Introduction
Diabetic cardiomyopathy (DCM) is a specific type of
cardiomyopathy that is not associated with coronary artery disease,
hypertension or other cardiac pathologies. The main pathological
changes associated with DCM include extensive myocardial cell
hypertrophy, apoptosis, necrosis and myocardial fibrosis (1–3).
Although modern medical technology has advanced greatly in other
areas, treatments for DCM remain outdated, involving the control of
blood glucose levels other comprehensive measurements. Developing a
more effective treatment for DCM thus remains a challenge.
Previous studies have indicated that the Notch
signaling pathway plays an important role in a variety of organisms
and cell types. The Notch protein is activated and cleaved by tumor
necrosis factor (TNF)-α converting enzyme (TACE) and the
γ-secretase complex, which release the Notch intracellular domain
(NICD) that then binds to the transcription factor, CSL (CBF-1 in
humans, suppressor of hairless in Drosophila, LAG in
Caenorhabditis elegans and RBP-Jκ in mice) (4–6).
Following the translocation of NICD into the nucleus, Notch
promotes the transcription of the target genes, hairy/enhancer of
split (Hes) and hairy/enhancer-of-split related with YRPW motif
(Hey), which not only regulates cell proliferation,
differentiation, apoptosis and adhesion, but also plays an
important role in embryonic development, cancer, neurodegenerative
sexual disorders, wound healing, angiogenesis and the inflammatory
response (7,8).
Notch1 is a member of the Notch family. In the
heart, Notch1 signaling not only regulates embryonic cardiac
development and differentiation, but also stimulates the
proliferation of immature cardiomyocytes (9,10).
In addition, Notch1 signaling regulates the response to myocardial
cell damage, and inhibition of Notch1 signaling can accelerate
cardiac myocyte hypertrophy, degeneration and necrosis, and may
eventually lead to myocardial apoptosis and fibrosis (11,12). On the other hand, the inhibition
of Notch1 expression can cause myocardial damage, which leads to
myocardial infarction (13). At
the embryonic stage, Notch1 signaling stimulates the proliferation
of immature cardiomyocytes, and after birth, the protein expression
of Notch1 gradually decreases in myocardial cells (10). Studies have indicated that Notch1
and phosphoinositide 3-kinase (PI3K)/AKT are able to interact and
are dependent on phosphatase and tensin homolog deleted on
chromosome 10 (PTEN) (14). In
the present study, we induced Notch1 signaling by transfecting
cardiomyocytes with a lentiviral vector containing Notch1
intracellular domain (N1ICD) in ain aim to protect the cells
against high glucose (HG)-induced damage. The HG-induced
cardiomyocyte damage was more severe when Notch1 signaling was
inhibited using DAPT. To date, the effects of Notch1 signaling in
DCM have not been fully determined. It is in the interest of the
research community to elucidate whether the regulation of Notch1
signaling has a protective effect on myocardial cells.
Based on the pathogenesis and pathological changes
associated with DCM, in this study, we performed experiments using
H9c2 cells exposed to HG to create a model of myocardial injury in
order to investigate the role of Notch1 in, and its effects on
HG-induced cardiomyocyte proliferation, apoptosis and myocardial
fibrosis.
Materials and methods
Cell culture
H9c2 cardiomyocytes were purchased from the Cell
Resource Center, Shanghai Institutes for Biological Sciences,
Chinese Academy of Sciences (Shanghai, China). The H9c2 cells were
cultured in culture plates with DMEM supplemented 10%
heat-inactivated fetal bovine serum (FBS). The mixture was placed
in a humidified incubator filled with 5% CO2 at 37°C.
The cell nutrition medium was changed every 2–3 days, and the cells
were subcultured once they covered 85–90% of the bottom of the
culture plates. The cells were cultured under normoxic conditions
with normal glucose (NG; 5.5 mmol/L-NG) for 2–3 days, before they
were divided into 6 groups as folllows: i) the control group: H9c2
cells were cultured under normoxic conditions for 72 h, without any
treatment; ii) the mannitol group: H9c2 cells were cultured in
mannitol (33 mmol/l; Genechem Co., Ltd., Shanghai, China) for 72 h
(mannitol was added to rule out the effect of osmolarity in cell
culture studies using high glucose); iii) the HG group: H9c2 cells
were cultured in HG (33 mmol/L-HG) for 72 h; iv) the DAPT group:
H9c2 cells were cultured in HG for 24 h, followed by the addition
of DAPT (50 µmol/l; Shanghai Genechem Co., Ltd.) and culture
for a further 48 h; v) the Notch1 group: H9c2 cells were
transfected with the lentiviral vector containing N1ICD
(lentivirus-N1ICD; Shanghai Genechem Co., Ltd.) for 8 h and were
then cultured in HG medium for a further 72 h; and vi) the mock
group: H9c2 cells were transfected with an empty lentiviral vector
for 8 h and were then cultured in HG medium for a further 72 h. The
resulting cells from each group were used in the following
experiments.
Assessment of cell viability
The above-mentioned H9c2 cells were incubated in
96-well plates with 3,000 cells in each well, and this was followed
by Cell Counting kit-8 (CCK-8) assay (CCK-8 solution, 10
µl/well). The absorbance at 450 nm was measured using a
Multiscan microplate reader at 1, 2, 3 and 4 h. The cell viability
for the control group was set at 100%, while the viability for the
other groups was expressed as a percentage of the control
group.
Measurement of cell apoptosis
The H9c2 cells were incubated in 6-well plates. The
cells were digested and the supernatant was transferred to a 1-ml
Eppendorf tube and centrifuged at 1,000 rpm at 4°C for 5 min. The
resultant supernatant was then discarded, and the precipitates were
re-suspended in 500 µl sterile phosphate-buffered saline
(PBS). The suspension was centrifuged twice at 1,000 rpm at 4°C for
5 min. The final precipitates were re-suspended in 200 µl
binding buffer, followed by the addition of 10 µl Annexin
V-FITC and 10 µl propidium iodide (PI; Nanjing KeyGen
Biotech Co., Ltd., Nanjing, China). The mixture was vortexed gently
in a dark environment at room temperature for 15 min. Another 300
µl binding buffer was added, and the apoptotic rate was
measured by flow cytometry within 1 h.
