Introduction
Dilated cardiomyopathy (DCM) is one of the most
prevalent inherited cardiomyopathies and is known to be one of the
leading causes of heart failure and sudden cardiac death, and
typically necessitates a heart transplant. DCM is a genetically
heterogeneous disease characterized by cardiac left ventricular
enlargement and systolic dysfunction (1–3).
In the general population, the prevalence of DCM is greater than 1
in 2,500 individuals (4). The
majority of patients present with sporadic DCM (SDCM); however,
genetic screening of first-degree relatives reveals that 20–35% of
cases are familial DCM. Familial DCM is defined as SDCM in two or
more closely related family members or, when involving a
first-degree relative of a patient with SDCM (5,6).
DCM is primarily caused by pathogenic gene mutations
inherited in a Mendelian autosomal dominant pattern (7–9).
However, in a small number of cases, autosomal recessive, X-linked,
or mitochondrial DNA inheritance characteristics have been observed
(10–14). Currently, over 50 genes have been
documented to cause DCM. Of these, the majority harbour missense
mutations located in sarcomere proteins, including cardiac
myosin-binding protein C (MYBPC3), cardiac alpha-myosin heavy chain
(MYH6), cardiac beta-myosin heavy chain (MYH7), troponin T type 2
(cardiac) (TNNT2), troponin I type 3 (cardiac) (TNNI3), tropomyosin
1 (alpha) (TPM1), myosin light chain 3 (MYL3), and actin, alpha,
cardiac muscle 1 (ACTC1). In addition, mutations in the nuclear
envelope proteins of nuclear lamin A/C (LMNA); cytoskeletal
proteins, including myopalladin (MYPN), desmin (DES) and vinculin
(VCL); calcium/sodium-handling proteins of sodium channel, voltage
gated, type V alpha subunit (SCN5A); desmosomal proteins, such as
desmoplakin (DSP) and RNA-binding motif protein 20 (RBM20); as well
as other types of genes are considered to be closely related to the
occurrence of DCM (Table I)
(4,15).
 | Table ISelected genes associated with
inherited cardiomyopathy. |
Table I
Selected genes associated with
inherited cardiomyopathy.
| Gene name | Protein name | Chromosome | Exons | OMIM | Inherited
cardiomyopathy phenotype |
|---|
| ABCC9 | ATP-binding
cassette, subfamily C, member 9 | 12p12.1 | 39 | 601439 | DCM |
| ACTN2 | Actinin, alpha
2 | 1q42-q43 | 21 | 102573 | HCM, DCM |
| CAV3 | Caveolin 3 | 3p25.3 | 2 | 601253 | DCM |
| DES | Desmin | 2q35 | 9 | 125660 | DCM, ARVC |
| LAMA4 | Laminin alpha
4 | 6q21 | 39 | 600133 | DCM |
| LAMP2 | Lysosome-associated
membrane protein 2 | Xq24 | 10 | 309060 | HCM, DCM |
| LDB3 | LIM domain-binding
3 | 10q23.2 | 16 | 605906 | HCM, DCM |
| LMNA | Lamin A/C | 1q22 | 12 | 150330 | DCM, ARVC |
| MYBPC3 | Cardiac
myosin-binding protein-C | 11p11.2 | 35 | 600958 | HCM, DCM |
| MYH6 | Myosin, heavy chain
6, cardiac muscle, alpha | 14q11.2-q12 | 39 | 160710 | HCM, DCM |
| MYH7 | Myosin, heavy chain
7, cardiac muscle, beta | 14q11.2-q12 | 40 | 160760 | HCM, DCM |
| MYL2 | Myosin light chain
2 | 12q23-q.24.3 | 7 | 160781 | HCM |
| MYL3 | Myosin light chain
3 | 3p21.2-p21.3 | 7 | 160790 | HCM |
| MYPN | Myopalladin | 10q21.1 | 20 | 608517 | DCM |
| PSEN2 | Presenilin 2 | 1q42.13 | 13 | 600759 | DCM |
| RBM20 | RNA-binding motif
protein 20 | 10q25.2 | 14 | 613171 | DCM |
| SCN5A | Sodium channel,
voltage-gated type V, alpha subunit | 3p22.2 | 29 | 600163 | DCM |
| SGCD |
Delta-sarcoglycan | 5q33.3 | 9 | 601411 | DCM |
| TMPO | Thymopoietin | 12q23.1 | 10 | 188380 | DCM |
| TCAP | Telethonin | 17q12-q21.1 | 2 | 604488 | HCM, |
| TNNC1 | Troponin C type 1
(slow) | 3p21.1 | 6 | 191040 | HCM, DCM |
| TNNI3 | Troponin I, cardiac
muscle | 19q13.4 | 8 | 191044 | HCM, DCM |
| TNNT2 | Troponin T, cardiac
muscle | 1q32 | 17 | 191045 | HCM, DCM |
| TPM1 | Tropomyosin 1
alpha | 15q22.1-q22.2 | 10 | 191010 | HCM, DCM |
| VCL | Vinculin | 10q22.1-q23 | 22 | 193065 | HCM, DCM |
Recent technical advances have allowed
high-throughput next-generation sequencing (NGS) to overcome the
limitations of traditional capillary Sanger sequencing (16,17). NGS has previously been used for
genetic diagnoses in clinical settings (18). In theory, genetic testing can
identify patients at risk of developing DCM prior to the onset of
clinical symptoms. The most frequently used Sanger sequencing
technique is, however, cumbersome and expensive. Comparatively, NGS
techniques are cost-efficient and generate a large amount of data
in one reaction (19,20). Moreover, the molecular genetics
data of patients with DCM are insufficient in China and have not
been reported previously in relation to the Yunnan population. To
provide new insight into the genetic profile of DCM in the Yunnan
population in China, in the current study, we employed NGS
screening to investigate the major DCM-causing genes in patients
diagnosed with the disease. To the best of our knowledge, this is
the first description of a gene mutation profile of patients with
DCM from Yunnan in southwestern China. In view of the fact that
genetic testing is a useful indicator for the clinical management
of disease, testing for pathogenic mutations may be beneficial to
the treatment of patients with DCM and may be used to predict
disease risk for their family members prior to the onset of
symptoms.
