Introduction
Hepatitis C virus (HCV) is a single-strand RNA virus
which belongs to the Flaviviridae family. The HCV genome is
approximately 9.6 kb in length with 5′ and 3′ non-coding regions
and a single open reading frame (1). The high mutation frequency of HCV
which often occurs during viral replication is due to the lack of
proofreading activity of RNA polymerase, thus contributing to the
genotyping of HCV. There are 6 genotypes and >90 subgenotypes of
HCV with regional differences (2). The geographical distribution of the
main HCV genotypes is as follows (3): 1a in USA, 2a and 2b in North America
and Japan, 3 in India, 4 in the Middle East and North Africa, 5 and
6 in Hong Kong, and 1b and 2a in China (4–10).
The differences in routes of HCV transmission based on differences
in regional genotypes may aid in epidemiological investigations and
source tracing (11).
HCV genotypes greatly influence the effects of
antiviral therapy and the sustained virological response (SVR)
(12). HCV genotyping is
important for designing an antiviral therapy plan and predicting
the effects of antiviral therapy. Pegylated interferon (PEG-IFN)-α
in combination with ribavirin (RBV) is the standard therapy for
chronic HCV infection. Previous studies have indicated that
patients with genotypes 2 and 3 have a higher SVR to PEG-IFN-α/RBV
therapy than patients with genotype 1 (13,14).
The differences in HCV genotypes significantly
contribute to the immune escape and sustained infection and these
differences have greatly impeded the development of HCV vaccines
(15). Thus, the identification
of HCV genotypes and subgenotypes in different regions is critical
to epidemiological investigation, diagnosis, vaccine development
and clinical therapy. There are currently 5 methods used for the
identification of the HCV genotype: nucleotide sequence analysis
(16), specific primer
amplification (17), probe
hybridization (18), restriction
fragment length polymorphism (19) and phylogenetic analysis (20). According to a new agreement of HCV
genotype naming rules published in 2005 (20), phylogenetic analysis was
considered the most accurate method for the identification of HCV
genotypes. With phylogenetic analysis, PCR amplification, fragment
sequencing and phylogenetic analysis are conducted on the specific
regions of HCV genes to identify HCV genotypes and
subgenotypes.
In the present study, the sequences from the core,
E1, E2 and NS5B regions reported previously (21) were selected and examined by
phylogenetic analysis to confirm whether the subgenotype
classification of specific regions based on phylogenetic analysis
could replace whole genome analysis in the identification of HCV
genotypes. The established classification based on specific regions
was then used in the analysis of the sequences from patients in
Sichuan province.
Materials and methods
Construction of phylogenetic trees based
on sequences from the HCV sequence database
The sequences of the core, E1, E2 and NS5B regions
and the total HCV genome, which have been reported in different
countries, were selected from the HCV sequence database (http://hcv.lanl.gov/content/sequence/NEWALIGN/align.html).
MEGA 5.0 was used to compare all the sequences, and phylogenetic
trees were constructed using the neighbor-joining method (MEGA
5.0), and the reliability of the trees was evaluated by the
bootstrap method with 1,000 replications.
Samples
A total of 153 blood samples were obtained from
HCV-positive patients with HCV RNA >104 IU/ml at
either Chengdu Infectious Diseases Hospital, the Affiliated
Hospital of Luzhou Medical College or the Second People's Hospital
of Yibin (both in Sichuan, Southwest China). All patients provided
written informed consent. Patients with hepatitis A, B and
autoimmune hepatitis were excluded from this study.
RNA extraction and RT-PCR
The Hepatitis C virus nucleotide quantitative
detection kit (20131201/1; Qiagen Shenzhen Co., Ltd., Shenzhen,
China) was used to isolate and measure the HCV RNA levels. HCV RNA
was amplified using a reverse transcription kit (AK5401 Takara
Biotechnology Co., Ltd., Dalian, China) according to the
manufacturer's instructions. The primers used and the amplification
procedure were as previously described (11). The sequencing for target fragments
was performed by BioSune Biotechnology Co., Ltd. (Shanghai,
China).
HCV sequence analysis
All HCV sequences were edited using BioEdit
software. To avoid potential laboratory errors, the Basic Local
Alignment Search Tool (BLAST) and phylogenetic analysis were used
to identify HCV genotypes. All the nucleotide sequences obtained in
this study were screened using the online BLAST search tool
(http://blast.ncbi.nlm.nih.gov/Blast.cgi) to search for
sequence similarities to previously reported sequences in the
database. The classification of HCV genotypes and subgenotypes was
according to the Consensus proposals for a unified system of
nomenclature of hepatitis C virus genotypes (20). The phylogenetic tree was
constructed using MEGA 5.0 software. The reference sequences were:
1b: CN. BJ.HQ639947; CN.BJ.JX961151; CN.HN.JX961093; CN.js.