Quantitative PCR (qPCR)
qPCR was performed using a 7500 Real-Time PCR system
(Applied Biosystems, Carlsbad, CA, USA), using All-in-One qPCR mix
(GeneCopoeia, Rockville, MD, USA). The qPCR reaction volume was 20
µl, containing 2 µl of cDNA. The experiment was
carried out according to the manufacturer's instructions. Relative
mRNA expression was calculated using the comparative threshold
cycle method, as previously described (15). Sequences for specific primers were
as follows: Hes-1 forward, 5′-TCAACACGACACCGGATAAA-3′ and reverse,
5′-TCAGCTGGCTCAGACTTTCA-3′; Hey-1 forward,
5′-GCGTTATCTGAGCATCATTGAAGG′ and reverse,
5′-CTGGGAAGCGTAGTTGTTGAGA-3′; GAPDH forward,
5′-GCAAGTTCAACGGCACAG-3′ and reverse,
5′-GCCAGTAGACTCCACGACAT-3′.
Western blot analysis
The cells were lysed with protein lysate, and 50 mg
protein from each group were analyzed using 10% SDS-PAGE under AU9
reducing conditions. The separated proteins were transferred onto a
nitrocellulose membrane (Millipore, Billerica, MA, USA) and the
membrane was blocked with 10% non-fat milk. The antibodies used
were as follows: anti-N1ICD (Beijing Zhongshan Golden Bridge
Biotechnology Co., Ltd., Beijing, China), anti-Bax, anti-B-cell
lymphoma-2 (Bcl-2; both from Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA), anti-transforming growth factor-β1 (TGF-β1) and
anti-connective tissue growth factor (CTGF; both from Wuhan Boster
Biological Engineering Co., Ltd., Wuhan, China),
anti-phosphorylated (p-) PI3K, anti-total (t-)PI3K, anti-p-AKT and
anti-t-AKT (all from Santa Cruz Biotechnology, Inc.) and
anti-β-actin (Beijing Zhongshan Golden Bridge Biotechnology Co.,
Ltd.). The reaction mixtures were incubated for 12 h at 4°C and
then for a further 4 h at room temperature in a shaker with the
secondary antibodies [goat anti-rabbit or goat anti-mouse IgG
(Zhongshan Goldenbridge Biotechnology Co., Beijing, China), goat
anti-rabbit or goat anti-mouse IgG (Histostain-Plus kits; ZsBio)].
The resulting mixture was washed 3 times with TBST, and the blots
were visualized by enhanced chemiluminescence.
Statistical analyses
All data were analyzed using SPSS 19.0 statistical
software (SPSS, Inc., Chicago, IL, USA). Experimental data are
