduction,
although this remained higher compared to the vehicle control group
and did not further enhance on the IA (P<0.01,
two-way ANOVA) (Fig. 3A and B).
However, the post-hoc analysis showed that only the low dose of
17-β-estradiol had significant enhancement on the IA
(P<0.05) (Fig. 3B).
The differences of the maximal amplitude of the
IA (IA(max)), which was evoked while the
current command was at +40 mV, was further studied among those
groups. The data showed that the IA(max) was 1,325±139
pA (n=27) in the low dose of 17-β-estradiol (0.1 ng/ml) group,
which was significantly higher than that of the IA(max)
(802±96 pA, n=16, P<0.05) in the vehicle control group
(17-β-estradiol at 0 ng/ml). By contrast, the IA(max) in
the high dose of 17-β-estradiol (1 ng/ml) group was 1,050±122 pA
(n=27), which was not different to that of the neurons tested in
the vehicle control group (P>0.05).
These data indicate that IA is capable of
being modulated by the long-term estrogen receptor stimulation at a
low dose of 17-β-estradiol, which enhanced the IA
amplitude.
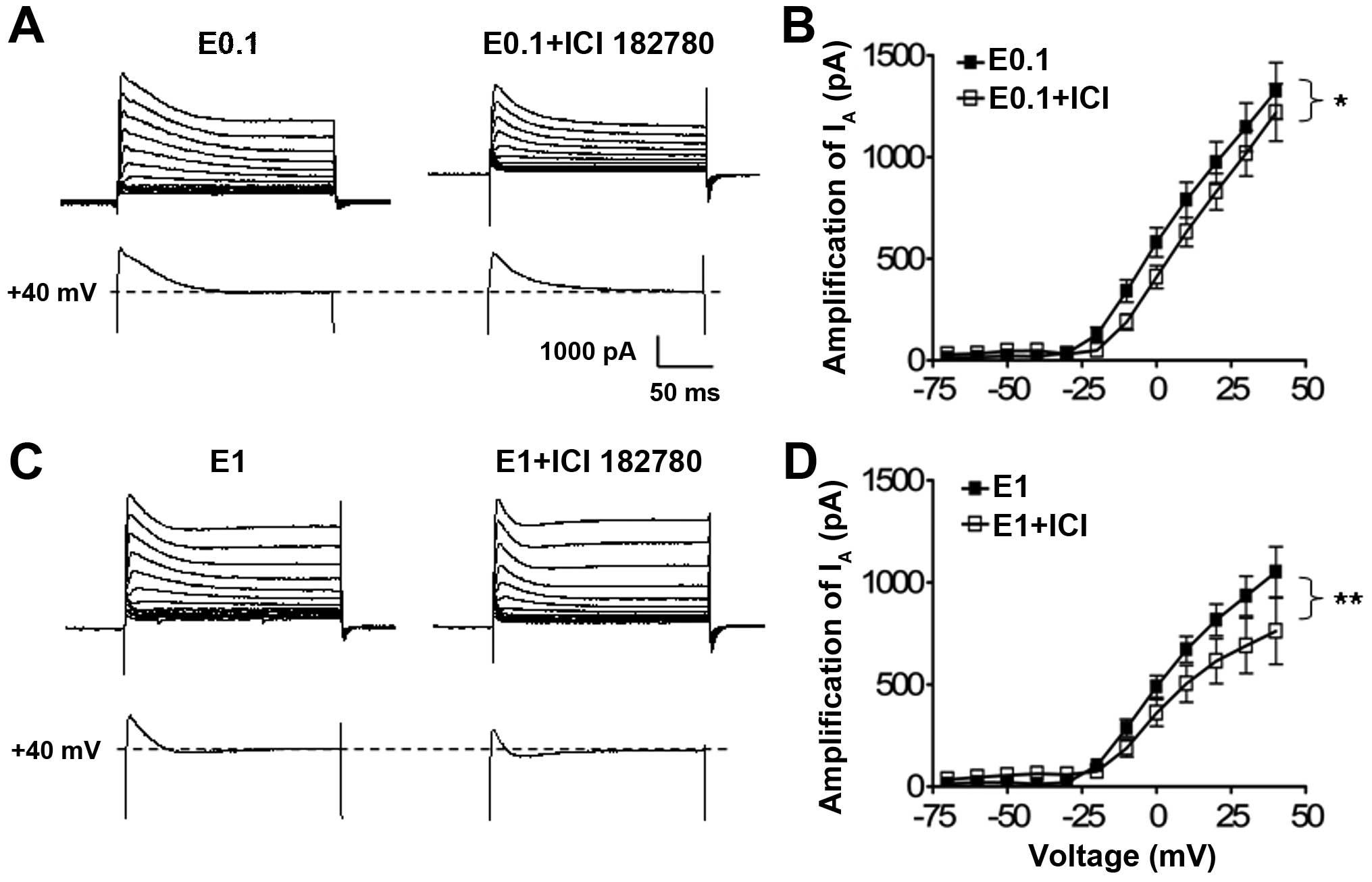
Effect of ICI 182,780 on the suppressive
action of low dose 17-β-estradiol on the IA currents in
cultured hippocampal neurons
As 17-β-estradiol treatment at the low and high
