Introduction
Diabetic retinopathy (DR) is the most common
complication of diabetes and the principal cause of blindness in
working-age individuals (1). The
early stages of DR are characterized by microvascular cell damage
(2). Microvascular cell damage is
associated with thickening of the capillary endothelial basement
membrane (BM) and pericyte apoptosis induced by hyperglycemia
(3). In cases of DR, apoptotic
cells have been found in all retinal layers, including retinal
endothelial cells (4).
Previous investigations into the molecular
mechanisms that cause DR have largely focused on vascular
endothelial growth factor (VEGF) (5,6).
This may be attributed partly to the fact that the prominent
clinical characteristics of DR have led to the general inference
that DR is entirely of a microvascular nature. Although significant
effort has been invested in elucidating the mechanisms that govern
destructive preretinal neovascularization in DR (7), considerably less is known about the
cellular processes that lead to increased retinal vascular
apoptosis. Mitochondrial uncoupling protein 2 (UCP2) is a novel
member of the mitochondrial anion carrier family, and displays 60%
sequence identity with the well-known thermogenic UCP1 from brown
adipose tissue (8). Previous
studies have suggested that UCP2 is involved in the control of
mitochondrial membrane potential (9) and the generation of reactive oxygen
species (ROS) (10). Recently, a
unifying hypothesis has emphasized the important role played by
increased mitochondrial ROS production in complications of
diabetes, including retinopathy (11). Previously, we demonstrated that
peroxisome proliferator-activated receptor γ (PPARγ)-mediated
changes to UCP2 involve the mitochondrial-ROS pathway, which is
associated with a decreased VEGF-to-pigment epithelium-derived
factor (PEDF) ratio caused by the effect that
angiotensin-converting enzyme inhibitor (ACEI) exerts on DR
(12). Moreover, we investigated
the inhibition of high glucose-induced apoptosis by UCP2 in human
umbilical vein endothelial cells (13). However, the more detailed
mechanism of UCP2 in vascular endothelial cell apoptosis in DR has
not been explored to date, to the best of our knowledge.
In the present study, using both UCP2-knockout mice
and HUVECs, we provide evidence that UCP2 plays an anti-apoptotic
role. UCP2-knockout mice exhibited retinal cell death and damage
in vivo, which was similar to db/db diabetic mice.
Additionally, UCP2 knockdown exaggerated high glucose (HG)-induced
apoptosis and caspase-3 activity in human umbilical vein
endothelial cells (HUVECs). Furthermore, we demonstrated that
adenovirus-mediated UCP2 overexpression exerted a protective effect
on apoptosis, and investigated the opening of the permeability
transition pore (PTP), cytochrome c release, ROS generation
and nitric oxide (NO) production in HUVECs. Thus, inducing UCP2
expression may represent an alternative therapeutic strategy in the
early stage of DR.
Materials and methods
Cell culture and adenovirus
transfection
HUVECs were obtained from ScienCell Research
Laboratories (San Diego, CA, USA). The cells were cultured in
endothelial cell medium (ECM) (ScienCell Research Laboratories)
with 5% (v/v) fetal bovine serum (FBS) at 37°C in an atmosphere
with 5% (v/v) CO2 and 95% humidity. When they reached
confluence, the cells were maintained in 1% (v/v) fetal calf serum
and exposed to normal amounts of glucose (5.5 mmol/l) or high
amounts of glucose (30 mmol/l) for 3–7 days, during which time the
medium was changed every 2 days. When HUVECs reached approximately
50% confluence in fresh serum-free medium, they were transiently
transduced with control adenovirus β-galactosidase (Ad-β-gal) or
with adenovirus overexpressing UCP2 (Genechem, Shanghai, China) at
multiplicities of infection (MOI) of 10. The cells were further
cultured in ECM with 5% (v/v) FBS after infection for 4 h and then
selected using 200 µm/ml puromycin (Thermo Fisher
Scientific, Waltham, MA, USA). The stable overexpression lines were
established when more than 95% of the transduced cells were found
to strongly express green fluorescent protein (GFP) under a
fluorescence microscope (BX51; Olympus, Tokyo, Japan).
Animals and sample preparation
All experiments in the present study comply with the
requirements of the Association for Research in Vision and
Ophthalmology Statement for the Use of Animals in Ophthalmic and
Vision Research. Eighteen-week-old male C57 mice weighing ~20 g and
db/db diabetic mice weighing ~20 g were obtained from the Shanghai
Laboratory Animal Center of Chinese Academy of Sciences (Jiuting,
Shanghai, China). UCP2-deficient mice weighing ~20 g were obtained
from the Shanghai Biomodel Organism Research Center (Jiuting,
Shanghai, China). This study was approved by the Ethics Committee
of Shanghai Jiao Tong University School of Medicine.
UCP2 siRNA transfection
Target sequences were aligned to the human genome
database in a BLAST search to ensure that the chosen sequences were
not highly homologous with those of other genes. Cells were seeded
in 6-well plates and cultured in drug-free medium (ScienCell
Research Laboratories, Carlsbad, CA, USA). When cells reached
90–95% confluence they were washed twice with phosphate-buffered
saline (PBS) and grown in 2 ml ECM without antibiotics. Using
Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA), the
indicated concentration (50 nM) of UCP2 siRNA oligo (Genechem) was
transfected into HUVECs according to the manufacturer's
instructions. Cells transfected with control siRNA served as a
negative control. Forty-eight hours later, the expression levels of
UCP2 were evaluated by western blot analysis. The cells transfected
with siRNA were used for experiments 48 h after transfection.