expressed as the means ± SD. Comparisons between groups were
compared by one-way analysis of variance (ANOVA). A value of
P<0.05 was considered to indicate a statistically significant
difference.
Results
Notch1 signaling is activated upon
HG-induced cardiomyocyte injury
To prove that Notch1 signaling plays an important
role in HG-induced cardiomyocyte injury, we examined alterations in
the expression of N1ICD and its target genes, Hes-1 and Hey-1. As
shown in Fig. 1, the N1ICD, Hes-1
and Hey-1 expression levels were lower in the HG, DAPT and mock
groups. The N1ICD, Hes-1 and Hey-1 expression levels were
significantly higher in the Notch1 group (cells transfected with
lentivirus-N1ICD prior to exposure to HG). However, in the DAPT
group, the N1ICD, Hes-1 and Hey-1 expression levels were
significantly decreased. Therefore, these data indicate that Notch1
signaling is activated upon HG-induced cardiomyocyte injury.
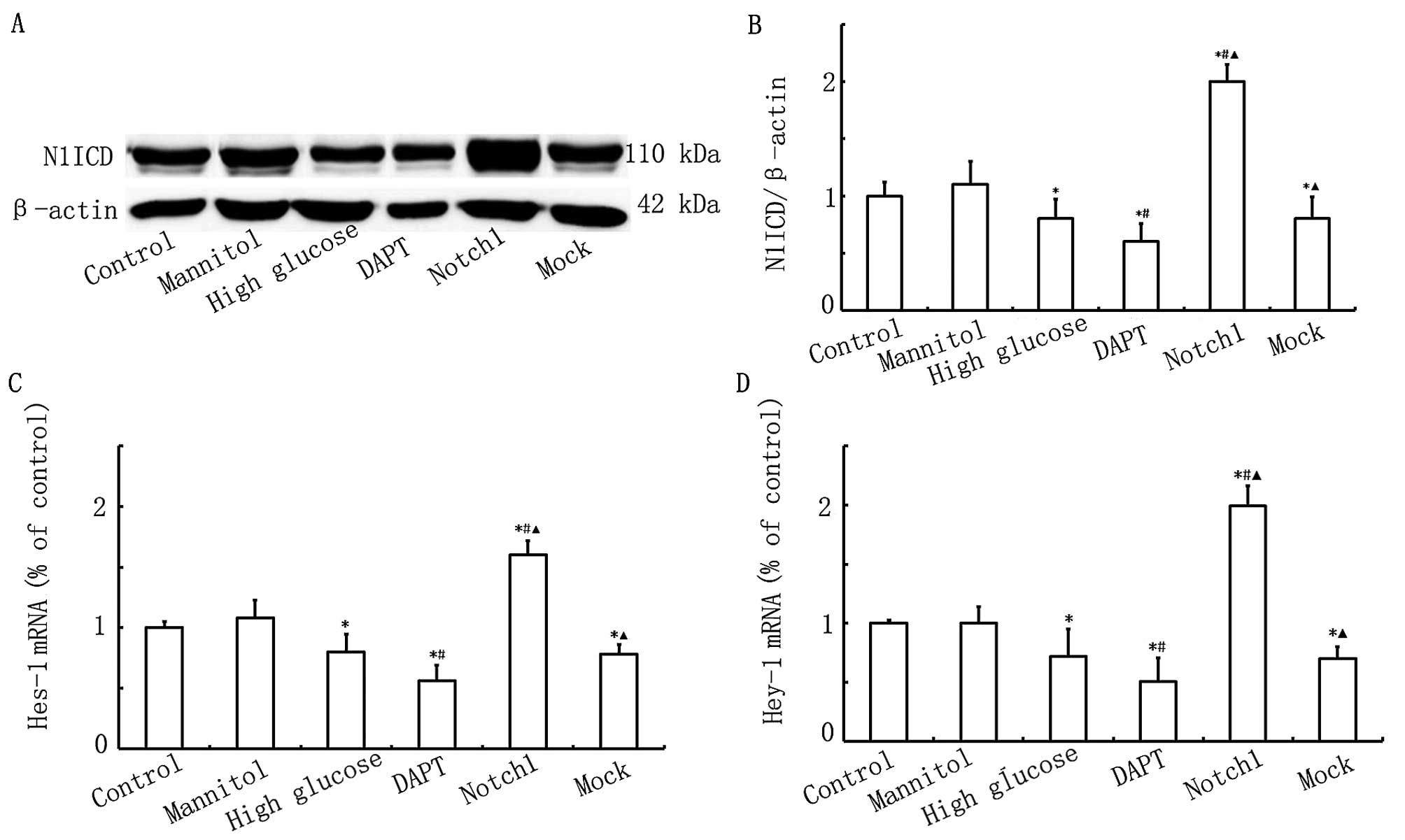 | Figure 1Notch1 signaling is activated upon
high glucose (HG)-induced myocardial cell injury. (A) Western blot
analysis of Notch1 intracellular domain (N1ICD) expression in the
control, mannitol, HG, DAPT, Notch1 and mock groups. (B)
Quantitative analysis of the N1ICD level. The mean density of N1ICD
in the control group was set as 100% (n=7, *P<0.05
vs. control group, #P<0.05 vs. HG group,
▲P<0.05 vs. DAPT group). (C and D) qPCR of
hairy/enhancer of split 1 (Hes-1) and hairy/enhancer-of-split
related with YRPW motif 1 (Hey-1) mRNA expression in the control,
mannitol, HG, DAPT, Notch1 and mock groups (n=7,
*P<0.05 vs. control group, #P<0.05 vs.
HG group, ▲P<0.05 vs. DAPT group). |
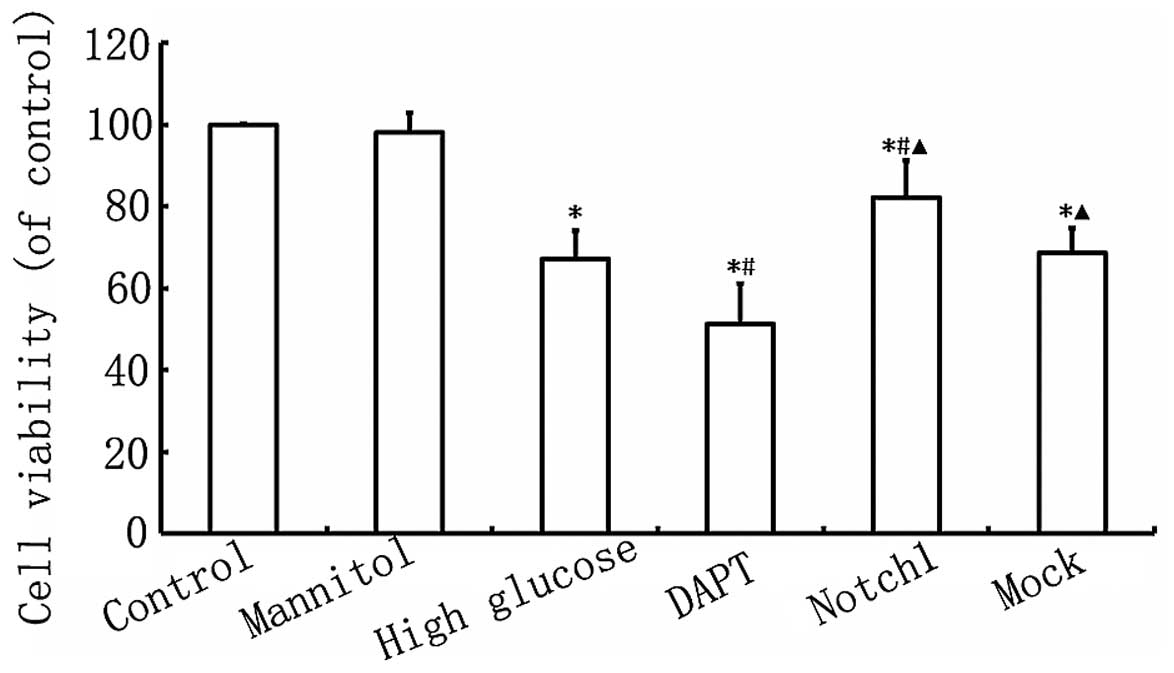
Notch1 signaling enhances H9c2 cell
viability following myocardial injury
As shown in Fig.
2, myocardial cell viability in the HG group was lower
(P<0.05) than that in the control group. Furthermore, myocardial
cell viability in the DAPT group was even lower than that in the HG
group (P<0.05), while myocardial cell viability was
significantly higher in the Notch1 group, compared with the HG
group (P<0.05; Fig. 2). These
findings indicate that the activation of the Notch1 signaling
pathway enhances the viability of H9c2 cardiomyocytes exposed to
HG.