JQ303617; CN.js.JQ303531; CN.KC844051; CN.YN. FJ462981;
JP.x.JT.D11355; 1b: USA.EU256090; 2a: JP.x.AY746460;
JP.x.HC-J6.D00944; 3a: CN.bj.HQ639941; CN.hb.KF292145;
CN.SH.HQ912953; CN.ZJ. HQ318890; Creteil.AM423014;.FR.FJ872277;
JP.NC_009824; 3b: 3b.CN.EU081507; 3b.CN. FJ462985;
3b.CN.HK.KC441470; CN.js.JQ303583; CN.sh. JQ065709; CN.yn.
EU081464; CN.YN. FJ462950; CN.YN. FJ462973; CN.YN.FJ462968;
CN.YN.FJ462999; 6a: CN. HK.DQ480520; CN.hk.KC441477;
CN.hk.KC441481; CN.js. JQ303556; JCN.js.JQ303557; 6a.CN.KC844037;
HK.x.6a33. AY859526; 6b: x.x.Th580.NC_009827; 6n: USA.DQ835768.
Results
NS5B could replace whole genome
sequencing (WGS) to classify HCV subgenotypes
Phylogenetic trees were constructed to identify the
correct method for the classification of HCV subgenotypes. All
analyzed sequences were from the HCV database, including whole
genome sequences (n=470), core region sequences (n=781), E1 region
sequences (n=899), E2 region sequences (n=627) and NS5B region
sequences (n=466). The results indicated that the phylogenetic tree
of the whole genome sequences divided all subgenotypes correctly
(Fig. 1). The analysis of the
NS5B region sequences revealed that subgenotype classification
based on the NS5B-tree could replace the WGS method as all analyzed
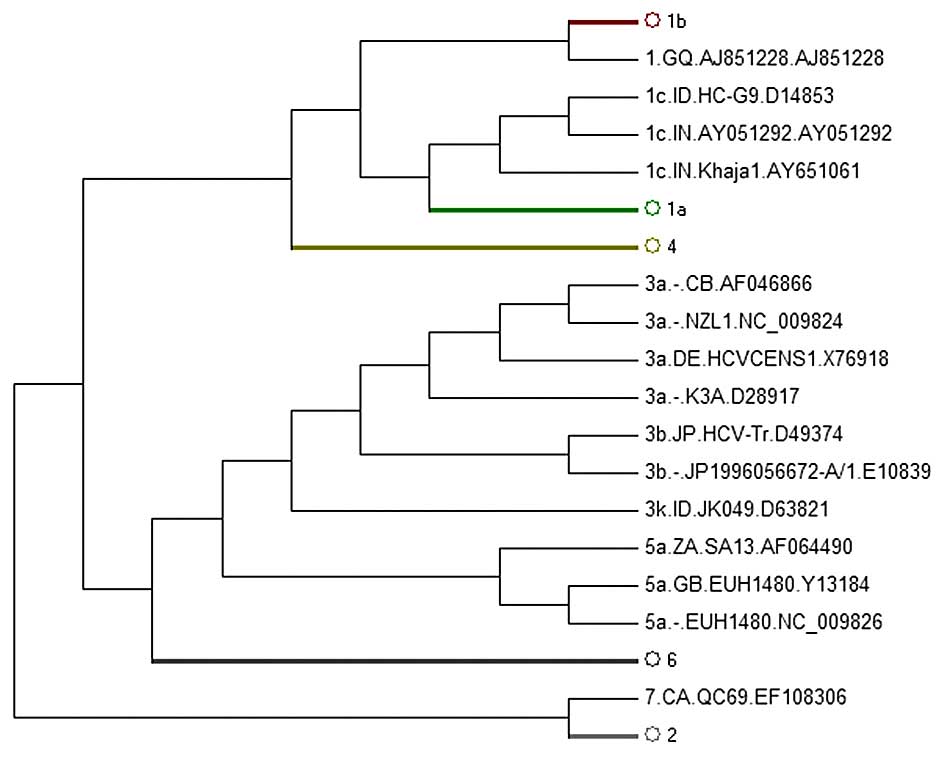
sequences were separated correctly (Fig. 2). We did not display the figure of
phylogenetic subtree strains of the 1b, 1a, 4, 2, 6 subgenotypes as
they did not reveal the abnormal sequences.