concentration had a modulation effect on the potassium
IA, whether the enhancement of the low dose of
17-β-estradiol on the potassium IA is mediated by the
estrogen receptors was further analyzed using ICI 182,780, an
estrogen receptor antagonist.
When neurons were co-cultured with 17-β-estradiol at
0.1 or 1 ng/ml and with ICI 182,780 (100 nM), the statistical
analysis of the I–V curves showed that antagonism of the estrogen
receptors with ICI 182,780 significantly suppressed the low
(P<0.05, n=34, two-way ANOVA) (Fig. 4A) and high (P<0.01, n=22,
two-way ANOVA) (Fig. 4B) dose
17-β-estradiol-induced potassium IA enhancement in
comparison with the 17-β-estradiol treatment alone groups (n=27 and
n=27, respectively) (Fig. 4B and
C).
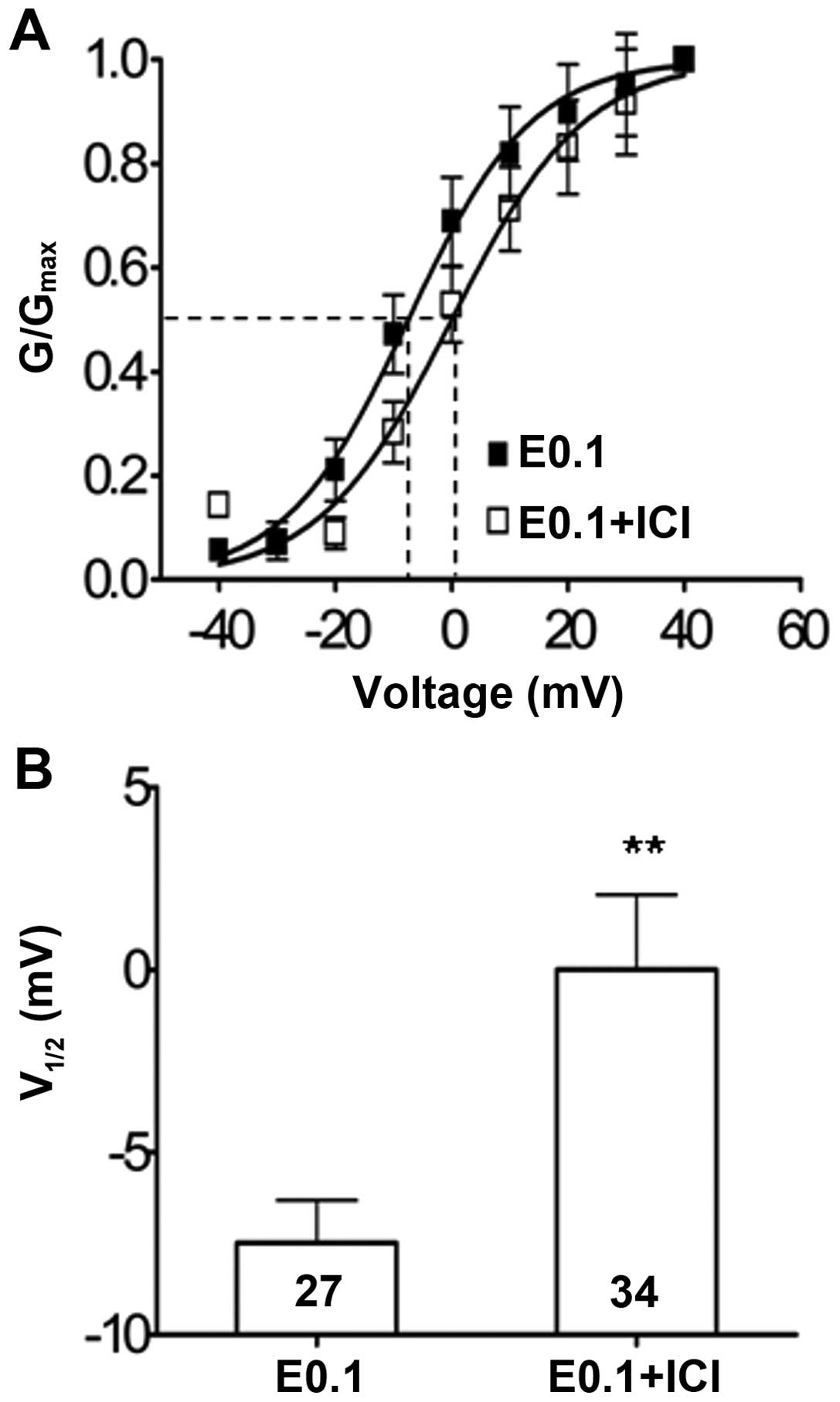
In addition to suppression of the IA
amplitude, the antagonism of the estrogen receptor with ICI 182,780
also significantly left-shifted the steady-state activation curve
of the IA currents, which represents the channel-opening
properties of the Kv4.2 potassium channels under the low dose
17-β-estradiol (0.1 ng/ml) culture condition. The IA
current half-activation potential (V1/2) underwent a
significant depolarization shift increase from −7.48±1.6 mV (n=27)
to 0.00±1.8 mV (n=34) (P<0.05) in the neuron groups with
17-β-estradiol (0.1 ng/ml) alone or co-treatment with ICI 182,780,
respectively (Fig. 5).
These data indicate that IA is capable of
being modulated by the long-term estrogen receptor stimulation at
the low dose of 17-β-estradiol, which enhanced the potassium Kv4.2
channel activities by enhancing IA amplitude and
channel-opening probability.
Discussion
The present experimental results demonstrated that
17-β-estradiol, the biologically active estrogen, had a regulative
action on abnormal epileptiform bursting activities in cultured
hippocampal neurons in a U-shape dose-dependent manner: Extremely
low or high concentrations of 17-β-estradiol enhance, but low
concentrations of 17-β-estradiol suppress, the epileptiform
bursting activities in the cultured hippocampal neurons. This
suppressive effect of 17-β-estradiol on the epileptiform bursting
activities is possibly due to, at least in part, the modulation of
the potassium Kv4.2 channel activities by activation of estrogen
receptors.
In the present study, the dose effect of estrogen
receptor stimulation on the epileptiform bursting activities in
cultured hippocampal neurons was examined. Estrogen has been
reported to have a differential effect on neuronal function.