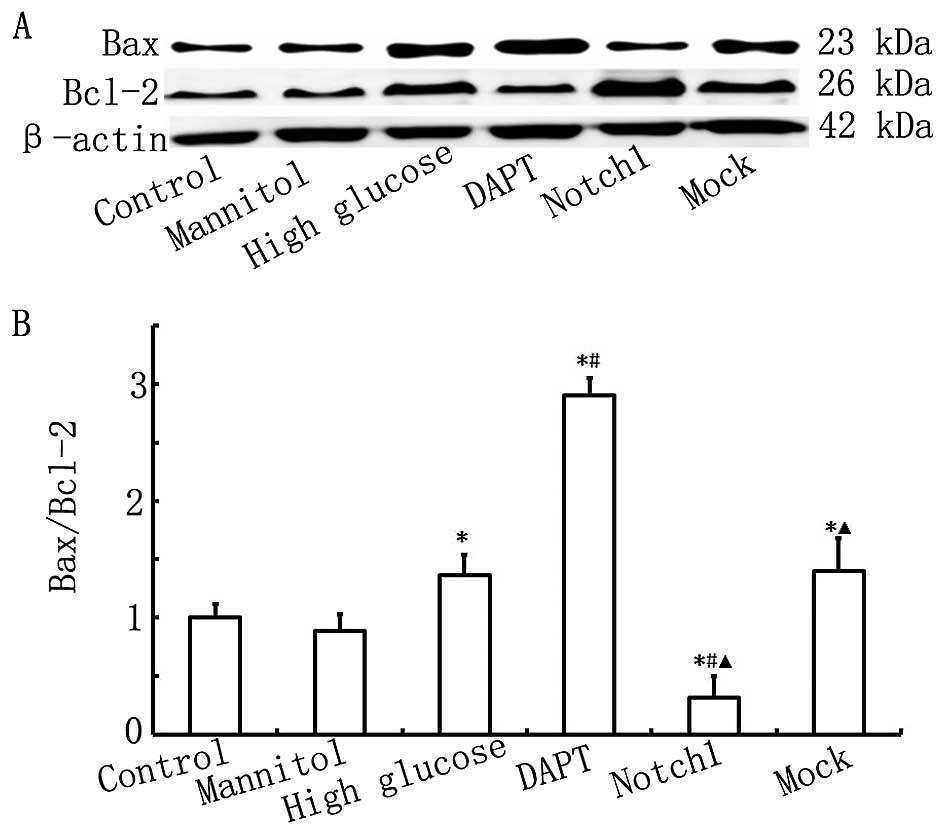
Notch1 signaling inhibits HG-induced H9c2
cell apoptosis
To confirm the protective effects of Notch1
signaling against HG-induced H9c2 cell damage, cell apoptosis was
determined by measuring the protein expression levels of Bax and
Bcl-2 by western blot analysis, and by Annexin V-FITC/PI double
staining and flow cytometry. The results from western blot analysis
and flow cytometry are presented in Figs. 3 and 4. As shown in Fig. 3, the ratio of Bax to Bcl-2 in the
HG group was higher than that in the control group (P<0.05).
Furthermore, the ratio of Bax to Bcl-2 in the DAPT group was even
higher than that in the HG group (P<0.05), while the ratio of
Bax to Bcl-2 was significantly lower in the Notch1 group compared
with that in the HG group (P<0.05). Our findings indicate that
exposure to HG induces H9c2 myocardial cell apoptosis and that this
is inhibited by the activation of Notch1 signaling.
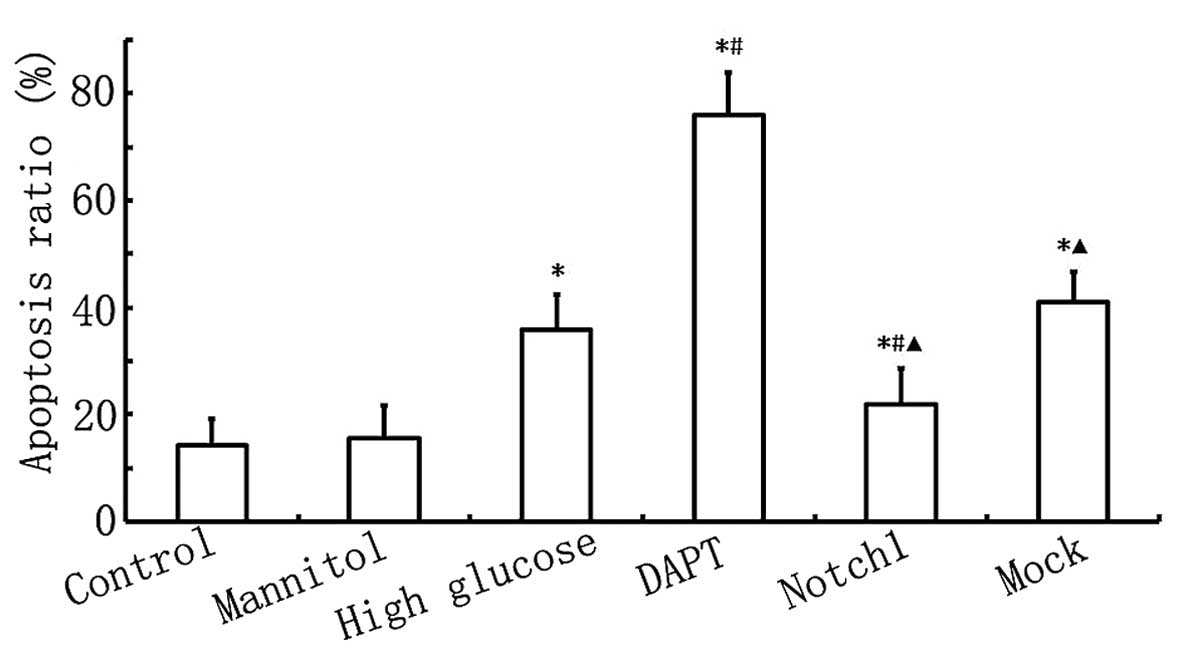
As shown in Fig.
4, the apoptotic rate in the HG group was higher than that in
the control group (P<0.05), and the apoptotic rate in the DAPT
group was higher than that in the HG group (P<0.05). When Notch1
signaling was activated following transfection with
lentivirus-N1ICD, the apoptotic rate decreased significantly and
was lower than that in the HG group (P<0.05). These results
further confirm that the activation of the Notch1 signaling pathway
inhibits HG-induced cardiomyocyte apoptosis.