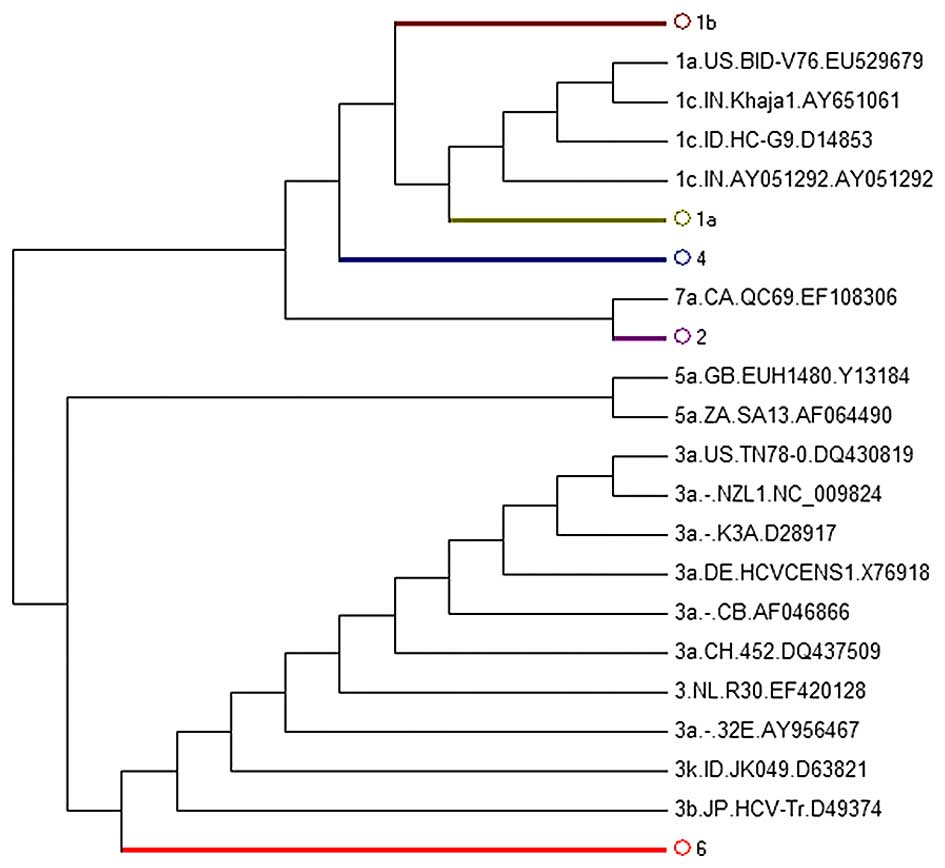
However, the phylogenetic trees of the other
regions, including the core, E1 and E2 regions, failed to
distinguish all subgenotypes. Among these, the core-tree failed to
separate subgenotypes 1c and 1a. 1c AY651061 was in 1a (Fig. 3B), whereas in the E1-tree, 1b
AY746882 was located in 1a (Fig.
4B) and 1a AY388455 was in 1b (Fig. 4C). It was also found that E2 could
not replace WGS for subgenotype classification as the E2-tree
failed to separate subgenotypes 1c and 1a; 1a EU529679 was located
in 1c (Fig. 5). We did not
display the figure of phylogenetic subtree strains of the 4, 3, 2,
5, 6 subgenotypes as they did not reveal the abnormal