17-β-estradiol has been reported to increase the excitability of
gonadotrophin-releasing hormone neurons (26), medial vestibular nucleus neurons
in brain stem (27) and
hippocampal neurons (28). By
contrast, estrogen receptor activation has also been reported to
decrease neuronal excitability by indirectly changing the local
neurotransmitter release (29),
particularly by changing the interaction with GABAergic neurons
(30,31). Our previous study also indicated
that on the same hippocampal neuron, a weak estrogen receptor
agonist, phenol red, could have a U-shape-like
activation-inhibition-activation effect on the epileptiform
bursting activities: Low and high concentrations of phenol red all
induced the epileptiform bursting activities in the cultured
hippocampal neurons, while the middle concentration (~28 µM)
of phenol red suppressed this activity (13). Similar to the previous reports, a
large proportion of the neurons cultured in the estrogen-free
medium had epileptiform bursting activities in the present study,
and the low dose 17-β-estradiol (~0.1 ng/ml) had a suppressive
effect, whereas the high dose 17-β-estradiol (1 ng/ml) had a
promoting effect on the neuronal excitability change, forming a
U-shape-like dose-dependent action on the neuronal excitation
change. This effect may explain the differential action of the
estrogen receptor stimulation caused increase or decrease of
neuronal excitability in various studies (28–31).
Neuron excitability is determined by various
factors; one of them is the open properties of the potassium
channels. Voltage-gated potassium (Kv) channels are important for
maintaining the membrane potentials, action potential shape, firing
adaptation and neuronal excitability in neurons (4). The Kv channels-mediated current
contains the transient and the sustained current (5,6).
Previous studies have demonstrated that the transient outward
IA has an important role in controlling the membrane
excitability and that it contributes to remodeling neuronal
excitation under pathological conditions (7,8).
In the present study, the modulatory effect of low dose
17-β-estradiol on the epileptiform bursting probability was blocked
by the selective Kv4.2 and 4.3 potassium channel blocker PaTx2,
indicating the involvement of the Kv4.2 and 4.3 potassium channels.
The effect of 17-β-estradiol on the voltage-gated fast transient
outward potassium current was further examined to improve the
understanding of how 17-β-estradiol affects neuronal excitability
and the probability of the epileptiform bursting activity
occurrence. Using whole cell clamp recordings, the results
demonstrated that 17-β-estradiol had a dose-associated modulatory
effect on the voltage-gated fast transient outward IA
curve and on the maximal current measured when the membrane
potential was transiently increased to +40 mV. The low and high
dose of 17-β-estradiol significantly increased the amplitude of the
IA. Notably, the low concentration of 17-β-estradiol at
0.1 ng/ml had the strongest effect on the increase of the amplitude
of IA, which is also significantly more than that of
17-β-estradiol at 1 ng/ml, showing a bell-shape
concentration-dependent manner on the facilitation of the
voltage-gated fast transient outward IA. These results
are consistent with the finding that a low dose of 17-β-estradiol
only had a significant suppressive effect on the epileptiform
bursting activities, which showed a U-shape concentration-dependent
inhibitory action. As the transient outward IA has an
important role in controlling the membrane excitability and
remodeling neuronal excitation under pathological conditions
(7,8), the present results indicate that
activation of estrogen receptors on modulating neuronal
epileptiform bursting activities may, through the mechanism of
modulating the transient outward IA, alter the cell
excitability.
Estrogen is one of the main hormones of female
mammals (however, it also exists in males), which has a complex and
wide physiological and pathophysiological effect, such as promoting
cell proliferation (32–34) as well as modulating the neuronal
excitability (3,27,28,35–37), and the effect of 17-β-estradiol,
an estrogen receptor agonist, observed in the present study is
possibly due to activation of the estrogen receptors, as the
facilitation effect on the transient outward IA in
cultured hippocampal neurons was inhibited by the estrogen receptor
antagonist ICI 182,780 (20,21) (Fig.
4). However, the results from the present study could not
distinguish which subtype of the estrogen receptors was mediating
this function, as ICI 182,780 is neither an α- nor β-estrogen
receptor subtype-selective antagonist. The reason why
17-β-estradiol has an effect on the transient outward
IA, as well as on the neuronal epileptiform bursting
activities, in a bell-shape (U-shape) mannor remains unknown. We
hypothesize that the estrogen receptor subtype may have a major
role in differentially modulating neuronal excitability with
regards to the firing properties. The involvement of either the α-
or β-subtype estrogen receptors in mediating this 17-β-estradiol
action requires further study using selective antagonists for α-
and β-estrogen receptor subtypes in the future.