Materials and methods
Subjects and clinical evaluation
All subjects (n=21; 15 male, 6 female; median age at
onset, 48.7 (±11.7) years; age range, 26–73 years) were recruited
from southern and western Yunnan, China, and had been previously
diagnosed with DCM in accordance with previously published
guidelines (21). At the clinical
interview, physical examinations, invasive examinations and
echocardiography (ECG) were performed to determine the phenotype of
each SDCM [left ventricular ejection fraction (LVEF) <50%; left
ventricular end-diastolic diameter (LVED) >112% of the predicted
value]. Written informed consent and additional clinical
information were obtained from each patient with DCM. This study
was approved by the Institutional Ethics Committee of the First
People's Hospital of Yunnan Province (Affiliated Hospital of
Kunming University of Science and Technology) and complied with the
principles of the Declaration of Helsinki.
Candidate gene sequencing
Peripheral whole blood lymphocyte samples (2 ml)
from each patient were collected in Vacutainer tubes coated with
EDTA (BD Biosciences, Franklin Lakes, NJ, USA) and stored at 4°C
until DNA extraction. Genomic DNA was extracted from each blood
sample using a commercially available genomic AxyPrep DNA miniprep
kit (Axygen Biosciences, Union City, CA, USA) following the
manufacturer's instructions. A total of 25 genes associated with
DCM (Table I) were selected as
candidate genes. The amplicons were captured using a
custom-designed multi-target gene library (Agilent Technology,
Santa Clara, CA, USA) to achieve sufficient sequences covering the
genes of interest, and the amplicons were then sequenced using a
Genome Analyzer IIx (Illumina, Inc., San Diego, CA, USA).
Sequence alignment and variant
calling
Low-quality reads were discarded following an
initial inspection with FASTX-tools (http://hannonlab.cshl.edu/fastx_toolkit/index.html),
and qualified sequences were aligned together with the human
reference genome assembly (GRCh37/hg19) in Burrows-Wheeler Aligner,
Smith-Waterman Alignment (BWA-SW, version 0.5.9) and Sequence
Alignment/Map (SAM; version 0.1.16) tools software package, as
previously described (22,23).
Sequence calls for variants were identified using Variant caller
(version 4.2.0), as previously described (24). Following variant detection, the
identified variants (splice, stop loss, synonymous, non-synonymous,
insertion, or deletion variants) were annotated with the ANNOVAR
annotation tool or using online Ion Reporter software (25;
https://ionreporter.lifetechnologies.com/ir/secure/home.html).
Molecular genetic analysis
The potential pathogenic role of variants in the
coding region was focused on in the present study, i.e. only the
statistical variation in the coding region. Putative pathogenic
mutations were considered to be pathogenic based on the following
criteria (26–29): i) the mutation has been reported
to be associated with the disease phenotype in the reference or
Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/docs/login.html); ii) the
mutation has a minor allele frequency (MAF) of <1% in the NCBI
dbSNP Build 137 (http://www.ncbi.nlm.nih.gov/projects/SNP), the 1000
Genomes Project (http://www.1000genomes.org/), and the National Heart,
Lung, and Blood Institute Exome Sequencing Project (https://esp.gs.washington.edu/drupal/)
databases; iii) the protein structure and function was
significantly altered, and the amino acid was highly conserved
across a number of species; iv) the mutation was analyzed and shown
to be pathogenic using the PolyPhen-2, SIFT, or MutationTaster
(30–32) algorithms; or v) novel mutation or
variants of uncertain significance had to be absent from unrelated
and healthy controls which were matched for ethnicity. The mutation
nomenclature was based on the nucleotide reference sequences: MYPN,
NM_032578; TNNT2, NM_001001432; MYH7, NM_000257; DES, NM_001927;
VCL, NM_014000.2; MYH6, NM_002471.3; RBM20, NM_001134363.1; TNNC1,
NM_003280.2; MYBPC3, NM_000256; and NM_000258.2 for MYL3.
Validation of selected mutations
All of the putative pathogenic mutations were
reconfirmed by traditional capillary Sanger sequencing (ABI 3130
Genetic Analyzer; Life Technology, Carlsbad, CA, USA) of the PCR
product for all suspected samples, using primers for preliminary
determined mutations (Table II).
The amplification of each mutation fragment was undertaken using
primeSTAR GXL DNA polymerase (Takara, Otsu, Japan, code no. R050A).
PCR conditions consisted of 1 cycle of 96°C for 5 min; 30 cycles of
96°C for 3 min, 54°C or 57°C for 30 sec, 68°C for 1 min (200
bp<fragment size<1000 bp) or 2 min (1000 bp<fragment
size<2000 bp); 1 extension cycle of 68°C for 5 min and holding
at 16°C. Control alleles (n=200) were taken from 100 unrelated
healthy subjects with normal phenotypes (matched for gender, age
and ethnicity) to exclude the possibility of rare polymorphisms of
the novel mutations and variants of uncertain significance.
| Table IIPCR primers for amplification of the
mutation sites. |
Table II
PCR primers for amplification of the
mutation sites.