sequences.
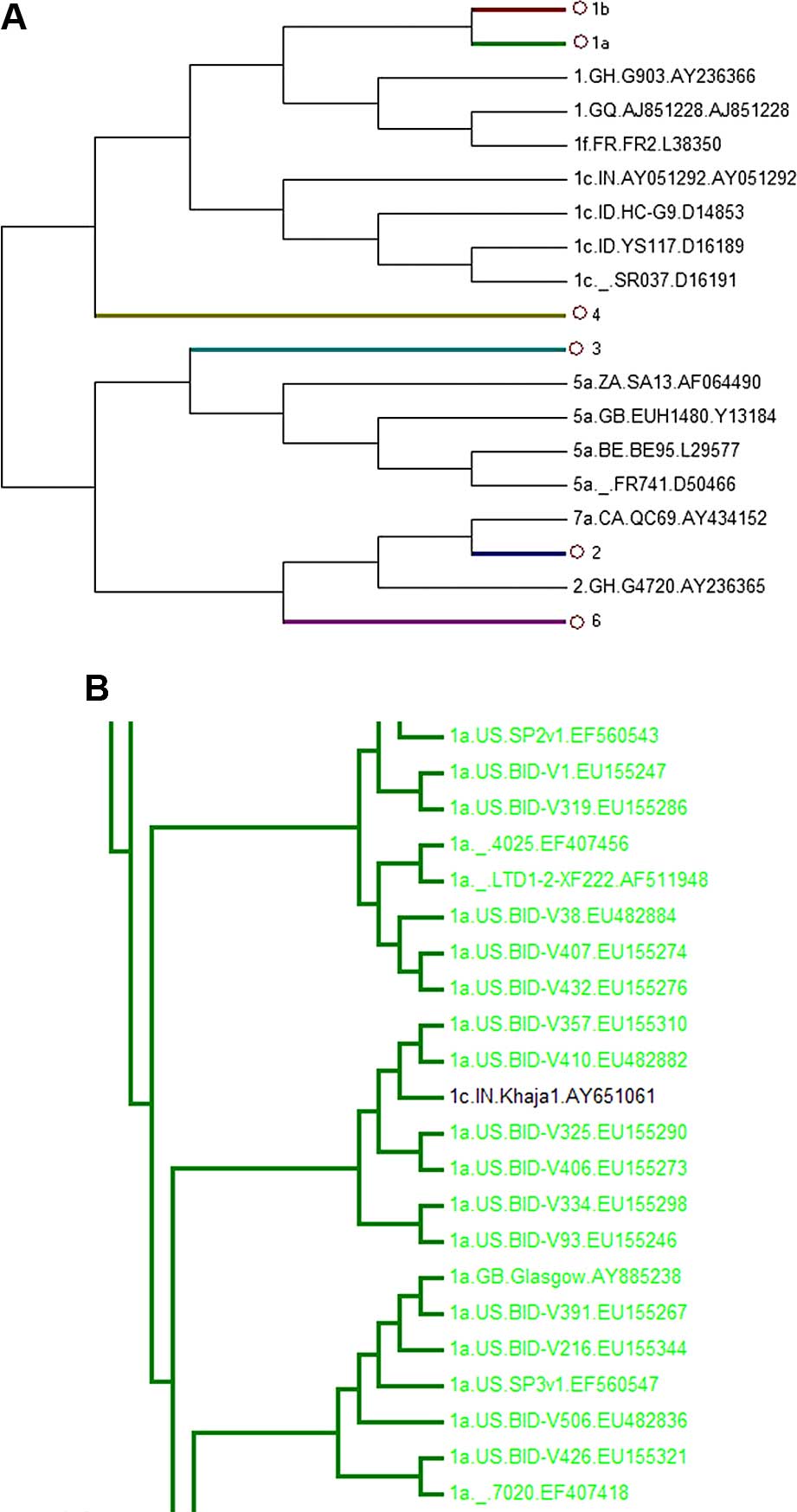 | Figure 3Phylogenetic tree of 781 HCV core
region sequences. Sequences were obtained from the HCV database
(http://hcv.lanl.gov/content/sequence/NEWALIGN/align.html).
(A) The whole phylogenetic tree. Ο1b, Ο1a, Ο4, Ο3, Ο2, Ο6 represent
1b, 1a, 4, 3, 2, 6 subgenotypes, respectively. (B) The partial
sub-tree of HCV subgenotype 1a that included abnormal sequences
which should be displayed in non-1a strains. |
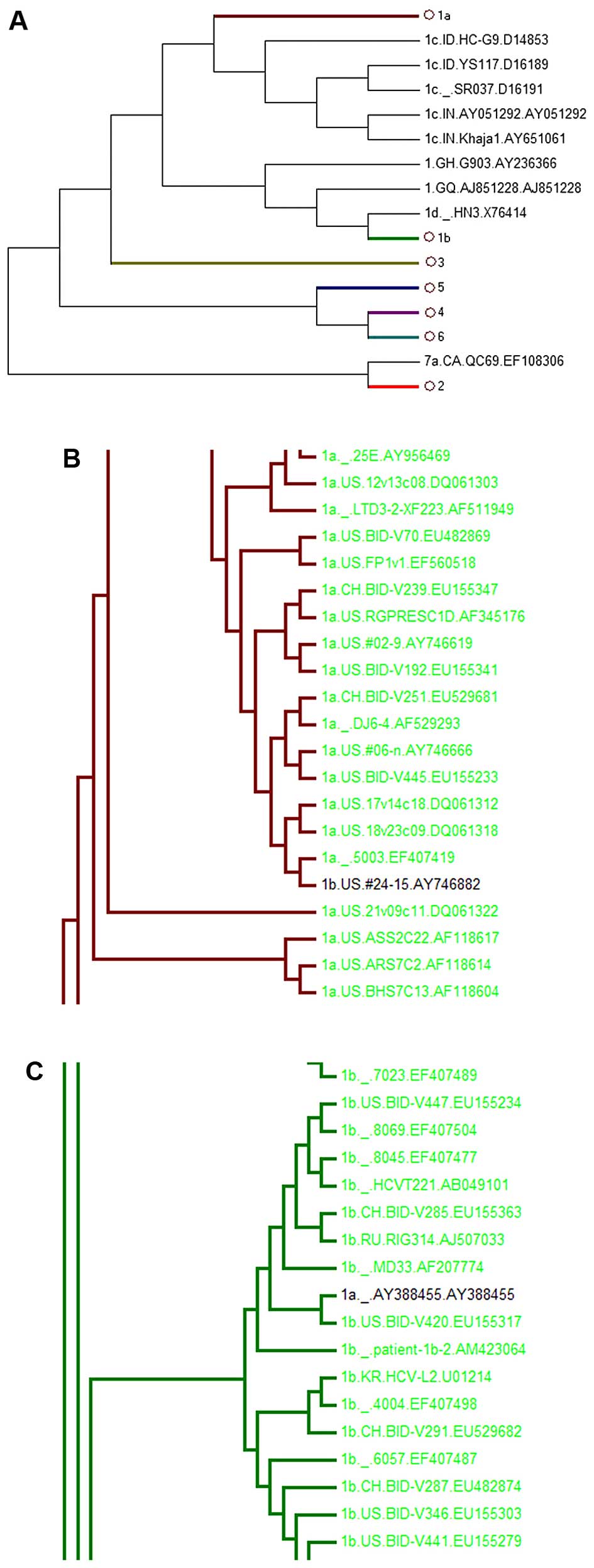 | Figure 4Phylogenetic tree of 899 HCV E1 region
sequences. Sequences were obtained from the HCV database
(http://hcv.lanl.gov/content/sequence/NEWALIGN/align.html).