Western blot analysis
Western blot analysis was performed, as previously
described (14). Briefly, the
primary antibodies used to probe the membranes included anti-UCP2
(1:500, cat. no. ab67241; Abcam, Cambridge, UK), anti-cytochrome
c (1:200, cat. no. 1896-1; Epitomics, Burlingame, CA, USA),
anti-caspase-3 (1:5,000, cat. no. ab32351; Abcam) and anti-β-actin
(1:1,000, cat. no. A1978; Sigma-Aldrich, St. Louis, MO, USA).
Experiments were repeated in triplicate.
Assessment of the release of cytochrome c
and subcellular localization of UCP2 protein expression
For the analysis of the release of cytochrome
c from mitochondria into the cytosol, proteins in the
cytosol and mitochondrial fractions were separated by SDS/PAGE.
Bands of proteins were transferred onto a PVDF membrane (Millipore,
Bedford, MA, USA). Cytochrome c was detected with an
enhanced chemiluminescence western blot analysis system (Amersham,
Buckinghamshire, UK) with monoclonal antibodies specific to
cytochrome c (1:200, cat. no. 1896-1; Epitomics). The same
method for western blot analyses was used on cytosolic and
mitochondrial preparations using antibody against human UCP2
(1:500, cat. no. ab67241; Abcam) and subunit IV of cytochrome
oxidase (1:500, cat. no. ab110272; Abcam).
Flow cytometric analysis
For quantification of cellular viability, cells were
double-stained with Annexin V and propidium iodide (PI) according
to the manufacturer's instructions [Annexin V-fluorescein
isothiocyanate (FITC) apoptosis detection kit (BD Biosciences,
Franklin Lakes, NJ, USA)]. The proportion of apoptotic and necrotic
cells was determined using FACSCalibur (Becton-Dickinson, San Jose,
CA, USA). Annexin-V is a marker of apoptosis, and PI reflects the
integrity of the cell membrane, thereby serving as a marker of
necrosis.
Caspase-3 activity
Caspase-3 activity was determined using a caspase-3
colorimetric protease assay kit (Beyotime Institute of
Biotechnology, Shanghai, China) following the manufacturer's
instructions. Briefly, the kit uses spectrophotometry to detect the
chromophore p-nitroaniline (pNA) after cleavage from the labeled
substrate DEVD-pNA (DEVD is the sequence recognized by caspases).
The free pNA was quantified using a spectrophotometer or a
microtiter plate reader at 405 nm. The ratio of absorbance from a
sample to that from a control allows for the determination of the
fold increase of caspase-3 activity.
Assessment of mitochondrial permeability
transition pore (mPTP) opening
To determine the effect of UCP2 overexpression on
mPTP opening, a previously validated cell model of mPTP opening was
used (15). A microplate
spectrofluorometer (SPECTRAmax GEM-INI-XS; Molecular Devices,
Sunnyvale, CA, USA) was used to study stimulation of the
fluorophore, tetramethylrhodamine methyl ester (TMRM), which
accumulates in mitochondria, and generation of ROS within
mitochondria. Culture medium was removed and replaced with Krebs
imaging buffer. Cells were then loaded with 3 mM TMRM for 15 min at
37°C and washed with Krebs imaging buffer (Guidechem, Shanghai,
China). The time taken to induce mitochondrial membrane
depolarization is recorded as a measurement of mPTP opening. This
was defined as the time taken to reach half the maximum TMRM
fluorescence intensity. Twenty transfected cells were randomly
selected for the induction and detection of mPTP opening from each
treatment group, and this was repeated in four independent
experiments, providing a total of 80 cells/treatment group. As a
positive control and in order to confirm that mitochondrial
membrane depolarization was indicative of mPTP opening, following
TMRM loading, a group of cells were pretreated for 10 min with the
mPTP inhibitor, cyclosporin A (CsA; 0.2 mM; Selleck Chemicals,
Houston, TX, USA), as previously described (16–18). The time taken to induce mPTP
opening was recorded.
Measurement of intracellular ROS
production and NO levels
In the present study, HUVECs were incubated with 10
µmol/ml carboxydichlorodihydrofluorescein diacetate
(DCFH2-DA; Molecular Probes, Inc., Eugene, USA) at 37°C. After 15
min incubation, the increase in DCFH2 oxidation was measured using
a FACSCalibur (Becton-Dickinson) (19,20). The NO level in HUVECs was measured
in situ using DAF-FM diacetate (Sigma-Aldrich), as
previously described (21).
Terminal deoxynucleotidyl
transferase-mediated dUTP nick-end labeling (TUNEL) assay
Mice were sacrificed by cervical dislocation. Eyes
were removed from four 8 week-old db/db mice, four age-matched
UCP2-KO mice and four age-matched normal mice were used as control
retinas. The eyes were immediately enucleated and the retina was
separated. The retina from one of the eyes was frozen in liquid
nitrogen and stored at −80°C. TUNEL was performed on frozen
sections using the DeadEnd™ TUNEL assay kit (Promega, Madison, WI,
USA) and were counterstained with PI, according to the
manufacturer's suggestions. Briefly, sections were hydrated with
alcohol (100, 95 and 70%), and then fixed in 3.7% paraformaldehyde.
