1. Introduction
Drug repositioning has become an increasingly
attractive alternative to traditional drug development. Two major
factors have contributed to this change, specifically cost and
time. Traditional drug development consists of three stages:
Discovery, preclinical and clinical testing. Each phase faces
unique challenges. The drug is first tested for efficacy using
in vitro or in silico modeling against specific
targets in the discovery stage. The drug is subsequently tested
with animal models in the preclinical stage, and human testing is
conducted in the first clinical stage. While certain drugs in
development may appear promising, there is no guarantee the drug
will be effective in humans (1).
Approximately 60% of the new oncology drugs under development
throughout the 1990's have successfully completed phase 2 programs
and subsequently failed in phase 3 clinical trials. Only 5% of all
agents developed for oncology applications in this time period have
ever reached application (2).
The cost of drug development has ballooned within
the United States (US) over the past decade, rising from $26
billion in 1998 to $50 billion in 2008 (3). The increase in cost is due in part
to the increase in time required to develop a drug and secure
approval from the US Food and Drug Administration (FDA); since the
1990's this time period has increased to 15 years (1). Further contributing to high
development costs is the increasingly strict regulatory environment
imposed by the FDA, resulting in high attrition rates for drugs in
development. Between 1999 and 2004, the probability of a drug in
the clinical stage reaching the market was only 16%. The drug
industry in general has become an increasingly unmanageable
environment (3).
In contrast to traditional drug development, the
process of repositioning, repurposing, redirecting or reprofiling
of existing approved drugs involves discovering uses beyond their
initial medical applications (4).
Such an approach has previously been commonly known as 'off-label'
use, such as when physicians make a decision to prescribe a
particular drug for unapproved indications, age groups, dosages or
forms of administration. Repositioning of existing and marketed
drugs reduces safety and pharmacokinetic uncertainty due to the
extensive pharmacokinetic and toxicological data already available
(5). Reliable information also
exists in formulation, bioactivities and pharmaceutical properties,
such as absorption, distribution, metabolism, excretion and
toxicity. As a result, the development process can be reduced to
just 3–12 years (4). Knowledge of
the traditional dosing and therapeutic profile of the repositioned
drugs can be readily integrated with current scientific advances to
provide a more informed framework on how patients may benefit from
these drugs and anticipation of untoward side effects.
Another benefit of drug repositioning research and
development is that it can accurately predict new drug targets as
primary sites of action, and also identify off-targets and
mediators of side effects (6).
Campillos et al (6)
compared side-effect information between unrelated drugs to infer
common protein targets. It was experimentally concluded that drugs
sharing a similar chemical structure and similar side effects
likely bind to associated targets, sometimes beyond the drug's
therapeutic indication. By contrast, Keiser et al (7) predicted new molecular targets of
drugs by comparing the similarity of the ligands targeted. The
advantage of their method is its ability to capture similarities
between drugs that are otherwise dissimilar in chemical structure.
For example, Rescriptor was indicated to bind the histamine H4
receptor, a target unrelated to its therapeutic function as a
reverse transcriptase inhibitor, but was consistent with the
painful rashes associated with the use of Rescriptor.
Drug repositioning studies can also compare gene
expression between drugs and disease states. Lamb et al
(8) illustrated the value of a
large publicly available database known as the 'Connectivity Map'
that could be used to study the associations between drugs, genes
and disease. The Connectivity Map can be applied without the use of
extensively precise experiments. Using this method, Sirota et
al (9) hypothesized that a
drug has a potential therapeutic use for a disease state and a drug
created opposite patterns of gene expression. Cimetidine's primary
therapeutic use was to treat stomach acidity and peptic ulcers due
to its antagonism of the histamine H2 receptor; however, the
Connectivity Map showed that cimetidine could be used to treat lung
adeno-carcinoma. This was confirmed experimentally through a
statistically significant reduction in tumor size. Thus, exploring
gene expression to characterize disease and drug signature is a
viable method to reposition drugs. Given the wealth of evidence
showing that convergent signaling pathways can elicit divergent
downstream cellular events in different diseased states, the
potential for repurposing existing drugs for novel therapeutic
indications appears immeasurable.
There are numerous examples of the success of drug
repositioning, but a notable example is the repurposing of
sildenafil. Sildenafil was developed by Pfizer to treat angina via
inhibition of phosphodiesterase-5 (4). Although sildenafil proved to be
ineffective in treating angina, researchers observed that
inhibition of phosphodiesterase-5 resulted in erections among male
subjects (4). Following this
discovery, Pfizer repositioned and successfully remarketed
sildenafil under the brand name Viagra, as a treatment for erectile
dysfunction (4).
The present review focuses on the use of the
drug-target interactome (DTome) database to illustrate the
developing field of drug repositioning, as applied to probenecid,
previously known as Benemid. Probenecid was initially introduced to
slow the elimination of the antibiotic penicillin via renal tubular
secretion (10,11). In the 1950's and 1960's the
'Probenecid Test' was used as a clinical tool to diagnose
depression and psychiatric disorders (10,11). Currently, probenecid is primarily
used as a long-term therapy for patients with refractory gout or in
patients who are unresponsive to first-line treatments (12). It is also used in the laboratory
to investigate intracellular calcium within in vitro cells
(11). However, new evidence
suggests that probenecid may have other therapeutic potentials. The
traditional therapeutic target of probenecid in the treatment of
gout will first be reviewed, followed by an overview on the
potential for repositioning probenecid as an anticancer and
anti-viral drug by exploring drug-protein and secondary
protein-protein interactions.
2. Gout
Gout is a metabolic disorder characterized by
chronic elevation in uric acid levels >6.8 mg/dl. A sustained
increase in uric acid causes hyperuricemia and results in the
formation and deposition, preferentially in joints, of monosodium
urate monohydrate crystals, otherwise known as tophi (13). Upon shedding, tophi are believed
to cause intense pain by triggering acute but self-limiting joint
inflammation (14). It is
estimated that the prevalence of gout has risen to 3.9% from 2.7%
between 1988 and 1994 based on the National Health and Nutrition
Examination Survey (15).