| Gene symbol | Nucleotide
changes | Exon | Primer (5′→3′) | Annealing
temperature (°C) |
|---|
| MYPN | c.1888G>A | 10 | Sense:
ACTCAGCCAACTCTACCACCAACC | 54 |
| | | Antisense:
ACTGGAGGGGGCTCTTTCACG | |
| TNNT2 | c.539G>C | 12 | Sense:
ACCTTCTCCCTATGCACACCT | 57 |
| | | Antisense:
CACAGCAGCTGGGAATCTCTT | |
| MYH7 | c.77C>T | 3 | Sense:
AGCCAGCTTCTGCTCACTCCAG | 57 |
| | | Antisense:
GCCACTTGTAAGGGTTGACGGT | |
| DES | c.1157G>A | 6 | Sense:
CAGGAGATGATGGAATACCGACAC | 54 |
| | | Antisense:
ACAGAAATGGACCACCCAGCAC | |
| VCL | c.625A>T | 6 | Sense:
CAGCAGGATACGCCATTCAGAGT | 57 |
| | | Antisense:
GACCAGAGCAGCAGCAACACA | |
| RBM20 | c.3545G>A | 13 | Sense:
GCAGAGAGAGGTACAGTGTGAAG | 57 |
| | | Antisense:
AAGTCTATGGGAAGATTAGGGGTTT | |
| MYBPC3 | c.3371G>T | 31 | Sense:
AGAGGCTCTCGGCATCAGGAAG | 54 |
| | | Antisense:
ACATAGATGCCCCCGTCAAAGG | |
| TNNC1 | c.8A>T | 1 | Sense:
GGGATAAACTTGATACGAACTCTG | 57 |
| | | Antisense:
GAGGAAGAGATGAAAAGAGAAAGG | |
| MYH6 | c.3139C>T | 24 | Sense:
TTTTGCTCTCTGTAGTTCCTCAC | 54 |
| | | Antisense:
TGCTCATCCTCAATCTTACTGTTC | |
| MYH6 | c.3758C>T | 27 | Sense:
ATGCCTTCTCTCTCTGTCTGCC | 54 |
| | | Antisense:
GTGAAATCATTGAGGGAGCGTT | |
| MYBPC3 | c.478C>T | 4 | Sense:
GCCCTTCAGTCTCAGCTTTTAGC | 54 |
| | | Antisense:
CTTGAGCCCTTAGCCCTGATACT | |
| MYL3 | c.377A>G | 4 | Sense:
AAAGTGCCTCGCGATGGTAGTTTG | 57 |
| | | Antisense:
GTCTGCCATTGAGGCTCCCTAATT | |
Statistical analysis
Statistical analysis was used to summarize the
clinical characteristics of the subjects; SPSS 16.0 software was
used. The mean values ± standard deviation (SD) are shown when
comparing the presence and absence of mutation groups. T-test
results of independent samples with p-values <0.05 were
considered to indicate statistically significant differences.
Results
Study population
Molecular screening was performed on a cohort of 21
unrelated patients in Yunnan, who were recruited with an original
clinical diagnosis of DCM. Of these patients, 15 were male and 6
were female, with a median age at onset of 48.7 (±11.7) years, and
an age range of 26–73 years. ECG revealed enlarged heart chambers
[LVED = 68.9±10.6; left ventricular end systolic diameter (LVESD) =
58.5±11.4] and a low LVEF (LVEF = 31.4±10.9); interventricular
septal thickness and left ventricular posterior wall thickness were
9.3 and 8.6 mm, respectively. The left atrium size was 45.7 mm. The
clinical characteristics of the patients with DCM are presented in
Table III.
| Table IIIClinical characteristics of patients
with dilated cardiomyopathy according to diagnosis. |
Table III
Clinical characteristics of patients
with dilated cardiomyopathy according to diagnosis.
| Characteristic | Cardiac screening
(%) | Genetic testing
| P-value |
|---|
| Mutation absence: 9
(42.8%) | Mutation presence:
12 (57.2%) |
|---|
| Gender, n (%) | | | | |