After washing, the slides were incubated in a mixture of TdT,
Mn2+, and TdT dNTP (Sangon Biotech, Shanghai, China) for
1 h at 37°C. The reaction was stopped with TdT Stop Buffer
(Trevigen, Gaithersburg, MD, USA) for 5 min. After washing with
deionized water, the slides were incubated with streptavidin-FITC
(diluted 1:200) solution for 20 min at room temperature. Slides
were counterstained, mounted, covered with coverslips, and
visualized by confocal microscopy (LSM 510; Carl Zeiss, Inc.,
Oberkochen, Germany). Apoptotic cells were identified as doubly
labeled with TdT fluorescein and PI, and only the nuclei which were
clearly labeled yellow were scored.
Transmission electron microscopy
Tissue processing, electron microscopy, morphometric
measurements [retinal capillary basement membrane thickness (BMT)],
and statistical analysis were performed as detailed in our previous
study (12). Eyes were removed
from four 8 week-old db/db mice, four age-matched UCP2-KO mice and
four age-matched normal mice were used as control. Briefly,
enucleated eyes were fixed in 2.5% (w/v) glutaraldehyde in 0.1 M
cacodylate buffer (pH 7.4) containing 0.2% (v/v) tannic acid,
washed in the same buffer, and post-fixed in 0.5% (v/v) osmium
tetroxide. Tissue sections were block stained with uranyl acetate,
lead stained, dehydrated through a graded series of ethanol, and
embedded in Epon. One-micrometer-thick sections were examined with
a JEM-1200EX transmission electron microscope (JEOL, Ltd.,
Akishima, Japan). Computer-assisted morphometric measurements (The
Image Center of Beijing University of Aeronautics and Astronautics,
Beijing, China) were performed on electron micrographs taken from
12 randomly selected capillaries of the outer plexiform layer from
four different tissue blocks of the same retina. Only
cross-sectioned capillaries were considered. A total of 96
capillaries were evaluated in each experimental group.
Statistical analysis
SPSS 17.0 was used to analyze the experimental data.
All experimental data are represented as the means ± standard
deviation (SD). One-way analysis of variance (ANOVA) followed by
Student-Newman-Keuls test was used to compare the effect of
treatment on the various parameters. Non-parametric data was
analyzed using the Chi-square test or Fisher's exact method. A
P-value <0.05 was considered to indicate a statistically
significant difference.
Results
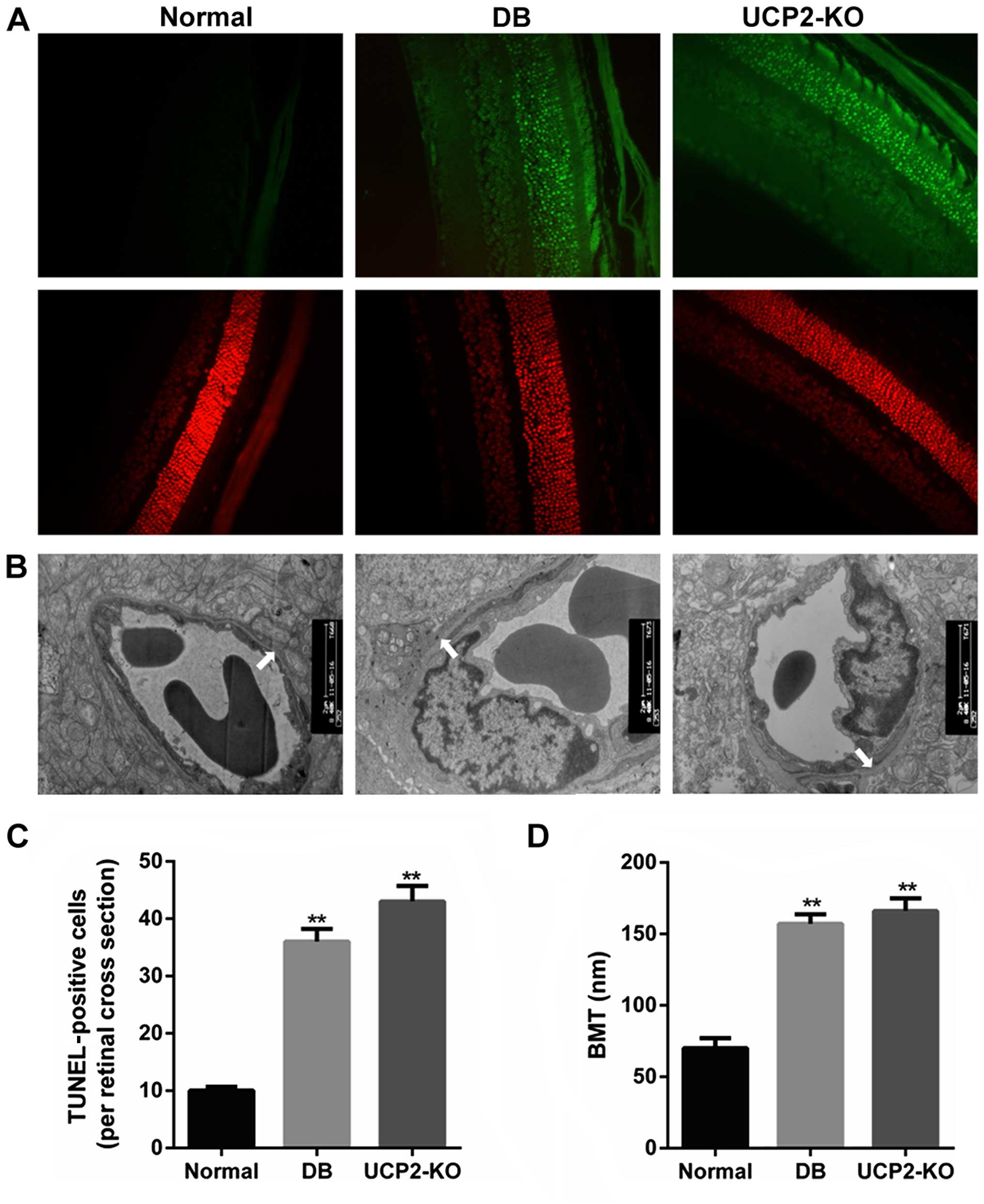
Retinal cell death and damage in
mice
Loss of cells in the ganglion cell layer was due to
apoptosis, and this was reflected as an increase in TUNEL in the
ganglion cell layer of both the UCP2-knockout mice and db/db mice.