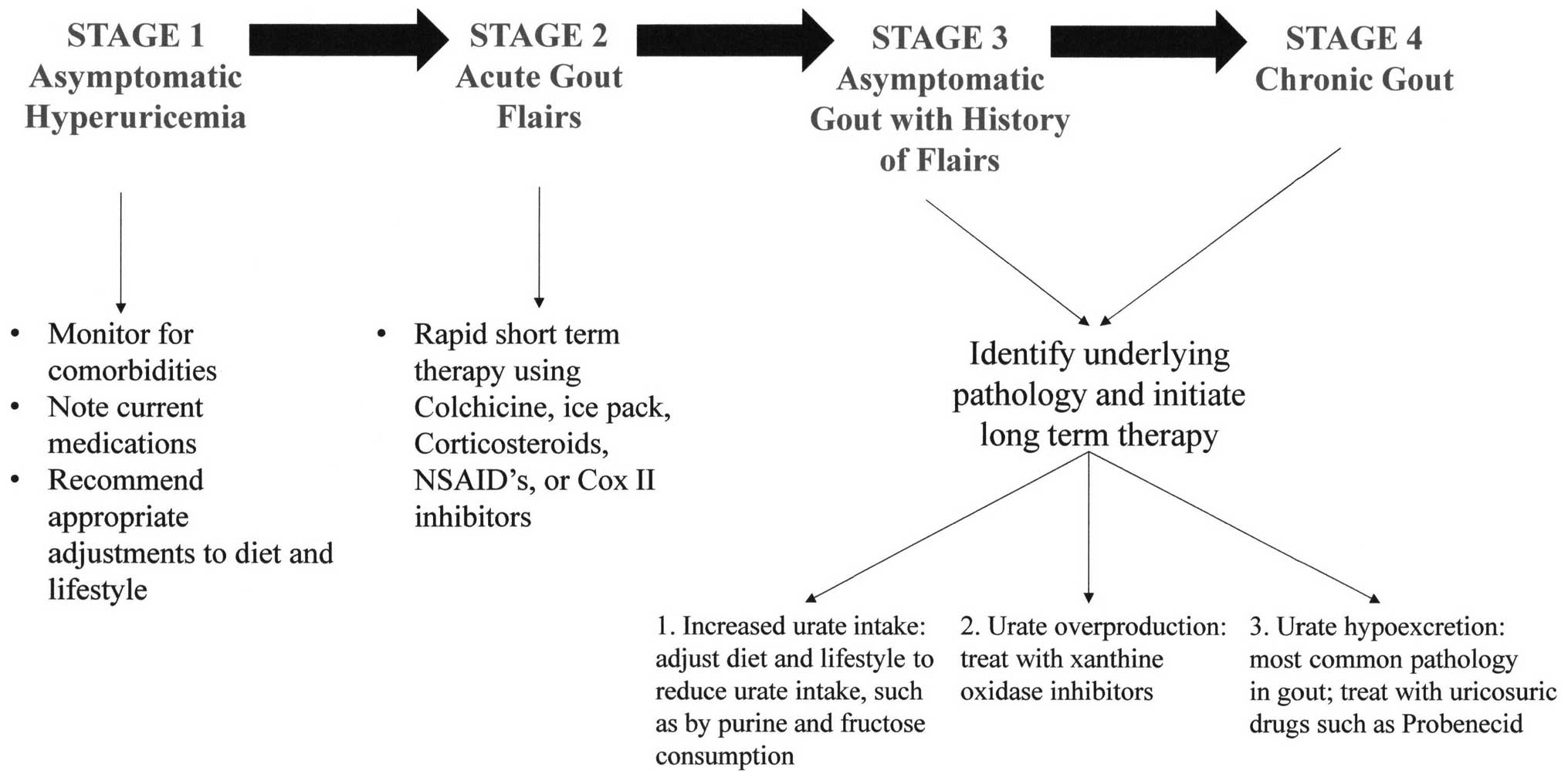
Gout has been proposed to progress in four stages
(Fig. 1) (16). Stage 1 is characterized by
asymptomatic hyperuricemia that has the potential to develop into
gout. Acute gout flairs are classified as stage 2. In stage 3,
periods are susceptible to additional flairs and otherwise
asymptomatic hyperuricemia prevail, whereas establishment of stage
4 is marked by the presence of chronic gout. The objective of
short-term therapy of gout is to manage acute episodes and minimize
the joint inflammation observed in stage 2. A popular short-term
treatment is colchicine, a non-selective inhibitor of inflammasome
activity and interleukin-1 production in leukocytes and migration
of leukocytes to affected areas, as well as release of histamine
from mast cells (14). In
contrast to short-term treatments, long-term therapy manages
patients in stages 3 and 4.
Hyperuricemia and gout can be attributed to the
overproduction, underexcretion or increased intake of urate
(16). As shown in Table I, urate-lowering therapy (ULT)
includes administration of drugs that act to prevent uric acid
formation, e.g., allopurinol or febuxostat, or enhance its
excretion, such as probenecid, as well as changes in the diet and
lifestyle. ULT drugs are prescribed as necessary to achieve
non-hyperuricemic blood plasma levels (17).
 | Table ILong-term therapies of gout. |
Table I
Long-term therapies of gout.
| Drug | Target | Effects | Concerns |
|---|
| Allopurinol | Purine analog
xanthine oxidase inhibitor | Reduced production
of uric acid | Allopurinol
hypersensitivity syndrome, renal impairment |
| Febuxostat | Xanthine oxidase
inhibitor | Reduced production
of uric acid | Liver impairment,
cost |
| Probenecid | Uricosuric effect
via competitive inhibition of URAT1a | Decreased
reabsorption of urate | Urate crystal
development in urinary tract, hypersensitivity |
Allopurinol, a purine analogue that lowers the
production of uric acid by competitively inhibiting the enzyme
xanthine oxidase is the ULT drug of choice (13,14,16,17). In patients that experience
Allopurinol Hypersensitivity Syndrome or have a history of renal
insufficiency, febuxostat, a xanthine oxidase inhibitor lacking a
purine core, is metabolized in the liver (13,14,16,17) and can safely be used in patients
with renal impairment, is a low-cost second option for patients who
are contraindicated for allopurinol.