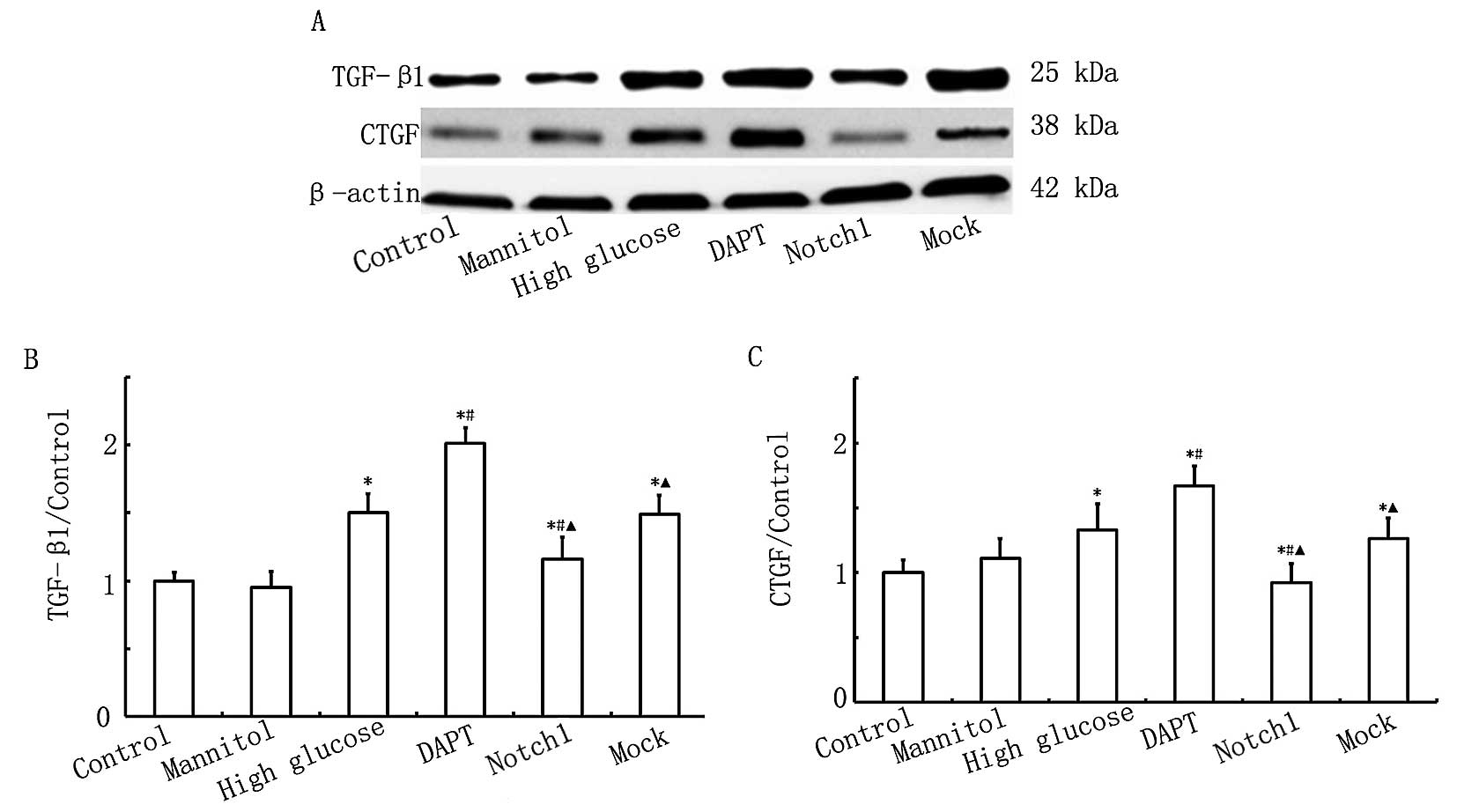
Activation of Notch1 signaling decreases
the expression of TGF-β1 and CTGF induced in H9c2 myocardial cells
by exposure to HG
To further confirm the protective effects of Notch1
signaling in H9c2 cells exposed to HG, the protein expression
levels of TGF-β1 and CTGF were measured by western blot analysis.
Both proteins are important factors in myocardial fibrosis
(16). As shown in Fig. 5, the expression levels of TGF-β1
and CTGF were higher (P<0.05) in the HG group compared with
those in the control group. The protein expression levels of TGF-β1
and CTGF were even higher (P<0.05) in the DAPT group compared
with those in the HG group, while these levels were significantly
lower (P<0.05) in the Notch1 group compared with those in the HG
group. The above findings indicate that exposure to HG increases
the expression of TGF-β1 and CTGF in H9c2 myocardial cells, and
that the activation of Notch1 signaling inhibits this increase in
the expression of pro-fibrotic factors.