We performed TUNEL on retinal sections to measure cell death in the
UCP2-knockout mice and db/db mice. Notably, we found that
UCP2-knockout mice exhibited a DR-like phenotype, similar to db/db
mice. Fig. 1A and C illustrate
increased TUNEL in the ganglion cell layer of UCP2-knockout mice
and db/db mice compared to their normal counterparts. Furthermore,
compared to the non-diabetic mice, the BMT was significantly
increased in the UCP2-knockout mice and db/db mice (P<0.01)
(Fig. 1B and D). However, there
was no significant difference in BMT between UCP2-knockout mice and
db/db mice. Therefore, these results demonstrate that knockdown of
UCP2 is a critical factor for retinal cell death and damage.
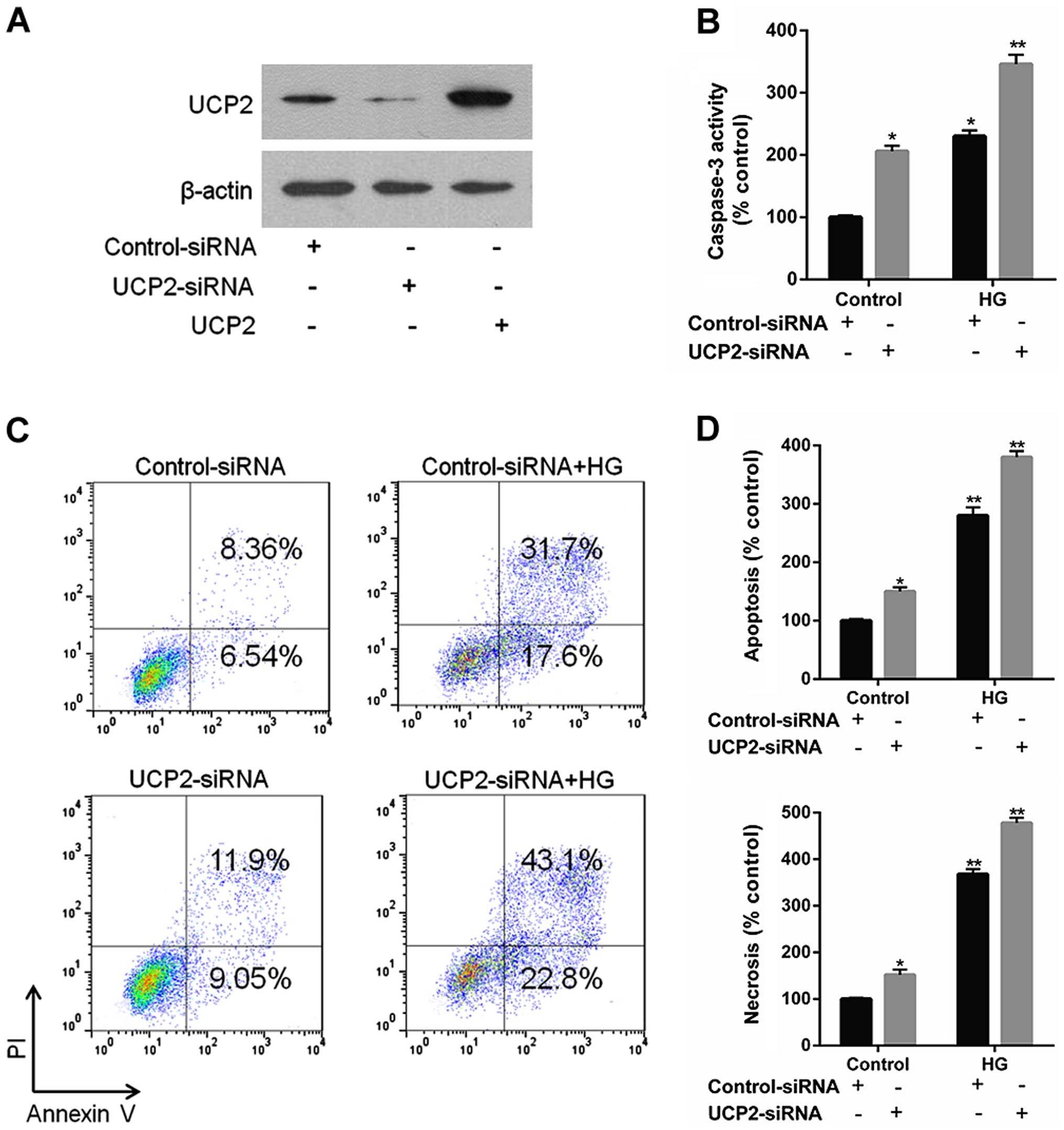
Knockdown of UCP2 exaggerates HG-induced
apoptosis and activated caspase-3 in HUVECs
UCP2 siRNA effectively reduced endogenous levels of
UCP2 protein compared to control siRNA (Fig. 2A). UCP2 siRNA increased caspase-3
expression to a significant extent, whereas control siRNA did not
(Fig. 2B). UCP2 siRNA exaggerated
HG-induced apoptosis compared to control-siRNA infected cells
(Fig. 2C and D). These data
demonstrate that endogenous UCP2 expression is important for
preventing apoptosis in HUVECs.