| Male | 15 (71.5) | 7 (77.8) | 8 (66.6) | |
| Female | 6 (28.5) | 2 (22.2) | 4 (33.4) | N/A |
| Age at onset
(years) | 48.7±11.7 | 57.1±9.1 | 42.3±9.3 | 0.002a |
| IVST (mm) | 9.3±1.9 | 10.2±1.6 | 8.6±1.7 | N/A |
| LVED (mm) | 68.9±10.6 | 74.4±12.6 | 64.8±6.6 | N/A |
| LVESD (mm) | 58.5±11.4 | 65.5±12.4 | 53.3±7.4 | N/A |
| LVPWT (mm) | 8.6±1.5 | 8.3±0.7 | 8.8±1.8 | N/A |
| LA (mm) | 45.7±9.5 | 50.3±10.8 | 42.3±7.1 | N/A |
| LVEF (%) | 31.4±10.9 | 26.6±9.0 | 35.0±11.1 | 0.078 |
Identification of DCM-related
mutations
Sequencing of the major DCM-causing genes (Table I) was performed. Subsequently, the
short reads mapped onto the reference genome (GRCh37/hg19) were
annotated with the ANNOVAR annotation tool or Ion Reporter
software. All successfully mapped sequence reads were analyzed to
detect sequence variants, including non-synonymous, synonymous,
insertion, and deletion variants. In all patients, the mean read
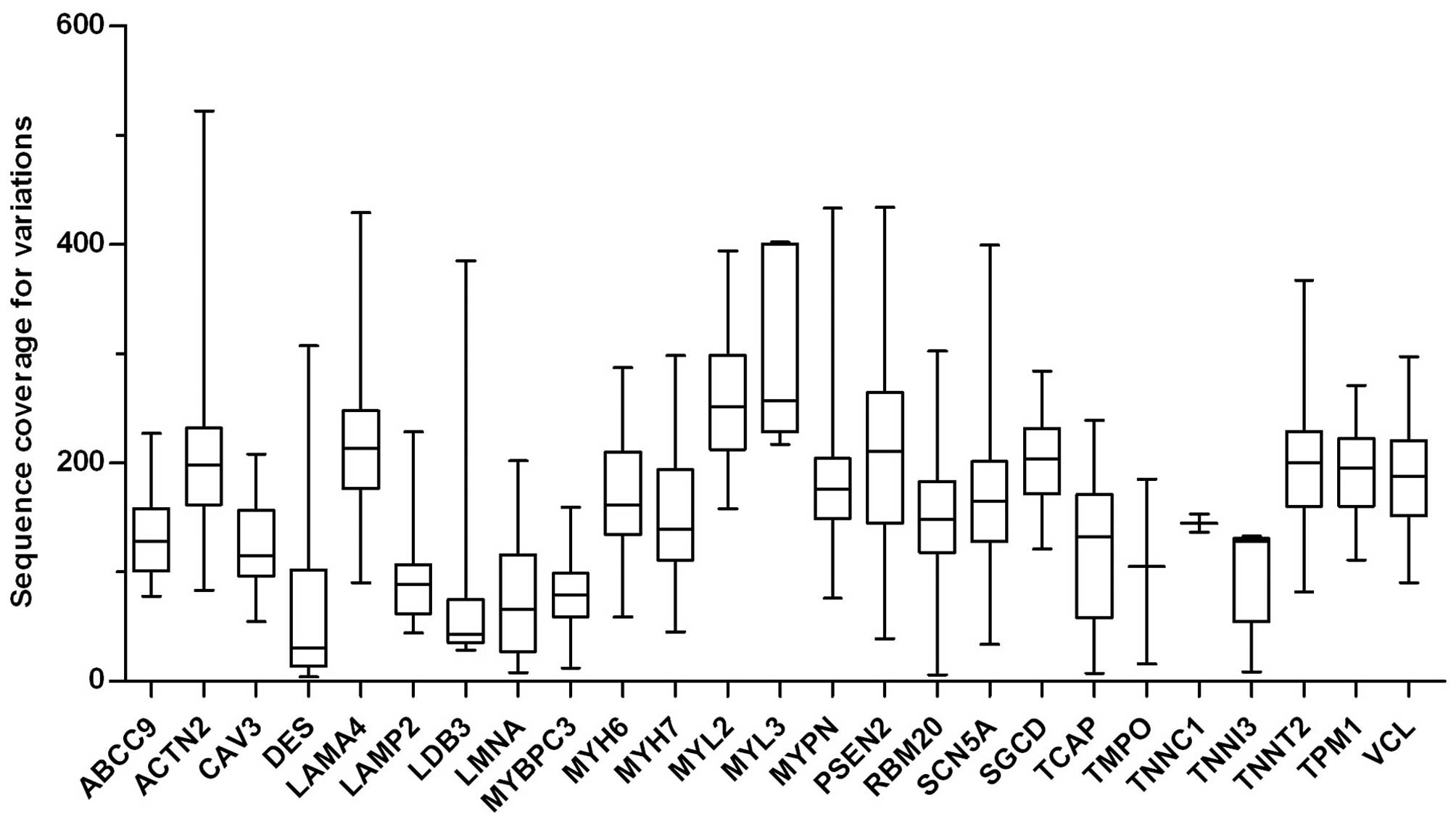
depth of coverage for all target gene variants was 170.8X (range, 4
to 522X; Fig. 1). Altogether, 243
missense mutations (data available upon request) were identified
with an average of 12 variants found per patient with DCM. On the
other hand, the potential of the variations were detected in the
coding region (CDS) of the target genes in 21 patients with DCM
(data available upon request).
The variants were selected from most of the
synonymous or non-synonymous variants and compared with the
reported data from NCBI dbSNP (MAF>0.01). Finally, 12 possibly
pathogenic heterozygous mutations from the candidate genes were
revealed (Table IV), and these
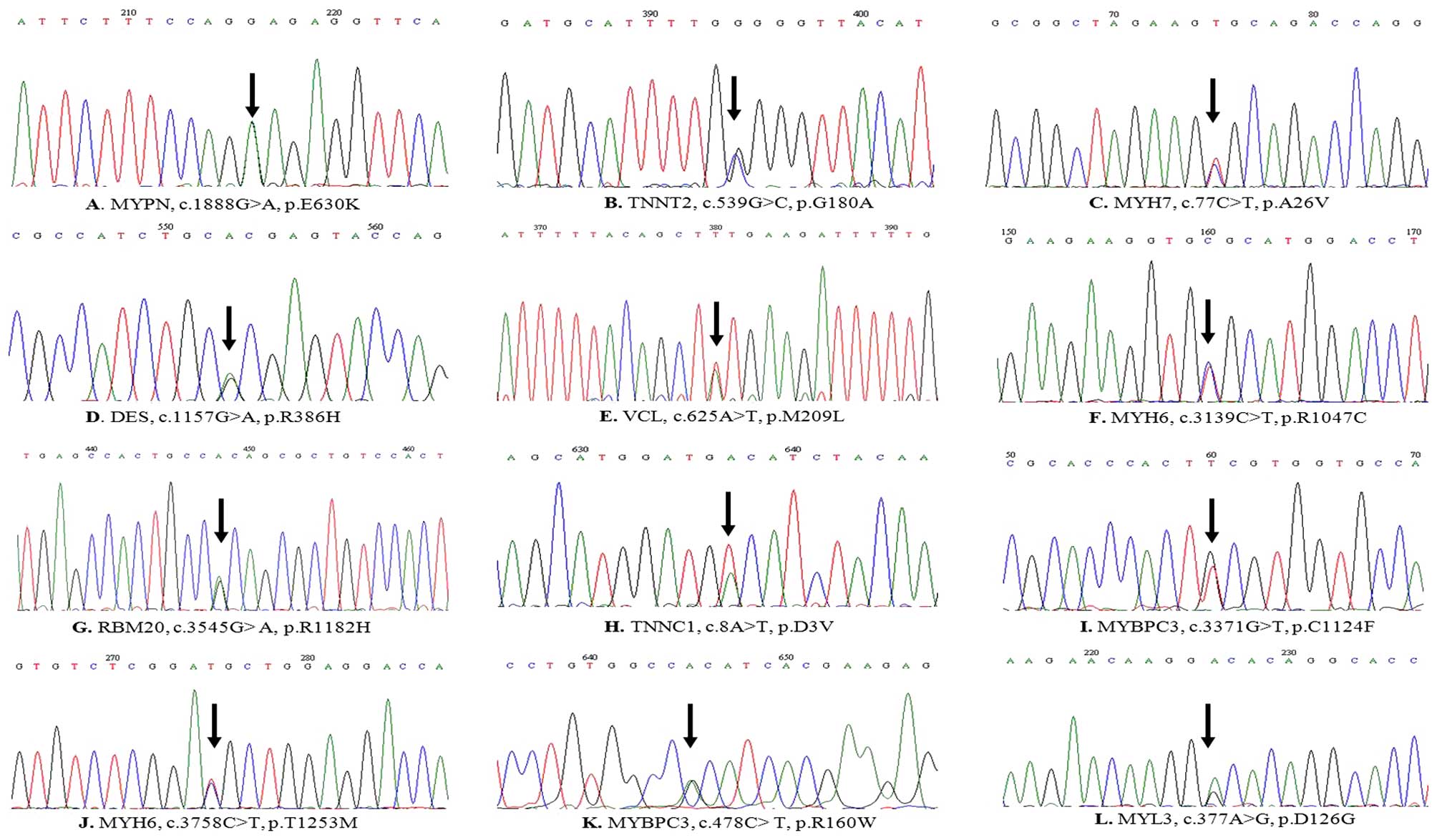
mutations were confirmed using traditional capillary Sanger
sequencing (Fig. 2). Of these, 2
mutations, p.A26V in MYH7 and p.R160W in MYBPC3, have
been previously documented (33),
and 3 variants of uncertain significance (VCL, p.M209L;
RBM20, p.R1128H; MYH6, p.T1253M) in dbSNP have been
documented in the 1000 Genomes Project database at MAF = 0.0001,
0.0049, and 0, respectively, in the Han Chinese population from
Beijing. Furthermore, it is worth noting that 7 novel mutations
(MYPN, p.E630K; TNNT2, p.G180A; DES, p.R386H;
MYBPC3, p.C1124F; MYH6, p.R1047C; MYL3,
p.D126G; and TNNC1, p.D3V) are not listed in the NCBI dbSNP
database, 1000 Genomes Project, the Human Gene Mutation database,
or Exome Sequencing Project database. These 7 novel mutations were
absent in the healthy control samples, and the corresponding
altered amino acids of the 7 novel mutations and 3 variants of
uncertain significance were highly conserved across a number of
species. This indicates that these altered amino acids share
related protein functions (data available upon request). Finally,
based on the online analysis using MutationTaster, PolyPhen2 and
SIFT, these mutations were predicted to be disease-causing, not