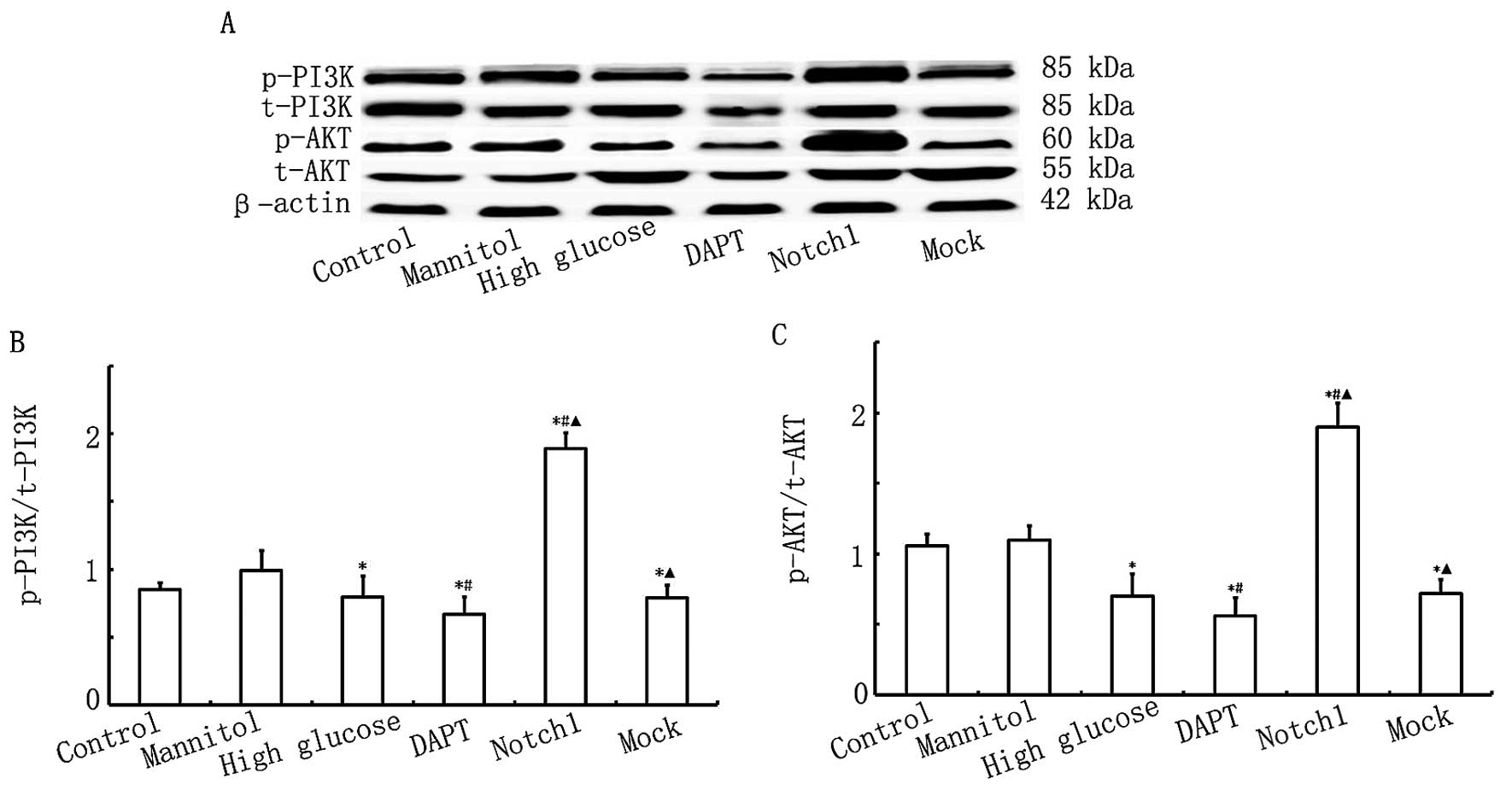
Overexpression of Notch1 activates the
PI3K/AKT pathway
To determine the cardioprotective effects of the
Notch1 signaling pathway in relation to HG-induced H9c2 cell
damage, we measured the expression levels of p-AKT/t-AKT and
p-PI3K/t-PI3K. As shown in Fig.
6, no significant differences were observed in the levels of
t-AKT between the different groups. Ηowever, AKT activity was lower
in the HG, DAPT and mock groups, as indicated by the downregulation
in the levels of p-AKT, which were lower in the DAPT group but
higher in the Notch1 group. As shown in Fig. 6, the levels of p-PI3K were lower
in the HG, DAPT and mock groups, and were particularly lowe in the
DAPT group, whereas in the Notch1 group, the level of p-PI3K was
significantly higher. These data suggest that the activation of
Notch1 signaling in turn activates the PI3K/AKT pathway. Our data
also suggest that the acvitaion of Notch1 signaling exerts
cardioprotective effects, possibly through cross-talk with the
PI3K/AKT signaling pathway.
Discussion
Our study demonstrated that HG-induced cardiomyocyte
apoptosis led to an increase in TGF-β1 and CTGF expression in the
H9c2 cells. Our findings also indicated that the over-expression of
Notch1 prevented HG-induced cardiomyocyte apoptosis and decreased
CTGF expression in the H9c2 cells exposed to HG; thus, Notch1 plays
a protective role and may reduce the severity of cardiac
fibrosis.
Despite the fact that the Notch1 signaling pathway
plays an important role in cardiovascular disease, to the best of
our knowledge, prior to this study, there was no in-depth report on
the role of Notch1 signaling in DCM. Our findings demonstrated that
the overexpression of Notch1 exerted therapeutic effects against
HG-induced H9c2 cardiomyocyte injury. Our data suggest that Notch1
lentiviral gene transduction into myocardial cells is feasible and
beneficial, and that it is possible to develop novel
Notch1-dependent therapeutic strategies for DCM.
Previous studies have confirmed that there is a
complex association between Notch1 signaling and the PI3K/AKT
pathway, and that the effects of the cross-talk between these
pathways are also quite complex (17–20). Our study demonstrated that
following the transfection of H9c2 cardiomyocytes with
lentivirus-N1ICD, the expression of Notch1 increased and this led
to the activation of the PI3K/AKT pathway. It has been demonstrated
that the Notch1 targt gene, Hes-1, activates the PI3K/AKT pathway
by negatively regulating PTEN. This downregulation is achieved
mainly through the upregulation of receptor tyrosine kinases (RTKs)
and the downregulation of PTEN. PI3K is phosphorylated, while
p-PI3K simultaneously gathers and phosphorylates AKT (21). p-AKT further regulates downstream
apoptotic factors, such as the Bcl-2 family, caspase-9 and -3, and
X-linked inhibitor of apoptosis protein (XIAP). Among them, the AKT
dependence of the Bcl-2 family is critical during the pathogenesis
of apoptosis (22–24).