(A) The whole phylogenetic tree. Ο1a, Ο1b, Ο3, Ο5, Ο4, Ο2, Ο6
represent 1a, 1b, 3, 5, 4, 2, 6 subgenotypes, respectively. (B) The
partial sub-tree of HCV 1a subgenotype strain that included
abnormal sequences which should be displayed in non-1a strains. (C)
The partial sub-tree of HCV 1b subgenotype strain that included
abnormal sequences which should be displayed in non-1b strains. |
HCV subgenotype analysis in infected
patients from Sichuan province
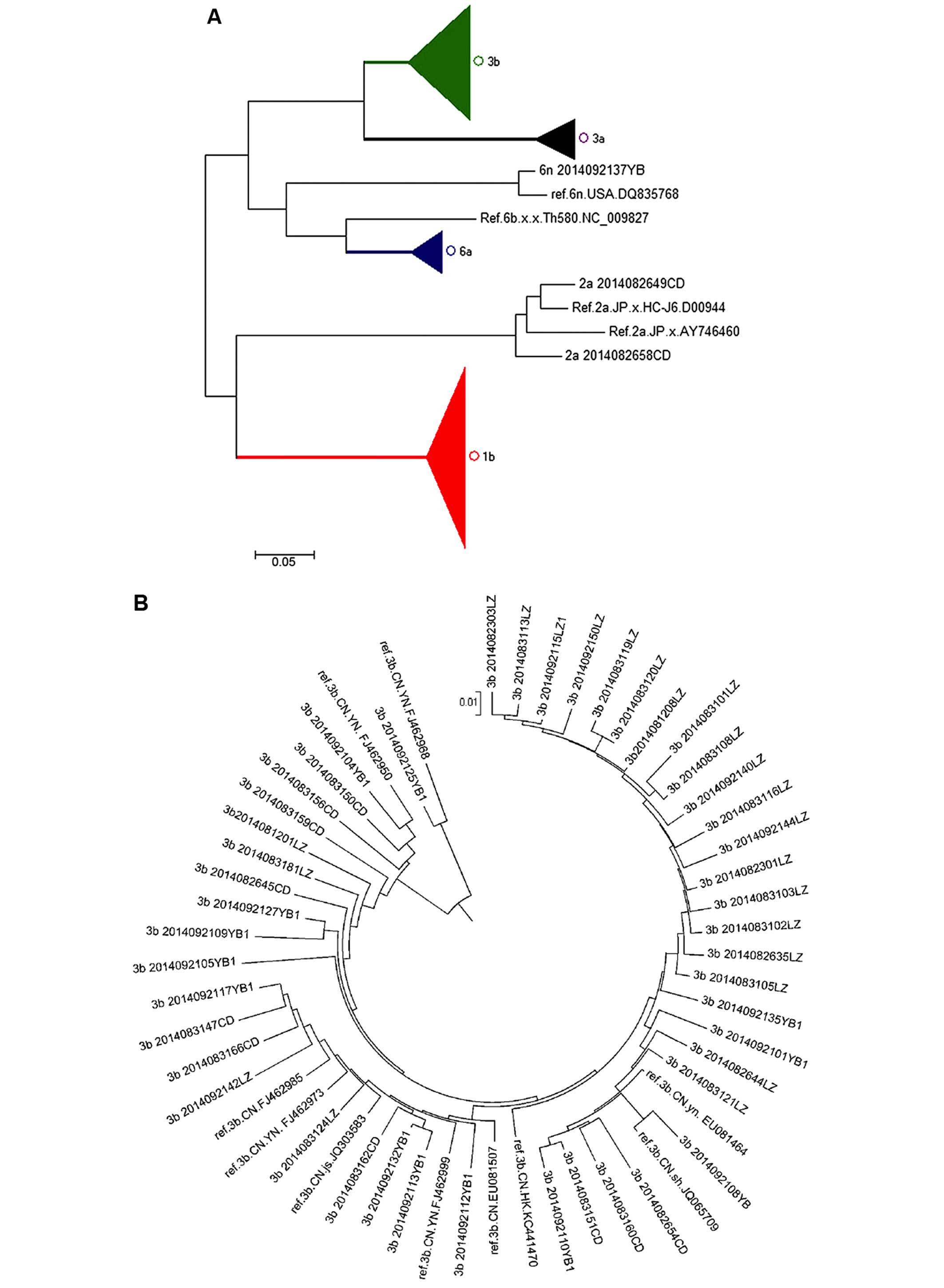
A total of 153 HCV samples obtained from infected
patients from Sichuan province were isolated and amplified by
RT-PCR. After sequencing the PCR products, all samples were
subjected to subgenotype classification using the neighbor-joining
method. Our results indicated that the dominant HCV subgenotype in
infected patients from Sichuan province was 1b (n=79, 51.6%), which
was closely related to sequences from patients in Yunnan, Henan,
Jiangsu and Zhejiang (Fig. 6E).
Subgenotype 3b was identified in 46 cases (30.1%) and was closest
to the sequences from patients in Yunnan, Fujian, Jiangsu and
Shanghai in the phylogenetic tree (Fig. 6B). There were 12 cases (7.8%)
belonging to subgenotype 3a and closely related to subgenotypes
from patients in Shanghai, Beijing, Henan, as well as in France and
Japan (Fig. 6C). There were 13
cases (8.5%) of subgenotype 6a in the present study which were
closely related to sequences from patients in Jiangsu and Hong Kong
(Fig. 6D). There were also 2
cases of subgenotype 2a and 1 case of subgenotype 6n, which were
closely related to sequences from in Japan and USA, respectively
(Fig. 6A).
Discussion
HCV is one of the main pathogens which induce
chronic liver disease, eventually leading to cirrhosis and
hepatocellular carcinoma (22).
There are almost 0.17 billion people infected with HCV, and this
accounts for 3% of the total global population (23). In China, there are 25–50 million
HCV-positive individuals (24),
and 3–4 million new cases diagnosed annually worldwide. Apart from
the 20% of patients who experience viral clearance, the majority of
HCV-infected individuals will remain positive for HCV for life and
they are more likely to develop chronic hepatitis, liver cirrhosis
and hepatocellular carcinoma than hepatitis B virus-infected