3. Probenecid as a uricosuric drug
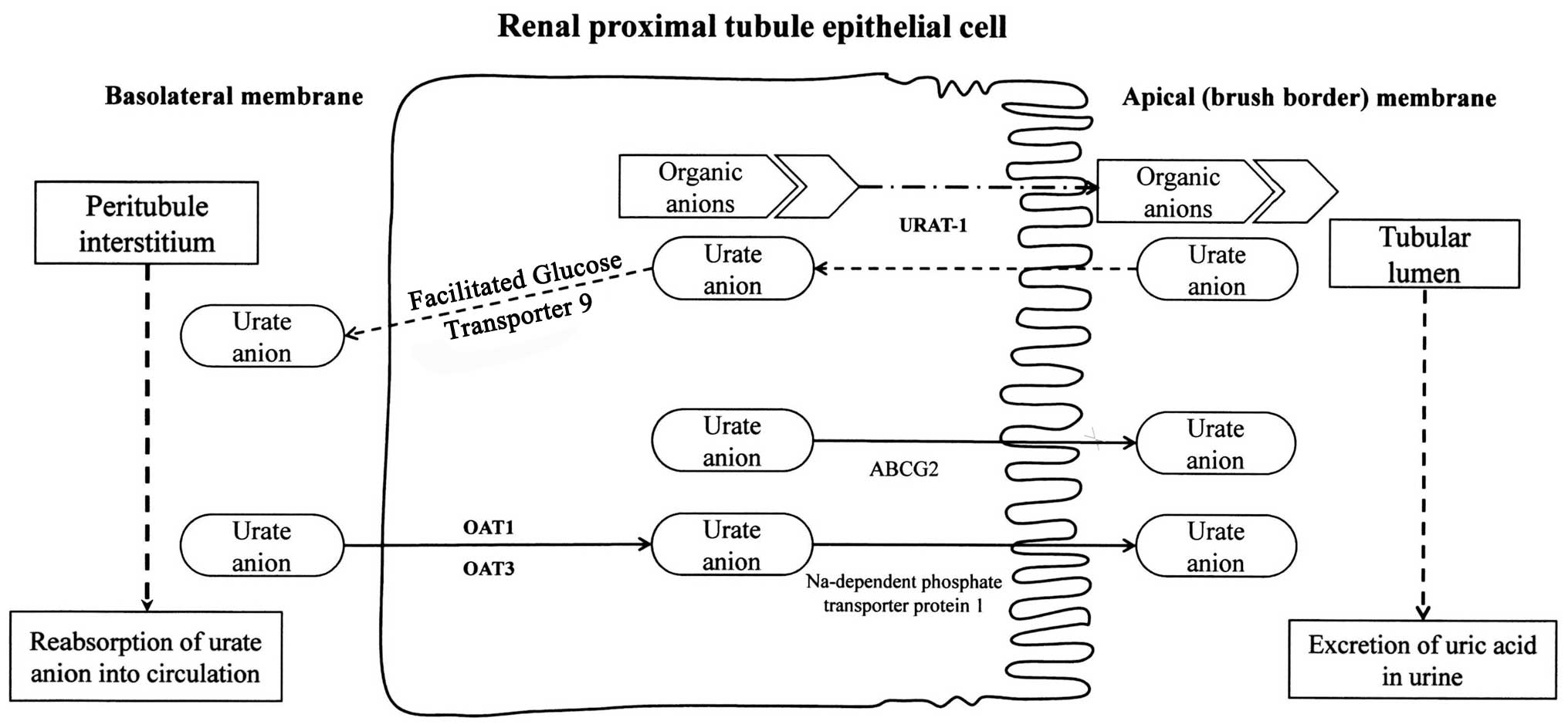
The process by which the human renal proximal tubule
extensively reabsorbs urate is blocked by uricosuric drugs,
including probenecid (12). It is
estimated that 90% of the daily load of urate is reabsorbed, and
this process is sustained by a family of organic anion transporters
(OATs) including the urate transporter 1 (URAT1), which are
targeted and inhibited by uricosuric drugs (18) (Fig.
2). URAT1 is a sodium-independent exchanger of chloride and
organic anions including urate (19,20). Therefore, probenecid can be used
as a long-term therapy of gout by minimizing post-secretory
reabsorption of urate in hyperuricemic patients. In addition to
URAT1, probenecid inhibits other organic anion transporters,
including OAT1, OAT2, OAT3 and OAT4, to varying degrees (19). While URAT1 transports urate across
the luminal membrane of the renal proximal tubule, it is believed
that OAT1 and OAT3 transport urate across the basolateral membrane.
For probenecid to have its intended cis-inhibitory effect it must
be excreted into the renal tubule. Once it is in the lumen of the
renal tubule, it can interact with URAT1 (20).
According to the Four Component Hypothesis (12), transport of urate begins with
glomerular filtration, reabsorption of filtered urate, secretion
and post-secretory reabsorption (12,21). In this mechanism, probenecid most
likely reduces post-secretory reabsorption by inhibiting URAT1.
Notably, urinary urate excretion was observed to increase by ≤84%
in 47 patients with chronic renal failure, suggesting loss of
function of OATs such as URAT1 that contribute to post-secretory
reabsorption of urate (21). The
paradoxical effect mimics that of probenecid, and therefore
patients with chronic renal failure are likely contraindicated for
probenecid and may have a reduced response to the drug.