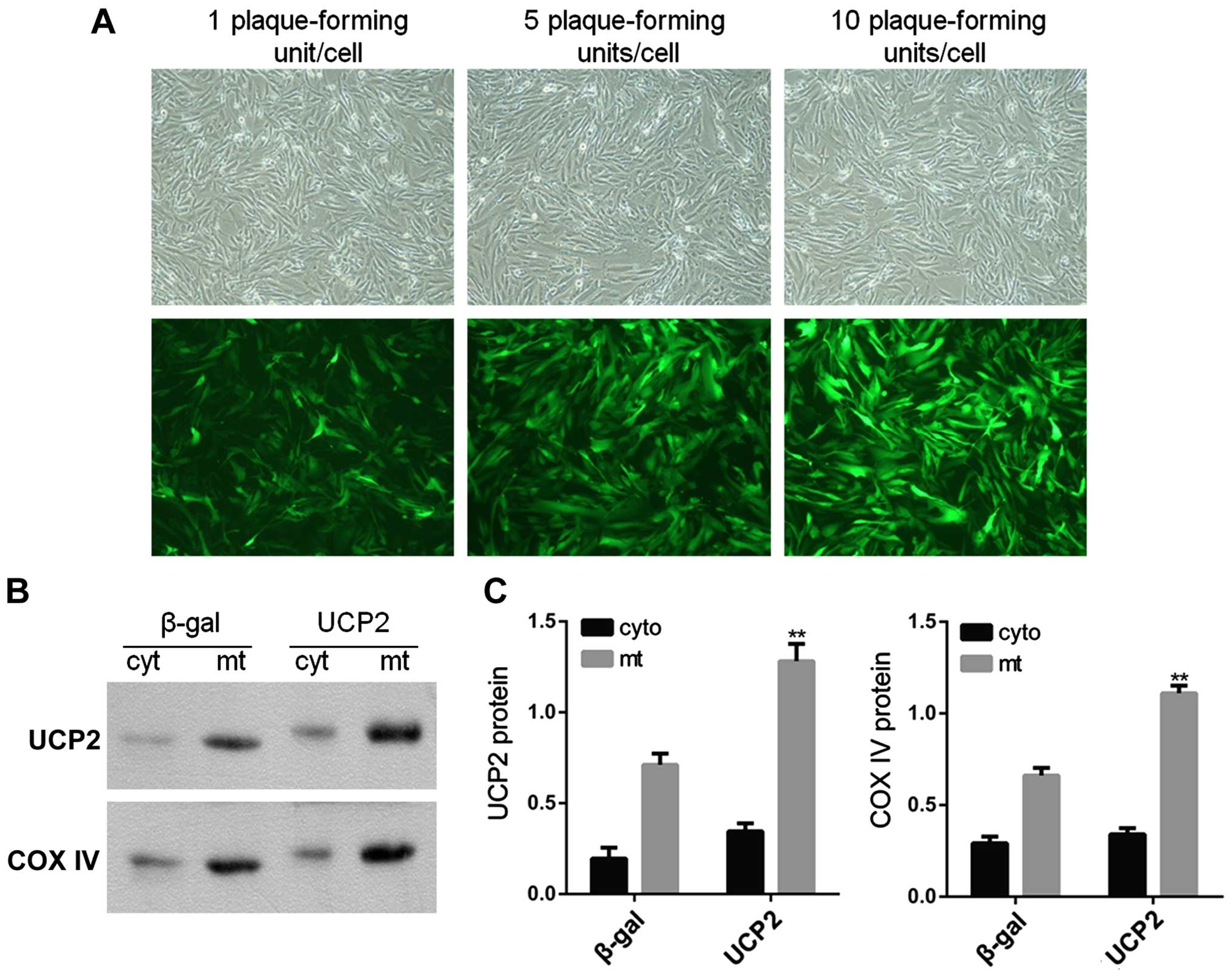
Subcellular localization of UCP2 protein
in UCP2-overex-pressing HUVECs
In the present study, HUVECs were infected with
adenovirus carrying UCP2 cDNA in sense orientation at an MOI of 1,
5 and 10 pfu/cell for 24 h. Immunofluorescence microscopy was then
used in order to observe UCP2 eGFP expression in infected cells. At
an MOI of 10 pfu/cell, UCP2 eGFP expression was detected in >95%
of cultured cells (Fig. 3A). UCP2
was overexpressed mainly within the mitochondria (Fig. 3B and C).