tolerated, or probably damaging (Table IV). These results suggest that
the 7 novel mutations and 3 variants of uncertain significance were
highly associated with DCM in the patients studied.
| Table IVSummary of the disease mutations
found in DCM patients. |
Table IV
Summary of the disease mutations
found in DCM patients.
| Patient no. | Gene | Gender | Nucleotide
change | Amino acid
change | SIFT | Ploy Phen-2 | Mutation
Taster | Previously
reported |
|---|
| DCM-1 | MYPN | M | c.1888G>A | p.E630K | T | B | DC | NO |
| DCM-10 | TNNT2 | M | c.539G>C | p.G180A | T | PD | DC | NO |
| DCM-12 | MYH7 | F | c.77C>T | p.A26V | NA | NA | NA | YES |
| DCM-21 | DES | F | c.1157G>A | p.R386H | NT | PD | DC | NO |
| DCM-29 | VCL | M | c.625A>T | p.M209L | T | B | DC | VUS |
| DCM-33 | MYH6 | M | c.3139C>T | p.R1047C | NT | PD | DC | NO |
| DCM-45 | RBM20 | M | c.3545G>A | p.R1182H | NT | B | DC | VUS |
| DCM-46 | TNNC1 | M | c.8A>T | p.D3V | NT | PD | DC | NO |
| DCM-7 | MYBPC3 | F | c.3371G>T | p.C1124F | T | B | DC | NO |
| DCM-55 | MYH6 | M | c.3758C>T | p.T1253M | T | PD | DC | VUS |
| DCM-11 | MYBPC3 | F | c.478C>T | p.R160W | NA | NA | NA | YES |
| DCM-13 | MYL3 | M | c.377A>G | p.D126G | NA | B | DC | NO |
Association of genotype and
phenotype
To elucidate the association of genotype and
phenotype in patients with DCM, a comparison of the clinical
characteristics, such as age at diagnosis and LVEF, was performed
between groups of patients with and without gene mutations
(Table III). No statistically
significant difference in LVEF was observed in the patients with
DCM based on the presence or absence of genetic mutations
(P=0.078>0.05). However, it was demonstrated that patients with
DCM harbouring DCM-associated mutations had been diagnosed with
their condition at a significantly younger age than patients with
DCM without the mutations (57.1±9.1 vs. 42.3±9.3 years;
P=0.002<0.05; Table
III).
Mutant spectra in patients with DCM
As shown in Table
IV, there were 12 probands (patients with DCM) harbouring one
mutation (12/21, ~57.2%), including the 7 novel mutations, 3
variants of uncertain significance, and 2 previously reported
mutations. From the patients with DCM carrying genetic variants (8
male, 4 female), the highest frequency of mutations was detected in
sarcomere genes (8/12, ~66.7%; MYH7, MYBPC3,
MYH6, TNNT2, TNNC1 and MYL3), followed
by cytoskeletal genes (3/12, ~25%; MYPN, DES and
VCL) and other types of genes (1/12, ~8.3%; RBM20). A
small number of mutated genes were identified with a high frequency
in the patients with DCM. To the best of our knowledge, the mutated
genes, MYBPC3 and MYH6, were the most frequently
observed genes with 2 out of 12 patients harbouring mutations in
these two genes. However, the other mutated genes were only found
in 1 patient each.