Probenecid is highly lipid soluble and also strongly
binds plasma proteins such as albumin (10). Humans metabolize probenecid by
oxidation of alkyl side chains and to a lesser extent by
glucuronide conjugation; however the oxidized metabolites have
nearly equal uricosuric effects as probenecid (10). Probenecid is eliminated from the
human body by the renal system, and its renal clearance is
dependent on pH and urine flow rate (10). Maximal renal clearance of
probenecid is established by alkaline urine and increasing urine
flow rate. Maintaining alkaline urine also minimizes the
development of urate crystals in the urinary tract when using
probenecid (22). Probenecid is
preferentially administered orally, and use can result in side
effects, although severe side effects are rare. The most severe
side effect is probenecid hypersensitivity, which is characterized
by fever, rash, nausea, vomiting and vasomotor collapse, which
occurred in 8 subjects in a study observing 2,502 patients using
probenecid (22). There was no
observed hepatic or renal toxicity observed, and to date no
fatalities have been caused by use of the compound.
Gastrointestinal problems, ranging from abdominal distress and
vomiting to diarrhea, appear to be the most common side effects
observed (22).
When comparing their effectiveness as urate lowering
drugs, there is no clear advantage of allopurinol over probenecid
(23). However, allopurinol is
usually preferred by physicians due to its reduced side-effect
profile and higher tolerability in comparison to probenecid. In a
study of 31 gout patients, it was shown that probenecid prescribed
concomitantly with allopurinol increased the overall hypouricemic
effect and reduced plasma urate levels by an additional 25% in
comparison to allopurinol alone (24). Notably, in addition to urate,
oxypurinol, the metabolite of allopurinol, is also a substrate of
URAT1. Probenecid blocks post-secretory uptake of oxypurinol and
reduces the efficacy of allopurinol. However, the uricosuric action
of probenecid was observed to compensate for this undesired effect
(24).
Based on the above evidence, probenecid is a viable
option for the long-term management of gout. However, xanthine
oxidase inhibitors, such as allopurinol have a smaller side-effect
profile and an increased tolerability, and thus maintain their
position as the primary drug of choice for gout. Thus, probenecid
is generally only used in patients with refractory gout or in
patients who cannot tolerate allopurinol or febuxostat. While it
has been traditionally prescribed as a monotherapy, the future
direction of probenecid may involve increased concomitant use with
other ULT medications, such as xanthine oxidase inhibitors.
4. Repositioning of probenecid using DTome
analysis
The observed pronounced effects probenecid exerts on
OATs, coupled with the fact that OATs have a vital role in the
distribution and excretion of several widely prescribed drugs
prompted us to query whether probenecid may have novel targets and
uses. Our approach was to use the DTome (25) database to investigate drug-drug,
drug-protein and second level protein-protein interactions for
probenecid (Fig. 3). These
interactions were subsequently visualized using Cytoscape (26), a program for network analysis
(Fig. 4). The network generated
from the DTome results was further expanded using Cytoscape tools,
which query public interaction databases. The aim was to develop a
network centered on the off-target interactions of probenecid with
genes involved with carcinogenesis, or altered in cancers, as well
as targets that may have an anti-viral effect. Results from the
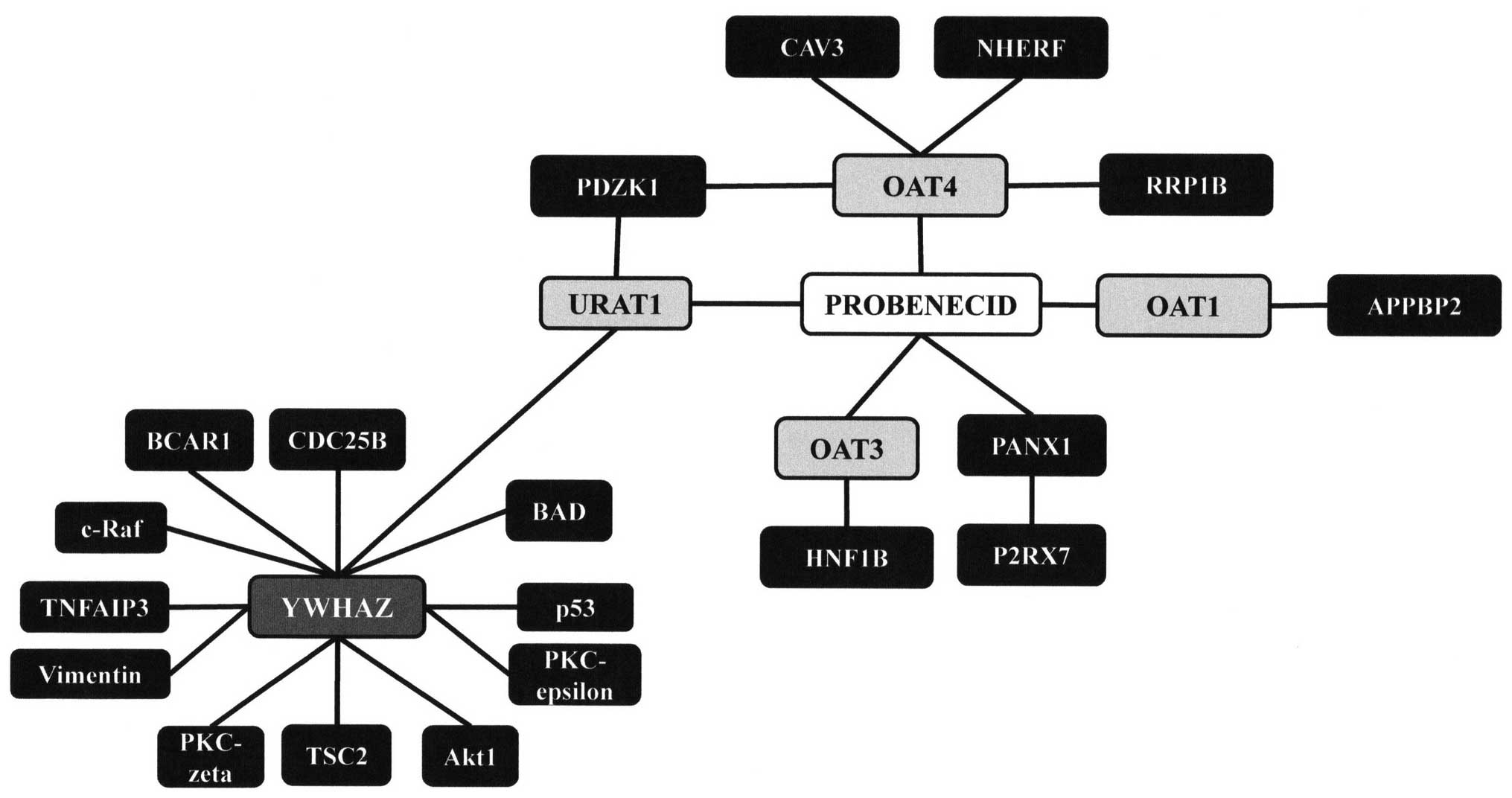
DTome database showed multiple proteins for which probenecid had
direct interactions. As expected, there is a direct interaction
between probenecid and URAT1, a member of the SLC22 family of
genes. This interaction has already been discussed as a path by
which probenecid can treat gout. Additionally, there were
interactions with OAT1, 3 and 4, all members of the SLC22 family of
genes, as well as a fourth protein pannexin 1 (PANX1), a protein
that is a structural component of gap junctions.