Potassium channels are important for post-excitatory
membrane repolarization and sustain different components of
hyperpolarizing after-potentials in intrinsically bursting cells.
The inhibition of K+ currents interferes with
repolarization and hyperpolarization, and increases
hyperexcitability and bursting activity (38). In the Kv channel family, Kv4 is
the major subtype mediating the transient outward IA
(9,10). In the hippocampus, dendrites of
CA1 pyramidal neurons contain a high density of transient A-type
potassium channels, and A-type K+ channels are crucial
modulators of information processing and synaptic plasticity in the
dendrites (9,10). A reduction in A-type K+
channel activity promotes burst firing in a location-dependent
manner (39). The changes in the
potassium currents alter shape and repetitive frequency of the
action potentials. The presence of the rapidly inactivating A-type
channels maintains the firing rate at a low frequency and broadens
the action potentials when neurons fire repetitively (40). These two changes diminish the
firing rate in the neurons. This indicates that the A-type
potassium current is a type of inhibitory effect in the normal
physiological condition in the hippocampus. In the present study,
the neurons with a high probability to have epileptiform bursting,
such as neurons in the estrogen-free culture group, also have a
high firing frequency of the action potentials in the burst, and
the lowest IA. Increasing the IA by low dose
of 17-β-estradiol significantly reduced the action potential firing
frequency along with the inhibited epileptiform bursting
probability and the suppressed bursting frequency. This result
indicates that there is a negative correlation between the
transient outward potassium current and the epileptiform bursting
properties, including bursting frequency and bursting action
potential frequency. This is consistent with the previous findings
that in pilocarpine seizure rats, the increased dendritic
excitability and the epileptic seizure generation is possibly due
to the decreased A-type potassium channels activities (12).
The results from the present study suggest that
estrogen receptor activation is important for modulating the
neuronal cell excitability and maintaining the normal neuronal
physiological conditions, including the firing properties of the
neurons, by modulating the potassium channel activities. As
estrogen receptor stimulation could either enhance the neuronal
excitability by directly modulating the intrinsic properties
(28,36) or, in the other direction, decrease
the neuronal excitability by indirectly changing the local
neurotransmitter release (29,35) particularly by interaction with
GABAergic neurons (29,30), the present experimental results
may indicate that, at least in part, the activation of estrogen
receptors, with a certain range of the estradiol concentration, in
modulating the neuronal bursting activities in the cultured
hippocampal neurons is possibly directly modulating the potassium
channel, particularly the Kv4.2 channel, properties by enhancing
the channel current and channel-opening probability.
In conclusion, the present study identified that the
estrogen level at a certain physiological concentration is
important in maintaining the potassium channel activities and in
turn influences the neuronal excitability to the extent of the
epileptiform activities. Thus, the present results indicate that
reduced activation of the transient outward potassium current by a
high estrogen level may be one of the causes for triggering
catamenial epilepsy, and provides us with a potential therapeutical
target to intervene with catamenial epilepsy in the future.
Acknowledgments
The present study was supported by grants from the
Nature Science Foundation of China (nos. 31129003, 81171224,
31271188 and 81301108) and the Science and Technology Commission of
Shanghai Municipality (nos. 13DJ1400302 and 13ZR1406500) to YW.,
X.L. and XW.
References
|
1
|
Newmark ME and Penry JK: Catamenial
epilepsy: A review. Epilepsia. 21:281–300. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Laidlaw J: Catamenial epilepsy. Lancet.
271:1235–1237. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Velísková J: The role of estrogens in
seizures and epilepsy: The bad guys or the good guys? Neuroscience.