Discussion
DCM is primarily caused by pathogenic genetic
mutations. For example, it has been suggested that mutations in
genes encoding contractile proteins result in functional changes
and lead to contractile dysfunction of cardiomyocytes (34,35). The majority of patients with DCM
exhibit autosomal dominant genetic disease, although there are
several reported cases with recessive, X-linked, and other patterns
of inheritance (11,12,14,15). Thus, the Heart Rhythm
Society/European Heart Rhythm Association expert consensus
statement recommends the performance of genetic testing on patients
with DCM. As regards clinical intervention for patients with DCM,
there is still no effective treatment, aside from heart
transplants. The identification of the molecular genetic basis of
the disease is important for determining appropriate strategies for
DCM prevention and management before the onset of symptoms.
The present study aimed to assess the prevalence of
known DCM-related genes and the association between the results of
cardiac screening and genetic testing on patients from Yunnan,
China. The screening of candidate genes (Table I) in 21 patients with DCM was
performed using NGS. Mutations were found in more than half of the
DCM patients (12/21, ~57.2%). In comparison with previous studies
(36–38), pathogenic mutations were present
at a higher frequency in Yunnan DCM patients than in patients from
other populations. Moreover, pathogenic mutations were not detected
in 9 patients with DCM, due to localisation of the mutations to
other untested genes, as has been previously suggested (39). MYH7-p.A26V (40,41) and MYBPC3-p.R160W (32,42) have been reported previously in
different patient groups with hypertrophic cardiomyopathy (HCM).
However, these mutations (MYH7-p.A26V and
MYBPC3-p.R160W) were originally found in patients with DCM,
once again illustrating the clinical and genetic heterogeneity of
patients with DCM. Of these two mutations, MYH7-p.A26V
results in an amino acid substitution located in the myosin heavy
chain affecting different functional domains of the head or the
head-rod junction of MYH7 (43).
MYBPC3-p. R160W results in changes in the charge of the
altered amino acid in the immunoglobulin-like domain, and this
mutation is associated with both HCM (32,42) and DCM.
Notably, the 7 novel mutations, p.E630K in
MYPN, p.G180A in TNNT2, p.R1047C in MYH6,
p.D3V in TNNC1, p.R386H in DES, p.C1124F in
MYBPC3, and p.D126G in MYL3, and the three variants
of uncertain significance (RBM20, p.R1182H; MYH6,
p.T1253M; and VCL, p.M209L) were not found in the genomes of
100 healthy controls with matched age, gender, and geographical
region (data available upon request). A cross-species alignment of
those sequences showed that their corresponding amino acids were
highly evolutionarily conserved (data available upon request).
Online bioinformatics software (PolyPhen-2, SIFT and
MutationTaster) was used to predict the functional effects of the
altered proteins in the DCM patients, resulting fromin? the seven
novel mutations and three variants of uncertain significance. The
results of the present analysis implied that these altered amino
acids damage protein functions, as these mutations were predicted
to localise to the functional region of the proteins (data
available upon request).
Of the patients with DCM recruited for this study
harbouring identified mutations, mutations in sarcomere genes and
cytoskeletal genes were the most common, with a prevalence of
~66.7% (8/12) and ~25% (3/12), respectively (18). Of the tested genes in the present
study, sarcomere (MYBPC3, MYH7, MYH6,
TNNT2, TNNC1 and MYL3) and cytoskeletal genes
(MYPN, DES and VCL) were the most frequently
mutated. It is worth noting that MYH6 (p.R1047C and
p.T1253M) and MYBPC3 (p.R160W and p.C1124F) gene mutations
were found in two patients; the other gene mutations (MYH7,
TNNT2, TNNC1, MYL3, MYPN, DES,
RBM20 and VCL) were identified in one patient each.
Comparatively, MYH6 and MYBPC3 gene mutations had not
occurred at such a high frequency in previously documented DCM
patients (2,44,37). This implies that genetic testing
on sarcomere and cytoskeletal genes is a valuable diagnostic tool
for individuals at a high risk for DCM. However, due to the
limitations caused by the small sample size in this study, it is
necessary to increase the sample size in future studies to confirm
the genetic basis of SDCM.
In the current study, the prevalence and
distribution of disease genes, the spectrum of gene mutations, and
clinical features were first reported in DCM patients in Yunnan
Province, China. We found a mutation in 57.2% of the tested DCM
patients, a total of 12 non-synonymous mutations, and of these,
seven novel mutations, were identified using NGS. MYBPC3 and
MYH6, both sarcomere protein-encoding genes, were the most
commonly identified. Therefore, our results indicate that targeted
gene sequencing is a feasible approach to the identification of
pathogenic mutations in DCM patients. Compared with whole genome
sequencing, whole exome sequencing, and Sanger sequencing, NGS
screening is efficient, fast, and cost-effective (45,46). We suggest that genetic testing on
DCM patients thus provides the most effective means to identify
at-risk family members, particularly those whose clinical features
are mild or ambiguous. It is also important for early diagnostic
clinical evaluation and the better management of family members at
risk for DCM.
However, there were several limitations to the
present study. The only information obtained for the control
subjects was age, gender, ethnicity and certain clinical
characteristics. Only 21 patients with DCM were recruited and
screened, thus resulting in a small sample size. Thus, additional
independent studies with a larger sample size are warranted in
order to confirm our results. In the current study, we have only
described the gene mutations of DCM patients; therefore, further
studies on the mechanisms of novel mutations are necessary in order
to elucidate the disease mechanisms at the level of cell or
transgenic animal models.