5. Repositioning of probenecid as an
antiviral drug
As mentioned earlier, probenecid was used in the
past to increase the plasma concentrations of medications such as
penicillin when such treatments were in limited supply. There is a
renewed interest in using probenecid to increase the efficacy of
medications for hard to treat conditions such as human
immunodeficiency virus (HIV) infection, in addition to other viral
infections. While probenecid targets several different organic
anion transporters, two of primary interest are SLC22A6/OAT1 and
SLC22A8/OAT3 (19). In a
randomized, two-way crossover study of 12 healthy subjects,
probenecid was observed to competitively inhibit the reabsorption
of ciprofloxacin, a quinolone antimicrobial agent. It was suggested
that ciprofloxacin was competitively blocked from OAT1 by the
actions of probenecid, although no conclusions could be made
regarding which transporters were involved (27). While they share a degree of
homology, OATs also have unexpected differences in substrate
binding and transport. For example, adefovir, tenofovir, cidofovir
and acyclovir all have strong affinities for OAT1 and complementary
weak affinities for OAT3 (28).
Several antivirals are substrates of either OAT1 or OAT3, and thus
have the potential to be manipulated by probenecid.
In addition to being in the proximal tubule of the
renal system, OAT1 and OAT3 have also been localized to the
modified cuboidal epithelial cells of the choroid plexus (CP)
(29,30). Notably, OAT3 is a sodium-dependent
transporter, and has been further localized to the apical side of
CP epithelial cells (31). These
transporters function as an efflux mechanism to remove organic
anions from the cerebral spinal fluid (CSF) into the blood. For
example, the first nucleoside used in acquired immune deficiency
syndrome therapy zidovudine (AZT) is rapidly removed from the CSF.
This process is mediated by OATs, including OAT1 and OAT3, although
the exact contributions of each OAT remain unknown (32).
Probenecid can be used to increase antiretroviral
retention and alter their pharmacokinetic behavior. With the above
newly discovered OATs, probenecid can achieve this by reducing loss
of antivirals from the plasma into the renal system and reducing
efflux of antivirals from the CSF. When combined with probenecid,
blood plasma and CSF concentrations of AZT significantly increased
(33). However, a previous study
has suggested that severely immunodeficient patients, such as HIV
patients, are more likely to experience side effects with
probenecid, limiting the dose that can be safely administered. In 8
HIV patients prescribed probenecid concomitantly with AZT, 6
experienced rashes, and 3 of those 6 had severe rashes. These
patients were prescribed 500 mg probenecid every 8 h (34).
Another study compared the efficacy and safety of a
reduced dose probenecid combined with Cidofovir, an acyclic
nucleotide analog of cytosine used to treat cytomegalovirus, an
opportunistic infection identified in severely immunosup-pressed
patients, such as HIV patients. In this randomized, open-label
comparison of the normal and reduced regimens of probenecid, the
lower dose of probenecid combined with cidofovir delivered
cidofovir without a significant change in its pharmacokinetics. The
lower dose also reduced the likelihood of side effects (35). Based on this evidence, the
pattern, timing and dose in which probenecid is administered
significantly influence the tolerability and likelihood of side
effects occurring. The present standard regimen of probenecid
should be restructured in severely immunosup-pressed patients to
improve the efficacy of concomitantly prescribed drugs and minimize
the occurrence of side effects. Furthermore, inhibition of an OAT,
such as OAT3, by probenecid may not result in chronic symptoms, as
OAT3-knockout mice did not have clear developmental defects in
major organ systems (30).
Probenecid can also be used concomitantly with
medications used to treat more common viral infections. Probenecid
can be combined with oseltamivir, an antiviral used to treat
influenza. When probenecid was prescribed four times daily, there
was a significant decrease in renal excretion of oseltamivir, such
that the dose of the antiviral could be decreased without
compromising its efficacy (36).
However, prescribing probenecid four times daily poses several
challenges, including increasing the likelihood of the patient to
develop side effects. Notably, in another study it was discovered
that probenecid inhibits the gene expression of OAT3. The classical
inhibition of OAT3 by probenecid significantly reduced the virus
load of influenza A (37). It is
hypothesized that by competitively inhibiting OAT3 and reducing its
gene expression, probenecid attenuates the transport of certain
viral factors required for infection. When probenecid was combined
with oseltamivir, the virus load of influenza A was further reduced
compared to probenecid alone (37).