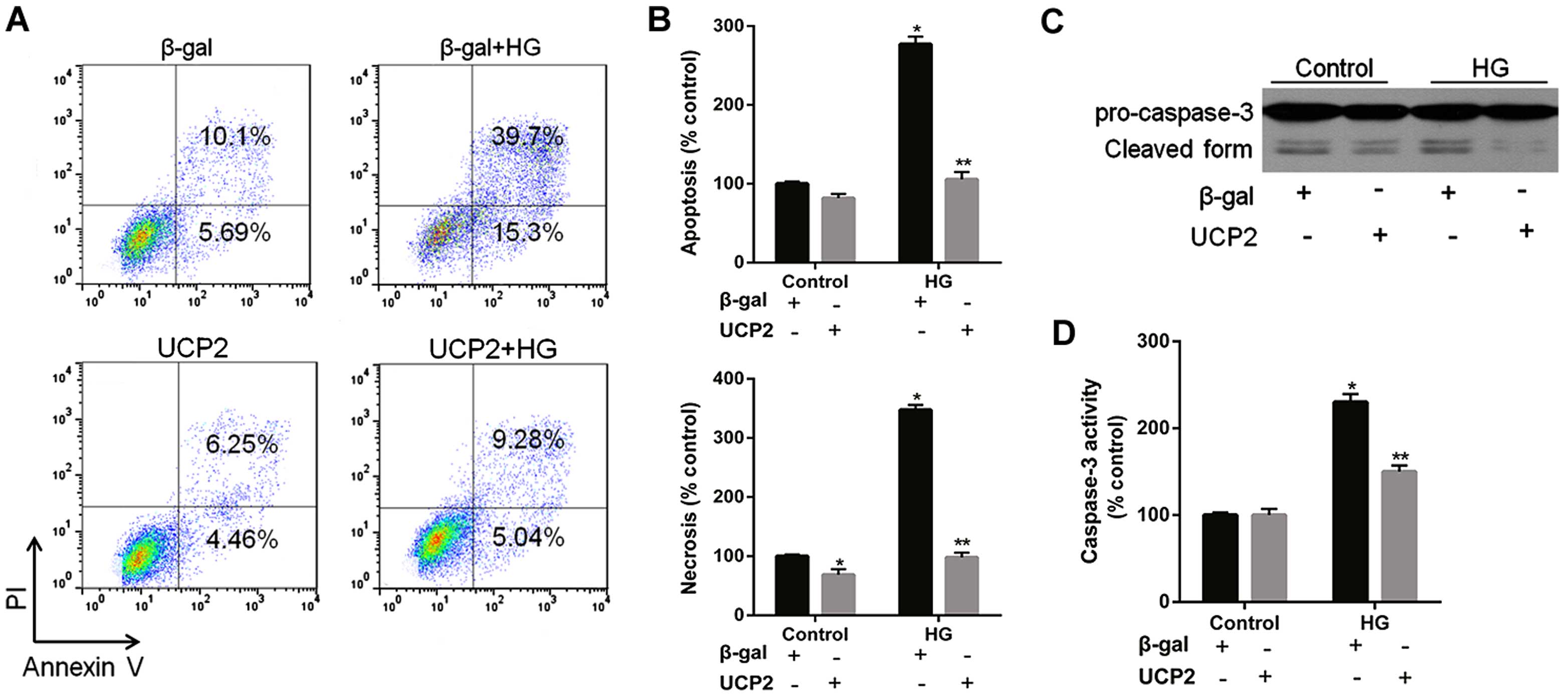
UCP2 overexpression inhibits HG-induced
apoptosis and caspase-3 activity in HUVECs
HG increased HUVEC apoptosis by 4.1-fold; UCP2
overexpression reduced HG-induced apoptosis by 65% in HUVECs
(Fig. 4A). FACScan analysis
revealed that UCP2 overexpression inhibited both HG-induced
apoptosis and necrosis of HUVECs (Fig. 4A and B). HG induced a significant
increase in caspase-3 activity in HUVECs (Fig. 4C and D). The HG-induced increase
in caspase-3 activity was inhibited by transfection with Ad-UCP2.
The generation of active caspase-3 from the proenzyme was enhanced
in the presence of HG (Fig. 4C and
D). HG-induced cleavage of caspase-3 into active subunits was
thus prevented in the presence of UCP2.
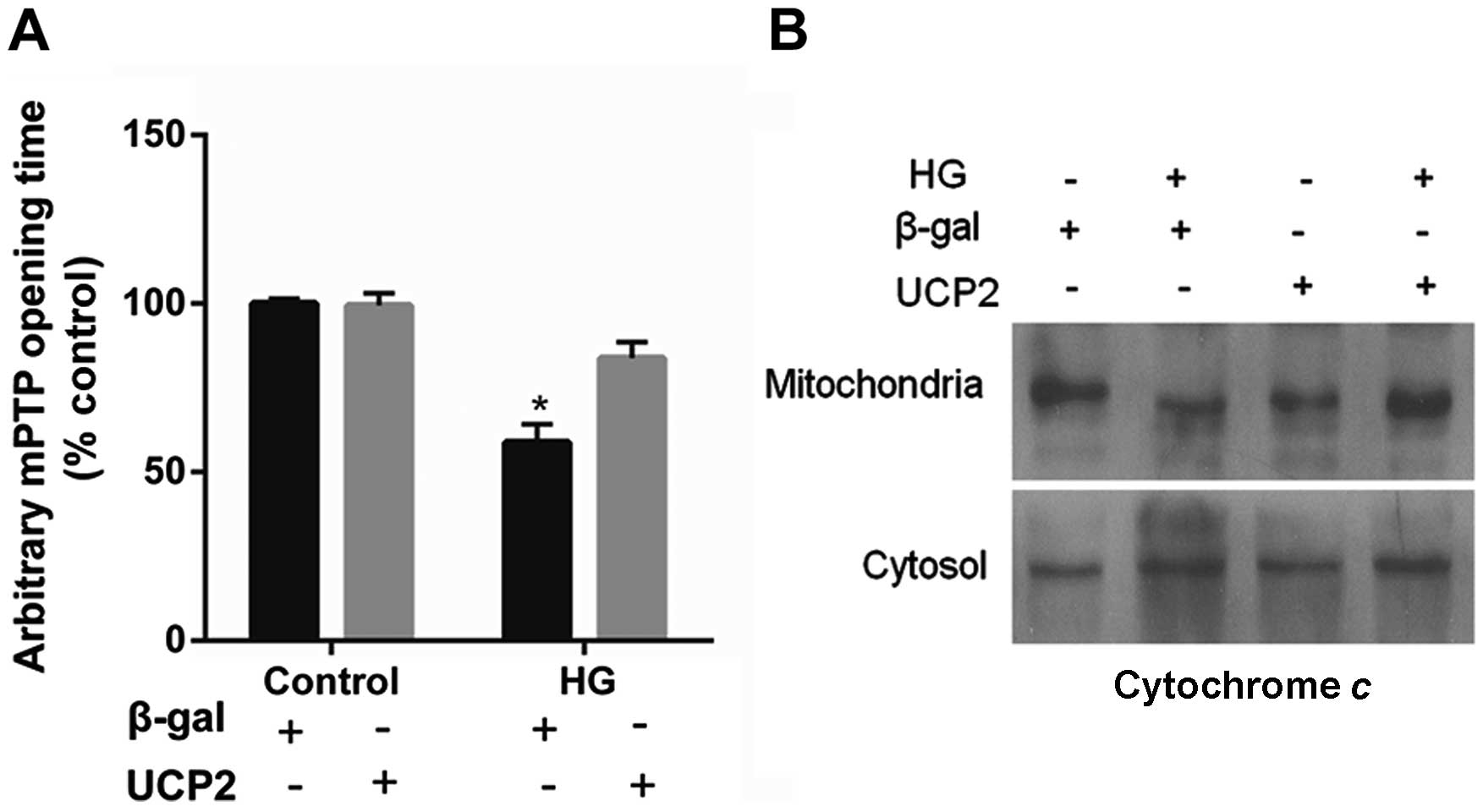
UCP2 overexpression delays mPTP opening
and blocks cytochrome c release
UCP2 overexpression in HUVECs decreased the