Acknowledgments
The authors acknowledge the contributions of the
participating patients in the dilated cardiomyopathy registry, and
the authors would like to thank the staff of The First Hospital of
Yunnan Province for providing their support. The present study was
supported by Major Program of Applied Basic Research of Yunnan
Province, China (grant no. 2013FC007).
References
|
1
|
Taylor MR, Carniel E and Mestroni L:
Cardiomyopathy, familial dilated. Orphanet J Rare Dis. 1:272006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hershberger RE and Siegfried JD: Update
2011: clinical and genetic issues in familial dilated
cardiomyopathy. J Am Coll Cardiol. 57:1641–1649. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Michels VV, Moll PP, Miller FA, Tajik AJ,
Chu JS, Driscoll DJ, Burnett JC, Rodeheffer RJ, Chesebro JH and
Tazelaar HD: The frequency of familial dilated cardiomyopathy in a
series of patients with idiopathic dilated cardiomyopathy. N Engl J
Med. 326:77–82. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hershberger RE, Hedges DJ and Morales A:
Dilated cardiomyopathy: the complexity of a diverse genetic
architecture. Nat Rev Cardiol. 10:531–547. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhao L, Xu JH, Xu WJ, Yu H, Wang Q, Zheng
HZ, Jiang WF, Jiang JF and Yang YQ: A novel GATA4 loss-of-function
mutation responsible for familial dilated cardiomyopathy. Int J Mol
Med. 33:654–660. 2014.
|
|
6
|
Zhou W, Zhao L, Jiang JQ, Jiang WF, Yang
YQ and Qiu XB: A novel TBX5 loss-of-function mutation associated
with sporadic dilated cardiomyopathy. Int J Mol Med. 36:282–288.
2015.PubMed/NCBI
|
|
7
|
Xu L, Zhao L, Yuan F, Jiang WF, Liu H, Li
RG, Xu YJ, Zhang M, Fang WY, Qu XK, et al: GATA6 loss-of-function
mutations contribute to familial dilated cardiomyopathy. Int J Mol
Med. 34:1315–1322. 2014.PubMed/NCBI
|
|
8
|
Zhang XL, Dai N, Tang K, Chen YQ, Chen W,
Wang J, Zhao CM, Yuan F, Qiu XB, Qu XK, et al: GATA5
loss-of-function mutation in familial dilated cardiomyopathy. Int J
Mol Med. 35:763–770. 2015.
|
|
9
|
Yuan F, Qiu XB, Li RG, Qu XK, Wang J, Xu
YJ, Liu X, Fang WY, Yang YQ and Liao DN: A novel NKX2-5
loss-of-function mutation predisposes to familial dilated
cardiomyopathy and arrhythmias. Int J Mol Med. 35:478–486.
2015.
|
|
10
|
Hershberger RE, Morales A and Siegfried
JD: Clinical and genetic issues in dilated cardiomyopathy: a review
for genetics professionals. Genet Med. 12:655–667. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Murphy RT, Mogensen J, Shaw A, Kubo T,
Hughes S and McKenna WJ: Novel mutation in cardiac troponin I in
recessive idiopathic dilated cardiomyopathy. Lancet. 363:371–372.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Roh JI, Cheong C, Sung YH, Lee J, Oh J,
Lee BS, Lee JE, Gho YS, Kim DK, Park CB, et al: Perturbation of
NCOA6 leads to dilated cardiomyopathy. Cell Rep. 8:991–998. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Man E, Lafferty KA, Funke BH, Lun KS, Chan
SY, Chau AK and Chung BH: NGS identifies TAZ mutation in a family
with X-linked dilated cardiomyopathy. BMJ Case Rep. 2013:pii:
bcr2012007529. 2013.
|
|
14
|
Charron P, Arad M, Arbustini E, Basso C,
Bilinska Z, Elliott P, Helio T, Keren A, McKenna WJ, Monserrat L,
et al: European Society of Cardiology Working Group on Myocardial
and Pericardial Diseases: Genetic counselling and testing in
cardiomyopathies: a position statement of the European Society of
Cardiology Working Group on Myocardial and Pericardial Diseases.
Eur Heart J. 31:2715–2726. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Posafalvi A, Herkert JC, Sinke RJ, van den
Berg MP, Mogensen J, Jongbloed JD and van Tintelen JP: Clinical
utility gene card for: dilated cardiomyopathy (CMD). Eur J Hum
Genet. 21:212013. View Article : Google Scholar
|
|
16
|
Mardis ER: Next-generation DNA sequencing
methods. Annu Rev Genomics Hum Genet. 9:387–402. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schuster SC: Next-generation sequencing
transforms today's biology. Nat Methods. 5:16–18. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ware SM: Genetic diagnosis in pediatric
cardiomyopathy: clinical application and research perspectives.
Prog Pediatr Cardiol. 31:99–102. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Biswas A, Rao VR, Seth S and Maulik SK:
Next generation sequencing in cardiomyopathy: towards personalized
genomics and medicine. Mol Biol Rep. 41:4881–4888. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Punetha J and Hoffman EP: Short read
(next-generation) sequencing: a tutorial with cardiomyopathy
diagnostics as an exemplar. Circ Cardiovasc Genet. 6:427–434. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mestroni L, Maisch B, McKenna WJ, Schwartz
K, Charron P, Rocco C, Tesson F, Richter A, Wilke A and Komajda M:
Guidelines for the study of familial dilated cardiomyopathies.
Collaborative Research Group of the European Human and Capital
Mobility Project on Familial Dilated Cardiomyopathy. Eur Heart J.
20:93–102. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li H and Durbin R: Fast and accurate
long-read alignment with Burrows-Wheeler transform. Bioinformatics.