6. Repositioning of probenecid as an
anticancer agent
Disruptions of the PANX1 gene have been associated
with melanoma tumor progression (38). The tumor progression associated
with alterations in the PANX1 gene may be due to direct factors, or
may be due to interaction with another protein, P2X purinoceptor 7
(P2RX7). It has been shown that P2RX7 is an activator of PANX1
(39); however, the two may also
interact in other ways. P2RX7 has been shown to activate tumor
necrosis factor-α (TNF-α), and may mediate apoptosis through that
pathway (40).
Querying public databases using Cytoscape, a
possible interaction was identified from a proteomics study in
mice, using codetermination techniques, between URAT1 and another
protein, YWHAZ. YWHAZ, also known as 14-3-3 protein ζ/δ, is a
protein that mediates signal transduction by binding to
phosphoserine-containing proteins (41). Modulation of signal transduction
via binding to phosophoserine-containing proteins suggests that it
can have a major role in modulating survival kinases. In addition
to interaction with survival kinases, such as Akt (42), studies have shown that this gene
interacts with the critical p53 tumor suppressor gene (43) as well as numerous other genes
associated with carcinogenesis. The expression and activity of the
YWHAZ protein are altered in numerous cancer cell lines. There has
been little research surrounding the URAT1-YWHAZ interaction, and
no research has been conducted for human cell lines. This
interaction may be extremely important in identifying the
off-target, chemotherapeutic properties of probenecid and could be
an important area for future study.
Additional interactions that may be involved with
development of cancers reported for the SLC22 family of proteins
(OAT1, OAT3, OAT4 and URAT1) include CAV3, PDZK1, RRP1B, HNF1B,
APPBP29 and NHERF (44). CAV3
belongs to the caveolin family of proteins serving as scaffolds for
the organization of the caveolae plasma membranes, which act as
reception and interface sites for signaling molecules in mammalian
cells. CAV3 has also been shown to interact with EGFR to initiate
events impinging on the regulation of survival kinases suggesting a
significant chemotherapeutic role (45). CAV3 reportedly interacts with
OAT4; however, structural domains and specific amino acid details
on their interaction remain to be determined. Future studies on the
molecular nature of CAV3:OAT4 binding could reveal insights on
probenecid as a potential chemotherapeutic agent. Similarly, PDZK1
has been shown to bind to URAT1 and OAT4, while NHREF has been
shown to bind with OAT4. Studies have shown that this association
increases the activity of OAT4 and possibly URAT1 (44). Of clinical relevance is the
observation that PDZK1 has a role in the regulation of breast
cancer resistance proteins (46).
Molecular connectivity reported to exist between RRP1B and OAT4 and
other members of the SLC22 family, respectively, HNF1B with OAT3
and OAT1 with APPBP29, all point to their participatory role in
carcinogenesis (25) and provide
the rationale for considering the repurposing of probenecid as a
drug with anticancer activities.
7. Conclusion
Drug repositioning is becoming an increasingly
attractive choice over traditional drug development as a result of
sky-rocketing cost and time of development. Three major
methodologies exist for drug repositioning: Exploring drug-protein
and secondary protein-protein interactions, indirect and downstream
drug-ligand interactions, or drug-gene interactions using a
'Connectivity Map'. This review explored the efficacy of drug
repositioning by testing drug-protein and secondary protein-protein
interactions of probenecid using the DTome database. Probenecid was
chosen as the focus of the present study due to decades of publicly
available research and data.
The drug-protein interactions of probenecid were
analyzed using DTome, and secondary protein-protein interactions
were probed thereafter. The primary drug-protein interactions
discovered included URAT1, OAT1, OAT3, OAT4 and PANX1. The accuracy
of this methodology was supported by the presence of URAT1 in the
results, which determined that a sodium-independent exchanger is
involved in the post-secretory reabsorption of urate. Extensive
study has proven that probenecid can block URAT1 and therefore be
used as a treatment for gout.
While URAT1 is the primary target of probenecid for
the treatment of gout, its other targets have been attributed with
its side effects. These targets, including the OATs and PANX1, were
further explored as possible avenues for drug repositioning. First,
probenecid had distinct drug-drug interactions due to its
interaction with OAT1 and OAT3. Specifically, it blocked
reabsorption of antivirals and anti-microbials, supporting its use
as an adjuvant in hard to treat infections, such as with HIV. The
use of probenecid can also be extended to the central nervous
system as an adjuvant to maintain antimicrobial and antiviral drug
CSF concentrations, which are normally rapidly depleted as a result
of OAT1 and OAT3. Notably, while probenecid can be used as an
adjuvant, studies have indicated that probenecid itself reduces the
viral load by attenuating viral factor uptake via OAT3. Further
studies are necessary to evaluate the efficacy and safety of
probenecid in patients, particularly severely immunosuppressed
patients.
Thus, probenecid has an significant future as a
potential antiviral medication. While it can improve the efficacy
and tolerability of other antivirals, probenecid itself can also
contribute to antiviral treatments by blocking theorized viral
factor uptake by OAT3. On a similar note, another study has
discovered that multidrug-resistance protein 1 (MRP1) is localized
to microglia and may contribute to the adenosine
triphosphate-dependent efflux of antivirals from the CSF (47). MRP1 is another protein that can
interact with probenecid, and may be another avenue for the
repositioning of probenecid as an antiviral drug. However, further
study is required to elucidate the contribution that channels and
transporters make to the process of viral infection.
Probenecid also had several drug-protein and
protein-protein interactions that prompted exploration of its use
as an anticancer agent. A primary interaction with PANX1 suggested
the use of probenecid in treatment of melanoma. PANX1 may
contribute to cancer progression itself or by the TNF-α mediated
pathway via its interaction with P2RX7. Notably, URAT1 also shows
an interaction with YWHAZ, a signal transduction protein that
interacts with survival kinases, such as Akt as well as the p53
tumor suppressor gene. While Akt and p53 are crucial to tumor
progression, the URAT1-YWHAZ pathway involves several more proteins
associated with carcinogenesis and collectively may be a gateway to
manipulating and managing tumor growth and progression. Finally,
the SLC22 family of proteins, including the OATs, have
protein-protein interactions that are significant cancer pathways.