patients; this poses a serious public health concern (25).
The different subgenotypes of HCV have various
biological and molecular epidemiological characteristics (26). The correct subgenotype
classification plays a very important role in diagnosis, clinical
therapy (27) and vaccine
development (28). WGS is the
most accurate method for the classification of HCV genotypes;
however, WGS cannot be widely used in clinical diagnosis as it is
expensive and time-consuming. Thus, the best choice is to use
specific regions instead of the whole genome.
HCV whole genome sequences selected from HCV
database, which were isolated from patients from different
countries and regions and had an annotated source of mature
peptides, were used for phylogenetic analysis by the
neighbor-joining method. The results of the analysis of the core,
E1, E2 and NS5B regions indicated that NS5B could replace WGS in
genotype classification, while the core region did not recognize
the 1c and 1a subgenotypes (1c AY651061 was located in 1a). E1 did
not distinguish subgenotypes 1a and 1b (1b AY746882 was in 1a and
1a AY388455 in 1b), E2 failed to identify subgenotypes 1a and 1c
(1a EU529679 in 1c). The results confirmed the role of the NS5B
region in HCV subgenotype classification. Attempts (9,17,21,29) have previously been made to replace
WGS by more simple and rapid methods for genotype classification.
The classified results could be confused by the 1a and 1c
subgenotypes when the core region and E1 region are applied in the
classification.
Research into HCV subgenotype distribution may prove
to be helpful in epidemiological studies (30). The distribution of genotypes and
subgenotypes in different regions were differs due to the
population mobility and increased numbers of drug users (drugs
injected). The dominant HCV subgenotype in China is 1b, but there
are differences between the northern and southern regions (2). The differences in genotypes from
patients from southern China are complex and a possible reason for
this is that the number of injection drug users has greatly
increased. The genotyping of 60 patients with chronic HCV from
Kunming, a city in southern China, indicated that the major
subgenotypes were 3b, 3a and 1b (31).