Myocardial interstitial fibrosis is a key risk
factor in the progression of DCM. Cardiac fibroblast proliferation
and activation lead to the secretion of a large amount of collagen
types I and III, resulting in the accumulation and imbalance of
those collagens in myocardial tissue (11). TGF-β1 has been the focus of recent
research and has been proven to be a regulatory element in many
tissues and cell fibrosis (25,26). It has been reported that TGF-β1
signal transduction plays an important role in a number of fibrotic
diseases by activating Smad protein (27,28). In addition, TGF-β1 is thought to
be involved in the transformation of fibroblasts by upregulating
the expression of TGF-β1 and CTGF in rat pulmonary fibrosis
(26). Activated TGF-β1
contributes to the pathogenesis of the fibrotic interstitium
observed in DCM. Furthermore, HG enhances the activity of the
transcriptional co-activator, p300, leading to the activation of
TGF-β via the acetylation of Smad2 (29).
During myocardial injury, several signaling pathways
function together and affect each other. The TGF-β1 pathway
activates the Notch1 signaling pathway, while the activated Notch1
signaling pathway inhibits the TGF-β1 pathway. Sassoli et al
(30) reported that the hormone
relaxin (RLX) downregulated the expression of TGF-β1 and type I
collagen by activating the Notch1 signaling pathway, which further
inhibited TGF-β1-mediated fibroblast myofibroblast transition.
However, the inhibitory effects of RLX decreased significantly
following intervention with the γ-secretase inhibitor, DAPT.
Furthermore, it has been suggested that the activation of the
PI3K/AKT pathway directly inhibits the expression of TGF-β1, and
thus inhibits myocardial fibrosis (31,32).
The present study confirmed that TGF-β1 and CTGF are
both expressed in H9c2 cells. The epxosure of H9c2 cells to HG led
to the upregulation of the expression of pro-fibrotic factors,
which may result in cardiac fibrosis. Our findings further
demonstrated that the overexpression of Notch1 in H9c2 cells led to
lower expression levels of CTGF, which were induced by TGF-β1.
These results suggest that Notch1 is involved in cardiac
remodeling, acting as a fibrogenic mediator of TGF-β1 and affecting
the cardiac myoblasts. We hypothesized that the mechanism
responsible for this is that the overexpression of Notch1 activated
the PI3K/AKT pathway, which further inhibited the expression of
TGF-β1 and eventually downregulated the expression of CTGF.
In recent years, the Notch1 signaling pathway has
attracted widespread attention (10–15,17–20,33). However, previous studies in this
field have mainly focused on the cellular (10,12,20) and animal levels (11,19,33) and no research has been carried out
at the human clinical trial level. In our study, human H9c2 cells
from were used to create a model of myocardial injury. Using this
model, we investigated protective effects of Notch1 against
myocardial injury as well as its mechanisms of action. That being
said, as our study was carried out at the in vitro level and
the in vivo environment is more complex, further studies are
warranted to determine whether Notch1 would have the same
protective effect in vitro.
In conclusion, this study explored the protective
effects of Notch1 signaling against HG-induced myocardial injury
in vitro, as well as its mechanism of action, and laid the
foundation for future in vitro experiments. Our data may aid
the development of novel treatment strategies for DCM. As the
processes of myocardial apoptosis and fibrosis are regulated by a
complex system of cytokines and signaling pathways, further
clinical studies are required to confirm the safety and efficacy of
any new treatment strategy for DCM.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (81041097) and the Natural Science
Foundation of Jiangxi Province (20142BAB205040). We thank Dr Wan
Zhang and Dr Junyi Zeng for expert technical assistance and
critical comments on the manuscript.
References
|
1
|
Fiordaliso F, De Angelis N, Bai A,
Cuccovillo I, Salio M, Serra DM, Bianchi R, Razzetti R, Latini R
and Masson S: Effect of beta-adrenergic and renin-angiotensin
system blockade on myocyte apoptosis and oxidative stress in
diabetic hypertensive rats. Life Sci. 81:951–959. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bell DS: Diabetic cardiomyopathy. Diabetes
Care. 26:2949–2951. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wen HL, Liang ZS, Zhang R and Yang K:
Anti-inflammatory effects of triptolide improve left ventricular
function in a rat model of diabetic cardiomyopathy. Cardiovasc
Diabetol. 12:502013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Leong KG and Karsan A: Recent insights
into the role of Notch signaling in tumorigenesis. Blood.
107:2223–2233. 2006. View Article : Google Scholar
|
|
5
|
Ji X, Wang Z, Geamanu A, Sarkar FH and
Gupta SV: Inhibition of cell growth and induction of apoptosis in
non-small cell lung cancer cells by delta-tocotrienol is associated
with notch-1 down-regulation. J Cell Biochem. 112:2773–2783. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cook KM and Figg WD: Angiogenesis
inhibitors: current strategies and future prospects. CA Cancer J
Clin. 60:222–243. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lefort K and Dotto GP: Notch signaling in
the integrated control of keratinocyte growth/differentiation and
tumor suppression. Semin Cancer Biol. 14:374–386. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sjölund J, Manetopoulos C, Stockhausen MT
and Axelson H: The Notch pathway in cancer: differentiation gone
awry. Eur J Cancer. 41:2620–2629. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
MacGrogan D, Nus M and de la Pompa JL:
Notch signaling in cardiac development and disease. Curr Top Dev
Biol. 92:333–365. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Collesi C, Zentilin L, Sinagra G and
Giacca M: Notch1 signaling stimulates proliferation of immature
cardiomyocytes. J Cell Biol. 183:117–128. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nemir M, Metrich M, Plaisance I, Lepore M,
Cruchet S, Berthonneche C, Sarre A, Radtke F and Pedrazzini T: The
Notch pathway controls fibrotic and regenerative repair in the
adult heart. Eur Heart J. 269:715–727. 2012.
|
|
12
|
Zhou XL, Wan L and Liu JC: Activated
Notch1 reduces myocardial ischemia reperfusion injury in vitro
during ischemic postconditioning by crosstalk with the RISK
signaling pathway. Chin Med J (Engl). 126:4545–4551. 2013.
|
|
13
|
Li Y, Hiroi Y and Liao JK: Notch signaling
as an important mediator of cardiac repair and regeneration after
myocardial infarction. Trends Cardiovasc Med. 20:228–231. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gutierrez A and Look AT: NOTCH and
PI3K-AKT pathways intertwined. Cancer Cell. 12:411–413. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li Y, Li B, Zhang C, Zhang J, Zeng M and
Zheng Z: Effect of NRG-1/ErbB signaling intervention on the
differentiation of bone marrow stromal cells into sinus node-like
cells. J Cardiovasc Pharmacol. 63:434–440. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Matsumoto Y, Niimi N and Kohyama K:
Characterization of fibrosis-promoting factors and siRNA-mediated
therapies in C-protein-induced experimental autoimmune myocarditis.