The interaction of OAT4 with CAV3 is involved in downregulation of
survival kinases. PDZK1 interacts with OAT4 and URAT1, and is
involved with the regulation of breast cancer resistance protein.
This multitude of interactions involves unique pathways among the
different targets of probenecid, and in certain instances involves
overlap of its targets as with the case of PDZK1. This network
suggests that the diverse set of cancer-related targets of
probenecid may combine synergistically in the clinical setting.
However, limited clinical studies have been performed to test these
pathways and more investigations are required.
The DTome analysis of the protein targets of
probenecid prompted the study on the possibility of repositioning
the drug as an antiviral or anticancer medication. The results were
numerous and promising; not only by identifying additional targets
previously considered to cause side effects such as the OATs, but
also pathways already used for therapeutic use such as URAT1
showing the potential for novel clinical use through new target
pathways (Table II).
Repositioning is best reserved for older and perhaps outdated
medicines, such as probenecid due to its ability to take maximum
advantage of a wealth of readily available study and data, thus
providing a good set of tools for a rapid and less expensive
approach to drug development.
| Table IIHypothesized associations between
targets of probenecid and potentially-related diseases. |
Table II
Hypothesized associations between
targets of probenecid and potentially-related diseases.
| Primary
targeta–f | Associated disease
process |
|---|
| URAT1a | Gout |
| OAT1 and 3b | General antiviral
adjuvant |
| CNS infections
adjuvant |
| Independent
antiviral effects via OAT3 |
| PANX1c | Melanomas |
| URAT1-PDZK1; | Breast cancer
resistance |
| OAT4-PDZK1d | |
| URAT1-YWHAZe | Carcinogenesis |
| OAT4-CAV3f | |
| OAT4-RRP1Bf | |
| OAT3-HNF1Bf | |
| OAT1-APPBP2f | |
References
|
1
|
Wilson JF: Alterations in processes and
priorities needed for new drug development. Ann Intern Med.
145:793–796. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu PY, LeBlanc M and Desai M: False
positive rates of randomized phase II designs. Control Clin Trials.
20:343–352. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kaitin KI: Deconstructing the drug
development process: The new face of innovation. Clin Pharmacol
Ther. 87:356–361. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ashburn TT and Thor KB: Drug
repositioning: Identifying and developing new uses for existing
drugs. Nat Rev Drug Discov. 3:673–683. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Padhy BM and Gupta YK: Drug repositioning:
Re-investigating existing drugs for new therapeutic indications. J
Postgrad Med. 57:153–160. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Campillos M, Kuhn M, Gavin AC, Jensen LJ
and Bork P: Drug target identification using side-effect
similarity. Science. 321:263–266. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Keiser MJ, Setola V, Irwin JJ, Laggner C,
Abbas AI, Hufeisen SJ, Jensen NH, Kuijer MB, Matos RC, Tran TB, et
al: Predicting new molecular targets for known drugs. Nature.
462:175–181. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lamb J, Crawford ED, Peck D, Modell JW,
Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, et
al: The Connectivity Map: Using gene-expression signatures to
connect small molecules, genes, and disease. Science.
313:1929–1935. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sirota M, Dudley JT, Kim J, Chiang AP,
Morgan AA, Sweet-Cordero A, Sage J and Butte AJ: Discovery and
preclinical validation of drug indications using compendia of
public gene expression data. Sci Transl Med. 3:96ra772011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cunningham RF, Israili ZH and Dayton PG:
Clinical pharmacokinetics of probenecid. Clin Pharmacokinet.
6:135–151. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Robbins N, Koch SE, Tranter M and
Rubinstein J: The history and future of probenecid. Cardiovasc
Toxicol. 12:1–9. 2012. View Article : Google Scholar
|
|
12
|
Roch-Ramel F and Guisan B: Renal transport
of urate in humans. News Physiol Sci. 14:80–84. 1999.
|
|
13
|
Terkeltaub R: Update on gout: New
therapeutic strategies and options. Nat Rev Rheumatol. 6:30–38.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rees F, Hui M and Doherty M: Optimizing
current treatment of gout. Nat Rev Rheumatol. 10:271–283. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhu Y, Pandya BJ and Choi HK: Prevalence
of gout and hyperuricemia in the US general population: The
National Health and Nutrition Examination Survey 2007–2008.
Arthritis Rheum. 63:3136–3141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Quillen DM: Crystal arthropathies:
Recognizing and treating 'the gouch'. Prim Care. 37:703–711. 2010.
View Article : Google Scholar
|
|
17
|
Hamburger M, Baraf HS, Adamson TC III,
Basile J, Bass L, Cole B, Doghramji PP, Guadagnoli GA, Hamburger F,
Harford R, et al: 2011 recommendations for the diagnosis and
management of gout and hyperuricemia. Phys Sportsmed. 39:98–123.
2011. View Article : Google Scholar
|
|
18
|
So A: Developments in the scientific and
clinical understanding of gout. Arthritis Res Ther. 10:2212008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Robertson EE and Rankin GO: Human renal
organic anion transporters: Characteristics and contributions to
drug and drug metabolite excretion. Pharmacol Ther. 109:399–412.
2006. View Article : Google Scholar
|
|
20
|
Enomoto A, Kimura H, Chairoungdua A,
Shigeta Y, Jutabha P, Cha SH, Hosoyamada M, Takeda M, Sekine T,
Igarashi T, et al: Molecular identification of a renal urate anion
exchanger that regulates blood urate levels. Nature. 417:447–452.