susceptibility of the cells to mPTP opening. UCP2 overexpression in
HUVECs delayed the time of mPTP opening by 1.5±0.2-fold when
compared to control values (P<0.01) (Fig. 5A). In addition, treatment with HG
resulted in the liberation of cytochrome c from mitochondria
to the cytosol (Fig. 5B). The
liberation of cytochrome c by HG was blocked in cells
overexpressing UCP2 (Fig.
5B).
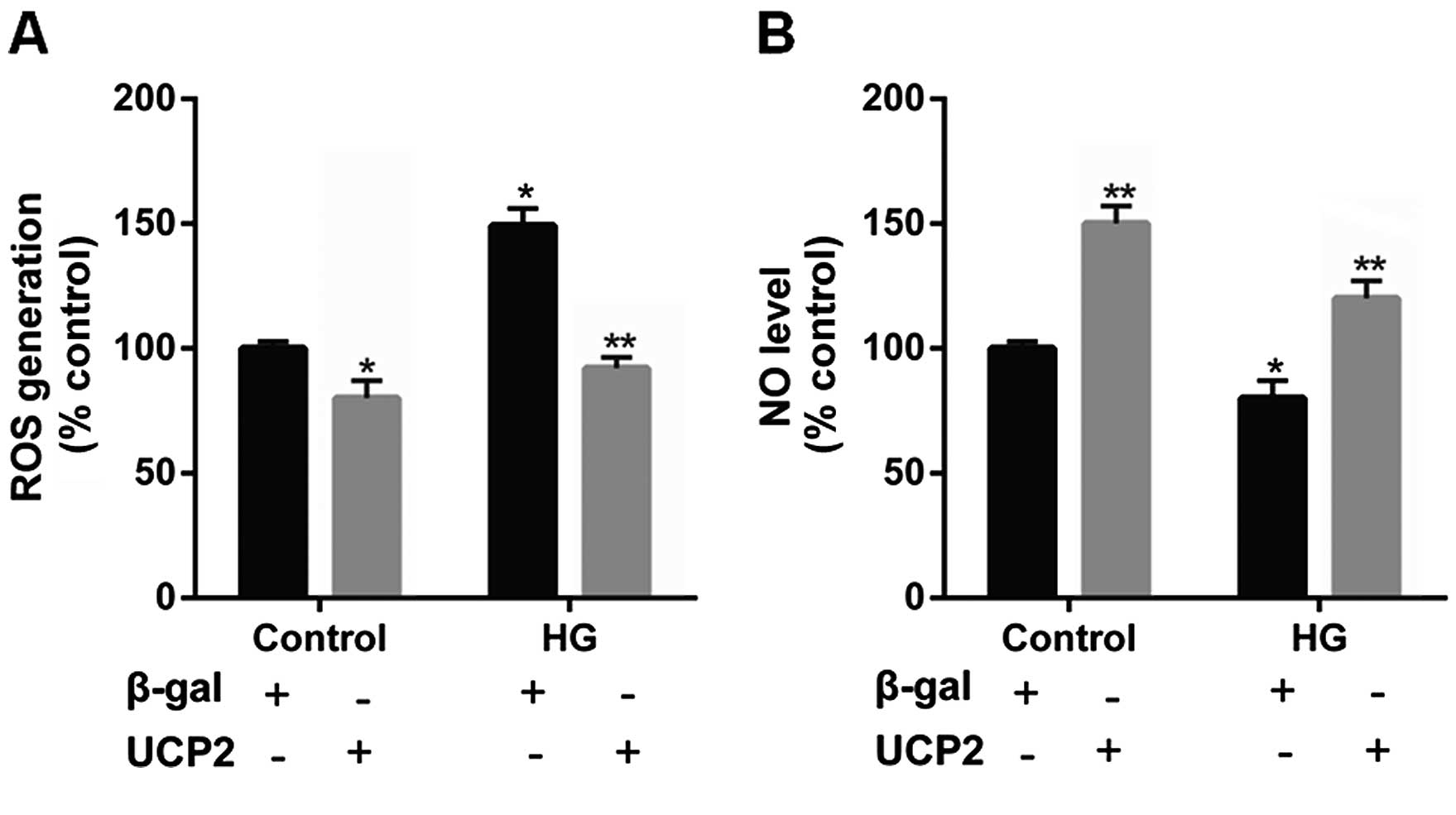
UCP2 overexpression suppresses
intracellular ROS production and increases the intracellular NO
levels in HUVECs
Previous research has indicated that UCP2 is
activated by ROS and attenuates excessive ROS production in the
form of a negative feedback mechanism (22–24). Thus, in the present study, the
levels of ROS were determined in HUVECs. Incubation with HG
significantly increased intracellular ROS generation. However, upon
UCP2 overexpression, we noted that the increase in ROS production
effected by HG was suppressed (Fig.