Cell Immunol. 279:70–77. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang H, Cheng H, Shao Q, Dong Z, Xie Q,
Zhao L, Wang Q, Kong B and Qu X: Leptin-promoted human extravillous
trophoblast invasion is MMP14 dependent and requires the cross talk
between Notch1 and PI3K/Akt signaling. Biol Reprod. 90:78–86. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xiao W, Chen X and He M: Inhibition of the
Jagged/Notch pathway inhibits retinoblastoma cell proliferation via
suppressing the PI3K/Akt, Src, p38MAPK and Wnt/β catenin signaling
pathways. Mol Med Rep. 10:453–458. 2014.PubMed/NCBI
|
|
19
|
Gude NA, Emmanuel G, Wu W, Cottage CT,
Fischer K, Quijada P, Muraski JA, Alvarez R, Rubio M, Schaefer E
and Sussman MA: Activation of Notch-mediated protective signaling
in the myocardium. Circ Res. 102:1025–1035. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang XM, Yao M, Liu SX, Hao J, Liu QJ and
Gao F: Interplay between the Notch and PI3K/Akt pathways in high
glucose-induced podocyte apoptosis. Am J Physiol Renal Physiol.
306:F205–F213. 2014. View Article : Google Scholar
|
|
21
|
Xie F, Su M, Qiu W, Zhang M, Guo Z, Su B,
Liu J, Li X and Zhou L: Kaempferol promotes apoptosis in human
bladder cancer cells by inducing the tumor suppressor, PTEN. Int J
Mol Sci. 14:21215–21226. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Katare RG, Kakinuma Y, Arikawa M, Yamasaki
F and Sato T: Chronic intermittent fasting improves the survival
following large myocardial ischemia by activation of BDNF/VEGF/PI3K
signaling pathway. J Mol Cell Cardiol. 46:405–412. 2009. View Article : Google Scholar
|
|
23
|
Tiwari RV, Parajuli P and Sylvester PW:
γ-Tocotrienol-induced autophagy in malignant mammary cancer cells.
Exp Biol Med (Maywood). 239:33–44. 2014. View Article : Google Scholar
|
|
24
|
Chang TH, Liu XY, Zhang XH and Wang HL:
Effects of dl-praeruptorin A on interleukin-6 level and Fas, bax,
bcl-2 protein expression in ischemia-reperfusion myocardium. Acta
Pharmacol Sin. 23:769–774. 2002.PubMed/NCBI
|
|
25
|
Yi X, Li X, Zhou Y, Ren S, Wan W, Feng G
and Jiang X: Hepatocyte growth factor regulates the TGF-β1-induced
proliferation, differentiation and secretory function of cardiac
fibroblasts. Int J Mol Med. 34:381–390. 2014.PubMed/NCBI
|
|
26
|
Zhang L, Li Y, Liang C and Yang W: CCN5
overexpression inhibits profibrotic phenotypes via the PI3K/Akt
signaling pathway in lung fibroblasts isolated from patients with
idiopathic pulmonary fibrosis and in an in vivo model of lung
fibrosis. Int J Mol Med. 33:478–486. 2014.
|
|
27
|
Wang M, Zhao D, Spinetti G, Zhang J, Jiang
LQ, Pintus G, Monticone R and Lakatta EG: Matrix metalloproteinase
2 activation of transforming growth factor-β1 (TGF-β1) and
TGF-β1-type II receptor signaling within the aged arterial wall.
Arterioscler Thromb Vasc Biol. 26:1503–1509. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Beaumont J, López B, Hermida N, Schroen B,
San José G, Heymans S, Valencia F, Gómez-Doblas JJ, De Teresa E,
Díez J and González A: microRNA-122 down-regulation may play a role
in severe myocardial fibrosis in human aortic stenosis through
TGF-β1 up-regulation. Clin Sci (Lond). 126:497–506. 2014.
View Article : Google Scholar
|
|
29
|
Bugyei-Twum A, Advani A, Advani SL, Zhang
Y, Thai K, Kelly DJ and Connelly KA: High glucose induces Smad
activation via the transcriptional coregulator p300 and contributes
to cardiac fibrosis and hypertrophy. Cardiovasc Diabetol.
13:892014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sassoli C, Chellini F, Pini A, Tani A,
Nistri S, Nosi D, Zecchi-Orlandini S, Bani D and Formigli L:
Relaxin prevents cardiac fibroblast-myofibroblast transition via
notch-1-mediated inhibition of TGF-β/Smad3 signaling. PLoS One.
8:e638962013. View Article : Google Scholar
|
|
31
|
Voloshenyuk TG, Landesman ES, Khoutorova
E, Hart AD and Gardner JD: Induction of cardiac fibroblast lysyl
oxidase by TGF-β1 requires PI3K/Akt, Smad3, and MAPK signaling.
Cytokine. 55:90–97. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chung EJ, Sohn YH, Kwon SH, Jung SA and
Lee JH: Lithium chloride inhibits TGF-β1-induced myofibroblast
transdifferentiation via PI3K/Akt pathway in cultured fibroblasts
from Tenon's capsule of the human eye. Biotechnol Lett.
36:1217–1224. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu J, Dong F, Jeong J, Masuda T and Lobe
CG: Constitutively active Notch1 signaling promotes
endothelial-mesenchymal transition in a conditional transgenic
mouse model. Int J Mol Med. 34:669–676. 2014.PubMed/NCBI
|