In the present study, the amplification, sequencing
and classification of the NS5B region were conducted in 153 samples
fromp patients from Sichuan province. The results indicated that 6
HCV subgenotypes, including 1a, 3b, 3a, 6a, 2a and 6n, were
circulating in patients from Sichuan province. The dominant
subgenotype was 1b (79 cases, 51.6%) and the other subgenotypes
were 3b (46 cases, 30.1%), 3a (12 cases, 7.8%), 6a (13 cases, 8.5%)
while 2a and 6n had only 2 cases and 1 case, respectively. Sichuan
and Yunnan are neighboring provinces and this revealed the fact
that the sequences from Sichuan were closley related to those from
patients in Yunnan province. Parts of sequences were related to
patients from Jiangsu, Shanghai and Fujian and this is possibly due
to the return of migrant workers. The fact that there were also
sequences from patients in the US, Japan and Hong Kong indicated
the broken of region limitation in HCV subgenotype distribution by
population mobility. The results of our research have highlighted
new issues regarding the prevention and management of HCV.
The clinical usage of antiviral drugs could be
guided by the HCV genotype (32).
However, the limitation of the present study was that the
classification based on the NS5B region could guide the usage of
drugs targeted directly to NS5B residues, as well as other target
drugs of other non-recombinant HCV genotypes, but this method has
no use in the guidance of recombinant genotypes.
Acknowledgments
The present study was supported by the Science and
Technology Bureau of Yibin City (2011SF007).
References
|
1
|
Reed KE and Rice CM: Overview of hepatitis
C virus genome structure, polyprotein processing, and protein
properties. Curr Top Microbiol Immunol. 242:55–84. 2000.
|
|
2
|
Gu L, Tong W, Yuan M, Lu T, Li C and Lu L:
An increased diversity of HCV isolates were characterized among 393
patients with liver disease in China representing six genotypes, 12
subtypes, and two novel genotype 6 variants. J Clin Virol.
57:311–317. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rong X, Xu R, Xiong H, Wang M, Huang K,
Chen Q, Li C, Liao Q, Huang J, Xia W, et al: a comparison between
2004–2007 and 2008–2011. Arch Virol. 159:3231–3237. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Grassi E and Aghemo A: How to optimize HCV
therapy in genotype 2 patients. Liver Int. 33(Suppl 1): 35–40.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Abe H, Aida Y, Ishiguro H, Yoshizawa K,
Seki N, Miyazaki T, Itagaki M, Sutoh S, Ika M, Kato K, et al: New
proposal for response-guided peg-interferon-plus-ribavirin
combination therapy for chronic hepatitis C virus genotype 2
infection. J Med Virol. 85:1523–1533. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Inamullah, Idrees M, Ahmed H,
Sajid-ul-ghafoor, Ali M, Ali L and Ahmed A: Hepatitis C virus
genotypes circulating in district Swat of Khyber Pakhtoonkhaw,
Pakistan. Virol J. 8(16)2011.PubMed/NCBI
|
|
7
|
Chakravarti A, Ashraf A and Malik S: A
study of changing trends of prevalence and genotypic distribution
of hepatitis C virus among high risk groups in North India. Indian
J Med Microbiol. 31:354–359. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Verbeeck J, Maes P, Lemey P, Pybus OG,
Wollants E, Song E, Nevens F, Fevery J, Delport W, Van der Merwe S
and Van Ranst M: Investigating the origin and spread of hepatitis C
virus genotype 5a. J Virol. 80:4220–4226. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lu L, Nakano T, He Y, Fu Y, Hagedorn CH
and Robertson BH: Hepatitis C virus genotype distribution in China:
predominance of closely related subtype 1b isolates and existence
of new genotype 6 variants. J Med Virol. 75:538–549. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang C, Wu N, Liu J, Ge Q, Huang Y, Ren
Q, Feng Q and He G: HCV subtype characterization among injection
drug users: implication for a crucial role of Zhenjiang in HCV
transmission in China. PLoS One. 6:e168172011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Du H, Qi Y, Hao F, Huang Y, Mao L, Ji S,
Huang M, Qin C, Yan R, Zhu X and Zhang C: Complex patterns of HCV
epidemic in Suzhou: evidence for dual infection and HCV
recombination in East China. J Clin Virol. 54:207–212. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wyles DL and Gutierrez JA: Importance of
HCV genotype 1 subtypes for drug resistance and response to
therapy. J Viral Hepat. 21:229–240. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fried MW, Shiffman ML, Reddy KR, Smith C,
Marinos G, Gonçales FL Jr, Häussinger D, Diago M, Carosi G,
Dhumeaux D, et al: Peginterferon alfa-2a plus ribavirin for chronic
hepatitis C virus infection. N Engl J Med. 347:975–982. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin JA, Chen YC, Cheng SN, Chen PJ, Chu
HC, Hsieh TY and Shih YL: Peginterferon alfa-2a plus ribavirin for
hemophilic patients with chronic hepatitis C virus infection in
Taiwan. J Formos Med Assoc. 113:727–733. 2014. View Article : Google Scholar
|
|
15
|
Waheed Y, Saeed U, Anjum S, Afzal MS and
Ashraf M: Development of global consensus sequence and analysis of
highly conserved domains of the HCV NS5B protein. Hepat Mon.