2002.PubMed/NCBI
|
|
21
|
Garyfallos A, Magoula I and Tsapas G:
Evaluation of the renal mechanisms for urate homeostasis in uremic
patients by probenecid and pyrazinamide test. Nephron. 46:273–280.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Boger WP and Strickland SC: Probenecid
(benemid); its uses and side-effects in 2,502 patients. AMA Arch
Intern Med. 95:83–92. 1955. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Scott JT: Comparison of allopurinol and
probenecid. Ann Rheum Dis. 25(Suppl 6): 623–626. 1966. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stocker SL, Graham GG, McLachlan AJ,
Williams KM and Day RO: Pharmacokinetic and pharmacodynamic
interaction between allopurinol and probenecid in patients with
gout. J Rheumatol. 38:904–910. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sun J, Wu Y, Xu H and Zhao Z: DTome: A
web-based tool for drug-target interactome construction. BMC
Bioinformatics. 13(Suppl 9): S72012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Landersdorfer CB, Kirkpatrick CM, Kinzig
M, Bulitta JB, Holzgrabe U, Jaehde U, Reiter A, Naber KG, Rodamer M
and Sörgel F: Competitive inhibition of renal tubular secretion of
ciprofloxacin and metabolite by probenecid. Br J Clin Pharmacol.
69:167–178. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Truong DM, Kaler G, Khandelwal A, Swaan PW
and Nigam SK: Multi-level analysis of organic anion transporters 1,
3, and 6 reveals major differences in structural determinants of
antiviral discrimination. J Biol Chem. 283:8654–8663. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nagle MA, Wu W, Eraly SA and Nigam SK:
Organic anion transport pathways in antiviral handling in choroid
plexus in Oat1 (Slc22a6) and Oat3 (Slc22a8) deficient tissue.
Neurosci Lett. 534:133–138. 2013. View Article : Google Scholar :
|
|
30
|
Sweet DH, Miller DS, Pritchard JB,
Fujiwara Y, Beier DR and Nigam SK: Impaired organic anion transport
in kidney and choroid plexus of organic anion transporter 3 (Oat3
(Slc22a8)) knockout mice. J Biol Chem. 277:26934–26943. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sykes D, Sweet DH, Lowes S, Nigam SK,
Pritchard JB and Miller DS: Organic anion transport in choroid
plexus from wild-type and organic anion transporter 3
(Slc22a8)-null mice. Am J Physiol Renal Physiol. 286:F972–F978.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Strazielle N, Belin MF and Ghersi-Egea JF:
Choroid plexus controls brain availability of anti-HIV nucleoside
analogs via pharmacologically inhibitable organic anion
transporters. AIDS. 17:1473–1485. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hedaya MA and Sawchuk RJ: Effect of
probenecid on the renal and nonrenal clearances of zidovudine and
its distribution into cerebrospinal fluid in the rabbit. J Pharm
Sci. 78:716–722. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Petty BG, Kornhauser DM and Lietman PS:
Zidovudine with probenecid: A warning. Lancet. 335:1044–1045. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wolf DL, Rodríguez CA, Mucci M, Ingrosso
A, Duncan BA and Nickens DJ: Pharmacokinetics and renal effects of
cidofovir with a reduced dose of probenecid in HIV-infected
patients with cytomegalovirus retinitis. J Clin Pharmacol.
43:43–51. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Holodniy M, Penzak SR, Straight TM, Davey
RT, Lee KK, Goetz MB, Raisch DW, Cunningham F, Lin ET, Olivo N, et
al: Pharmacokinetics and tolerability of oseltamivir combined with
probenecid. Antimicrob Agents Chemother. 52:3013–3021. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Perwitasari O, Yan X, Johnson S, White C,
Brooks P, Tompkins SM and Tripp RA: Targeting organic anion
transporter 3 with probenecid as a novel anti-influenza a virus
strategy. Antimicrob Agents Chemother. 57:475–483. 2013. View Article : Google Scholar :
|
|
38
|
Penuela S, Gyenis L, Ablack A, Churko JM,
Berger AC, Litchfield DW, Lewis JD and Laird DW: Loss of pannexin 1
attenuates melanoma progression by reversion to a melanocytic
phenotype. J Biol Chem. 287:29184–29193. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Iglesias R, Locovei S, Roque A, Alberto
AP, Dahl G, Spray DC and Scemes E: P2X7 receptor-Pannexin1 complex:
Pharmacology and signaling. Am J Physiol Cell Physiol.
295:C752–C760. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Clark AK, Staniland AA, Marchand F, Kaan
TK, McMahon SB and Malcangio M: P2X7-dependent release of
interleukin-1beta and nociception in the spinal cord following
lipopolysaccharide. J Neurosci. 30:573–582. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li Z, Peng H, Qin L, Qi J, Zuo X, Liu JY
and Zhang JT: Determinants of 14-3-3σ protein dimerization and
function in drug and radiation resistance. J Biol Chem.
288:31447–31457. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Powell DW, Rane MJ, Chen Q, Singh S and
McLeish KR: Identification of 14-3-3zeta as a protein kinase B/Akt
substrate. J Biol Chem. 277:21639–21642. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Waterman MJ, Stavridi ES, Waterman JL and
Halazonetis TD: ATM-dependent activation of p53 involves
dephosphorylation and association with 14-3-3 proteins. Nat Genet.
19:175–178. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
44
|
Miyazaki H, Anzai N, Ekaratanawong S,
Sakata T, Shin HJ, Jutabha P, Hirata T, He X, Nonoguchi H, Tomita
K, et al: Modulation of renal apical organic anion transporter 4
function by two PDZ domain-containing proteins. J Am Soc Nephrol.
16:3498–3506. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Couet J, Sargiacomo M and Lisanti MP:
Interaction of a receptor tyrosine kinase, EGF-R, with caveolins.
Caveolin binding negatively regulates tyrosine and serine/threonine
kinase activities. J Biol Chem. 272:30429–30438. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shimizu T, Sugiura T, Wakayama T, Kijima
A, Nakamichi N, Iseki S, Silver DL and Kato Y: PDZK1 regulates
breast cancer resistance protein in small intestine. Drug Metab
Dispos. 39:2148–2154. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dallas S, Zhu X, Baruchel S, Schlichter L
and Bendayan R: Functional expression of the multidrug resistance
protein 1 in microglia. J Pharmacol Exp Ther. 307:282–290. 2003.
View Article : Google Scholar : PubMed/NCBI
|