26:589–595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R: 1000 Genome
Project Data Processing Subgroup: The Sequence Alignment/Map format
and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Koboldt DC, Zhang Q, Larson DE, Shen D,
McLellan MD, Lin L, Miller CA, Mardis ER, Ding L and Wilson RK:
VarScan 2: Somatic mutation and copy number alteration discovery in
cancer by exome sequencing. Genome Res. 22:568–576. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang K, Li M and Hakonarson H: ANNOVAR:
functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tester DJ and Ackerman MJ: Genetic testing
for potentially lethal, highly treatable inherited
cardiomyopathies/channelopathies in clinical practice. Circulation.
123:1021–1037. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Maron BJ and Semsarian C: Prevention of
sudden death for patients with cardiomyopathies another step
forward. J Am Coll Cardiol. 59:501–502. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chiu C, Tebo M, Ingles J, Yeates L, Arthur
JW, Lind JM and Semsarian C: Genetic screening of calcium
regulation genes in familial hypertrophic cardiomyopathy. J Mol
Cell Cardiol. 43:337–343. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Richards CS, Bale S, Bellissimo DB, Das S,
Grody WW, Hegde MR and Lyon E: ACMG recommendations for standards
for interpretation and reporting of sequence variations: Revisions
2007. Genet Med. 10:294–300. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kumar P, Henikoff S and Ng PC: Predicting
the effects of coding non-synonymous variants on protein function
using the SIFT algorithm. Nat Protoc. 4:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schwarz JM, Rödelsperger C, Schuelke M and
Seelow D: MutationTaster evaluates disease-causing potential of
sequence alterations. Nat Methods. 7:575–576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang S, Zou Y, Fu C, Xu X, Wang J, Song L,
Wang H, Chen J, Wang J, Huan T and Hui R: Worse prognosis with gene
mutations of beta-myosin heavy chain than myosin-binding protein C
in Chinese patients with hypertrophic cardiomyopathy. Clin Cardiol.
31:114–118. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Robinson P, Griffiths PJ, Watkins H and
Redwood CS: Dilated and hypertrophic cardiomyopathy mutations in
troponin and alpha-tropomyosin have opposing effects on the calcium
affinity of cardiac thin filaments. Circ Res. 101:1266–1273. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mirza M, Marston S, Willott R, Ashley C,
Mogensen J, McKenna W, Robinson P, Redwood C and Watkins H: Dilated
cardiomyopathy mutations in three thin filament regulatory proteins
result in a common functional phenotype. J Biol Chem.
280:28498–28506. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mller DV, Andersen PS, Hedley P, Ersbll
MK, Bundgaard H, Moolman-Smook J, Christiansen M and Kber L: The
role of sarcomere gene mutations in patients with idiopathic
dilated cardiomyopathy. Eur J Hum Genet. 17:1241–1249. 2009.
View Article : Google Scholar
|
|
37
|
Hershberger RE, Norton N, Morales A, Li D,
Siegfried JD and Gonzalez-Quintana J: Coding sequence rare variants
identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312
patients with familial or idiopathic dilated cardiomyopathy. Circ
Cardiovasc Genet. 3:155–161. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hershberger RE, Parks SB, Kushner JD, Li
D, Ludwigsen S, Jakobs P, Nauman D, Burgess D, Partain J and Litt
M: Coding sequence mutations identified in MYH7, TNNT2, SCN5A,
CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic
dilated cardiomyopathy. Clin Transl Sci. 1:21–26. 2008. View Article : Google Scholar
|
|
39
|
Lakdawala NK, Funke BH, Baxter S, Cirino
AL, Roberts AE, Judge DP, Johnson N, Mendelsohn NJ, Morel C, Care
M, et al: Genetic testing for dilated cardiomyopathy in clinical
practice. J Card Fail. 18:296–303. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu W, Liu W, Hu D, Zhu T, Ma Z, Yang J,
Xie W, Li C, Li L, Yang J, et al: Mutation spectrum in a large
cohort of unrelated Chinese patients with hypertrophic
cardiomyopathy. Am J Cardiol. 112:585–589. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Matsushita Y, Furukawa T, Kasanuki H,
Nishibatake M, Kurihara Y, Ikeda A, Kamatani N, Takeshima H and
Matsuoka R: Mutation of junctophilin type 2 associated with
hypertrophic cardiomyopathy. J Hum Genet. 52:543–548. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zou Y, Wang J, Liu X, Wang Y, Chen Y, Sun
K, Gao S, Zhang C, Wang Z, Zhang Y, et al: Multiple gene mutations,
not the type of mutation, are the modifier of left ventricle
hypertrophy in patients with hypertrophic cardiomyopathy. Mol Biol
Rep. 40:3969–3976. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang J, Xu SJ, Zhou H, Wang LJ, Hu B, Fang
F, Zhang XM, Luo YW, He XY, Zhuang SW, et al: A novel mutation of
the beta myosin heavy chain gene responsible for familial
hypertrophic cardiomyopathy. Clin Cardiol. 32:E16–E21. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Carniel E, Taylor MR, Sinagra G, Di
Lenarda A, Ku L, Fain PR, Boucek MM, Cavanaugh J, Miocic S, Slavov
D, et al: Alpha-myosin heavy chain: a sarcomeric gene associated
with dilated and hypertrophic phenotypes of cardiomyopathy.
Circulation. 112:54–59. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Herman DS, Hovingh GK, Iartchouk O, Rehm
HL, Kucherlapati R, Seidman JG and Seidman CE: Filter-based
hybridization capture of subgenomes enables resequencing and
copy-number detection. Nat Methods. 6:507–510. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sikkema-Raddatz B, Johansson LF, de Boer
EN, Almomani R, Boven LG, van den Berg MP, van Spaendonck-Zwarts
KY, van Tintelen JP, Sijmons RH, Jongbloed JD and Sinke RJ:
Targeted next-generation sequencing can replace Sanger sequencing
in clinical diagnostics. Hum Mutat. 34:1035–1042. 2013. View Article : Google Scholar : PubMed/NCBI
|