138:837–844. 2006. View Article : Google Scholar
|
|
4
|
Takeda M, Tsuboi Y, Kitagawa J, Nakagawa
K, Iwata K and Matsumoto S: Potassium channels as a potential
therapeutic target for trigeminal neuropathic and inflammatory
pain. Mol Pain. 7:52011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Iverson LE, Tanouye MA, Lester HA,
Davidson N and Rudy B: A-type potassium channels expressed from
Shaker locus cDNA. Proc Natl Acad Sci USA. 85:5723–5727. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Solc CK, Zagotta WN and Aldrich RW:
Single-channel and genetic analyses reveal two distinct A-type
potassium channels in Drosophila. Science. 236:1094–1098. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sonner PM and Stern JE: Functional role of
A-type potassium currents in rat presympathetic PVN neurones. J
Physiol. 582:1219–1238. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sonner PM, Filosa JA and Stern JE:
Diminished A-type potassium current and altered firing properties
in presympathetic PVN neurones in renovascular hypertensive rats. J
Physiol. 586:1605–1622. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Covarrubias M, Wei AA and Salkoff L:
Shaker, Shal, Shab, and Shaw express independent K+
current systems. Neuron. 7:763–773. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Serôdio P, Vega-Saenz de Miera E and Rudy
B: Cloning of a novel component of A-type K+ channels
operating at subthreshold potentials with unique expression in
heart and brain. J Neurophysiol. 75:2174–2179. 1996.
|
|
11
|
Hoffman DA, Magee JC, Colbert CM and
Johnston D: K+ channel regulation of signal propagation
in dendrites of hippocampal pyramidal neurons. Nature. 387:869–875.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bernard C, Anderson A, Becker A, Poolos
NP, Beck H and Johnston D: Acquired dendritic channelopathy in
temporal lobe epilepsy. Science. 305:532–535. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu X, Chen B, Chen L, Ren WT, Liu J, Wang
G, Fan W, Wang X and Wang Y: U-shape suppressive effect of phenol
red on the epileptiform burst activity via activation of estrogen
receptors in primary hippocampal culture. PLoS One. 8:e601892013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hoffman GE, Moore N, Fiskum G and Murphy
AZ: Ovarian steroid modulation of seizure severity and hippocampal
cell death after kainic acid treatment. Exp Neurol. 182:124–134.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Carroll JC, Rosario ER and Pike CJ:
Progesterone blocks estrogen neuroprotection from kainate in
middle-aged female rats. Neurosci Lett. 445:229–232. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fatehi M, Kombian SB and Saleh TM:
17beta-estradiol inhibits outward potassium currents recorded in
rat parabrachial nucleus cells in vitro. Neuroscience.
135:1075–1086. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bosch MA, Kelly MJ and Rønnekleiv OK:
Distribution, neuronal colocalization, and 17beta-E2 modulation of
small conductance calcium-activated K(+) channel (SK3) mRNA in the
guinea pig brain. Endocrinology. 143:1097–1107. 2002.PubMed/NCBI
|
|
18
|
Farkas I, Varju P and Liposits Z: Estrogen
modulates potassium currents and expression of the Kv4.2 subunit in
GT1-7 cells. Neurochem Int. 50:619–627. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen B, Jiang M, Zhou M, Chen L, Liu X,
Wang X and Wang Y: Both NMDA and non-NMDA receptors mediate
glutamate stimulation induced cofilin rod formation in cultured
hippocampal neurons. Brain Res. 1486:1–13. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Howell A, Osborne CK, Morris C and
Wakeling AE: ICI 182,780 (Faslodex): Development of a novel, 'pure'
antiestrogen. Cancer. 89:817–825. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wong JK, Le HH, Zsarnovszky A and Belcher
SM: Estrogens and ICI182,780 (Faslodex) modulate mitosis and cell
death in immature cerebellar neurons via rapid activation of
p44/p42 mitogen-activated protein kinase. J Neurosci. 23:4984–4995.
2003.PubMed/NCBI
|
|
22
|
Bosmans F, Rash L, Zhu S, Diochot S,
Lazdunski M, Escoubas P and Tytgat J: Four novel tarantula toxins
as selective modulators of voltage-gated sodium channel subtypes.
Mol Pharmacol. 69:419–429. 2006. View Article : Google Scholar
|
|
23
|
Qi J, Wang Y, Jiang M, Warren P and Chen
G: Cyclothiazide induces robust epileptiform activity in rat
hippocampal neurons both in vitro and in vivo. J Physiol.