12:e61422012.
|
|
16
|
Weck K: Molecular methods of hepatitis C
genotyping. Expert Rev Mol Diagn. 5:507–520. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yamashita K, Ohtsuka N, Kagawa S and
Matsuoka A: Improved method for detection and subtyping of HCV.RNA
by nested polymerase chain reaction. Rinsho Byori. 43:1147–1152.
1995.In Japanese. PubMed/NCBI
|
|
18
|
Stuyver L, Rossau R, Wyseur A, Duhamel M,
Vanderborght B, Van Heuverswyn H and Maertens G: Typing of
hepatitis C virus isolates and characterization of new subtypes
using a line probe assay. J Gen Virol. 74:1093–1102. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Buoro S, Pizzighella S, Boschetto R,
Pellizzari L, Cusan M, Bonaguro R, Mengoli C, Caudai C, Padula M,
Egisto Valensin P and Palù G: Typing of hepatitis C virus by a new
method based on restriction fragment length polymorphism.
Intervirology. 42:1–8. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Simmonds P, Bukh J, Combet C, Deléage G,
Enomoto N, Feinstone S, Halfon P, Inchauspé G, Kuiken C, Maertens
G, et al: Consensus proposals for a unified system of nomenclature
of hepatitis C virus genotypes. Hepatology. 42:962–973. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cai Q, Zhao Z, Liu Y, Shao X and Gao Z:
Comparison of three different HCV genotyping methods: Core, NS5B
sequence analysis and line probe assay. Int J Mol Med. 31:347–352.
2013.
|
|
22
|
Modi AA and Liang TJ: Hepatitis C: a
clinical review. Oral Dis. 14:10–14. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Asselah T and Marcellin P: New
direct-acting antivirals' combination for the treatment of chronic
hepatitis C. Liver Int. 31(Suppl 1): 68–77. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rao H, Wei L, Lopez-Talavera JC, Shang J,
Chen H, Li J, Xie Q, Gao Z, Wang L, Wei J, et al: Distribution and
clinical correlates of viral and host genotypes in Chinese patients
with chronic hepatitis C virus infection. J Gastroenterol Hepatol.
29:545–553. 2014. View Article : Google Scholar :
|
|
25
|
Ashfaq UA, Javed T, Rehman S, Nawaz Z and
Riazuddin S: An overview of HCV molecular biology, replication and
immune responses. Virol J. 8(161)2011. View Article : Google Scholar
|
|
26
|
Zein NN: Clinical significance of
hepatitis C virus genotypes. Clin Microbiol Rev. 13:223–235. 2000.
View Article : Google Scholar
|
|
27
|
Chevaliez S, Bouvier-Alias M, Brillet R
and Pawlotsky JM: Hepatitis C virus (HCV) genotype 1 subtype
identification in new HCV drug development and future clinical
practice. PLoS One. 4:e82092009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jang JY and Chung RT: New treatments for
chronic hepatitis C. Korean J Hepatol. 16:263–277. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lu J, Tao W, Li R, Xiang Y, Zhang N, Xiang
X, Xie Q and Zhong J: Construction and characterization of
infectious hepatitis C virus chimera containing structural proteins
directly from genotype 1b clinical isolates. Virology. 443:80–88.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhou X, Chan PK, Tam JS and Tang JW: A
possible geographic origin of endemic hepatitis C virus 6a in Hong
Kong: Evidences for the association with Vietnamese immigration.
PLoS One. 6. pp. e248892011, View Article : Google Scholar
|
|
31
|
Tao J, Liu J, Pu D and Lei H: Efficacy of
interferon alpha with ribavirin for treatment of chronic hepatitis
C. Zhonghua Gan Zang Bing Za Zhi. 19:683–685. 2011.PubMed/NCBI
|
|
32
|
Wendt A, Adhoute X, Castellani P, Oules V,
Ansaldi C, Benali S and Bourlière M: Chronic hepatitis C: future
treatment. Clin Pharmacol. 6:1–17. 2014.PubMed/NCBI
|