571:605–618. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang Y, Qi JS, Kong S, Sun Y, Fan J, Jiang
M and Chen G: BDNF-TrkB signaling pathway mediates the induction of
epileptiform activity induced by a convulsant drug cyclothiazide.
Neuropharmacology. 57:49–59. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhuang JL, Wang CY, Zhou MH, Duan KZ and
Mei YA: TGF-β1 enhances Kv2.1 potassium channel protein expression
and promotes maturation of cerebellar granule neurons. J Cell
Physiol. 227:297–307. 2012. View Article : Google Scholar
|
|
26
|
Rønnekleiv OK, Bosch MA and Zhang C:
17β-oestradiol regulation of gonadotrophin-releasing hormone
neuronal excitability. J Neuroendocrinol. 24:122–130. 2012.
View Article : Google Scholar
|
|
27
|
Grassi S, Frondaroli A, Scarduzio M, Dutia
MB, Dieni C and Pettorossi VE: Effects of 17beta-estradiol on
glutamate synaptic transmission and neuronal excitability in the
rat medial vestibular nuclei. Neuroscience. 165:1100–1114. 2010.
View Article : Google Scholar
|
|
28
|
Zadran S, Qin Q, Bi X, Zadran H, Kim Y,
Foy MR, Thompson R and Baudry M: 17-Beta-estradiol increases
neuronal excitability through MAP kinase-induced calpain
activation. Proc Natl Acad Sci USA. 106:21936–21941. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Saleh TM, Connell BJ, McQuaid T and Cribb
AE: Estrogen-induced neurochemical and electrophysiological changes
in the parabrachial nucleus of the male rat. Brain Res. 990:58–65.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Blurton-Jones M and Tuszynski MH: Estrogen
receptor-beta colocalizes extensively with parvalbumin-labeled
inhibitory neurons in the cortex, amygdala, basal forebrain, and
hippocampal formation of intact and ovariectomized adult rats. J
Comp Neurol. 452:276–287. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhou J, Pfaff DW and Chen G: Sex
differences in estrogenic regulation of neuronal activity in
neonatal cultures of ventromedial nucleus of the hypothalamus. Proc
Natl Acad Sci USA. 102:14907–14912. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Frankfurt M, Gould E, Woolley CS and
McEwen BS: Gonadal steroids modify dendritic spine density in
ventromedial hypothalamic neurons: A Golgi study in the adult rat.
Neuroendocrinology. 51:530–535. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Woolley CS and McEwen BS: Estradiol
mediates fluctuation in hippocampal synapse density during the
estrous cycle in the adult rat. J Neurosci. 12:2549–2554.
1992.PubMed/NCBI
|
|
34
|
Calizo LH and Flanagan-Cato LM: Estrogen
selectively regulates spine density within the dendritic arbor of
rat ventromedial hypothalamic neurons. J Neurosci. 20:1589–1596.
2000.PubMed/NCBI
|
|
35
|
Velísková J and Velísek L: Beta-estradiol
increases dentate gyrus inhibition in female rats via augmentation
of hilar neuropeptide Y. J Neurosci. 27:6054–6063. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Edwards HE, Burnham WM, Mendonca A, Bowlby
DA and MacLusky NJ: Steroid hormones affect limbic afterdischarge
thresholds and kindling rates in adult female rats. Brain Res.
838:136–150. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Herzog AG: Catamenial epilepsy:
Definition, prevalence pathophysiology and treatment. Seizure.
17:151–159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ptacek LJ and Fu YH: Channelopathies:
Episodic disorders of the nervous system. Epilepsia. 42(Suppl 5):
35–43. 2001. View Article : Google Scholar
|
|
39
|
Johnston D, Hoffman DA, Magee JC, Poolos
NP, Watanabe S, Colbert CM and Migliore M: Dendritic potassium
channels in hippocampal pyramidal neurons. J Physiol. 525:75–81.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Connor JA and Stevens CF: Prediction of
repetitive firing behaviour from voltage clamp data on an isolated
neurone soma. J Physiol. 213:31–53. 1971. View Article : Google Scholar : PubMed/NCBI
|