6A).
It has previously been noted that NO produced by
eNOS exerts anti-inflammatory effects on the vascular wall, and
inhibits the migration and proliferation of vascular smooth muscle
cells (25). Thus, in the present
study we detected the intracellular NO level in HUVECs. We noted
that intracellular NO levels were decreased by HG, whereas UCP2
overexpression increased intracellular NO levels (Fig. 6B).
Discussion
Therapeutic strategies to treat complications
associated with DR previously consisted predominantly of
controlling systemic vascular deregulation (26–29). Although laser photocoagulation and
targeted treatments, such as locally administered corticosteroids,
are currently available, their off-target effects underscore the
need to explore novel therapeutic approaches (30,31). In the present study, we provide
evidence that UCP2 plays a critical role in vascular cell apoptosis
induced by HG in DR.
A previous study has shown that loss of retinal
capillary endothelial and mural cells and focal BM thickening has
been noted in streptozotocin-induced diabetic rats (32). Similarly, in the present study we
performed TUNEL assay on retinal sections to measure cell death in
the UCP2-knockout mice, db/db mice and C57 mice. Notably,
UCP2-knockout mice exhibited DR-like damage, similar to db/db mice.
We noted an increase in TUNEL in the ganglion cell layer of
UCP2-knockout mice and db/db mice compared to the wild-type mice.
Furthermore, compared with non-diabetic mice, the BMT was also
significantly increased in the UCP2-knockout mice and db/db mice.
There was no significant difference between UCP2-knockout mice and
db/db mice. These results suggest that UCP2 exerts a protective
effect in the early stages of DR.
Endothelial apoptosis is an important early event in
the pathogenesis of DR, and increased intracellular oxidative
stress plays an important causative role in the pathogenesis of
endothelial apoptosis (33–36). Moreover, caspase-3 activation is a
crucial step in the process of apoptosis. Activation of caspase-3
is essential for cell execution and occurs immediately prior to the
development of morphological changes consistent with apoptosis
(37). Hence, a cell with
activated caspase-3 is considered destined to die. In agreement
with previous research on other cell types (38), in the present study, we noted that
suppression of UCP2 expression using UCP2-specific siRNA
exaggerated caspase-3 activation and apoptosis induced by HG.
Conversely, overexpression of UCP2 inhibited HG-induced endothelial
apoptosis and caspase-3 activation. In addition, UCP2
overexpression decreased ROS generation, PTP opening, and
cytochrome c release induced by HG. These data demonstrate
that endogenous UCP2 expression is important for preventing
apoptosis in HUVECs.
The endothelium acts not only as a barrier, but also
as a regulator of vascular tone and smooth muscle cell growth,
migration, and proliferation. Vascular tone is modulated through
the release of relaxing and contracting substrates. Of these, NO is
physiologically the most important regulator of vascular tone.
Although a previous study demonstrated that the vascular benefit of
UCP2 was due to increased NO bioavail-ability after the inhibition
of ROS production in HG-treated primary mouse aortic endothelial
cells (39), in the present study
we showed that HG significantly increased intracellular ROS
production and decreased intracellular NO levels in HUVECs. UCP2
overexpression reversed these effects. Thus, we suggest that UCP2
reduces ROS production and increases NO levels.
Acknowledgments
The present study was supported by grants from the
Research Fund for the National Nature Science Funding of China
(nos. 30930097, 81273424 and 81170862), the Major National Science
and Technology Projects during the 12th Five-Year Plan (no.
011ZX09302-007-02).
References
|
1
|
Cerani A, Tetreault N, Menard C, Lapalme
E, Patel C, Sitaras N, Beaun F, Leboeuf D, De Guire V, Binet F, et
al: Neuron-derived semaphorin 3A is an early inducer of vascular
permeability in diabetic retinopathy via neuropilin-1. Cell Metab.
18:505–518. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Barot M, Gokulgandhi MR, Patel S and Mitra
AK: Microvascular complications and diabetic retinopathy: recent
advances and future implications. Future Med Chem. 5:301–314. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Geraldes P, Hiraoka-Yamamoto J, Matsumoto
M, Clermont A, Leitges M, Marette A, Aiello LP, Kern TS and King
GL: Activation of PKC-delta and SHP-1 by hyperglycemia causes
vascular cell apoptosis and diabetic retinopathy. Nat Med.
15:1298–1306. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jiang Y, Zhang Q, Soderland C and Steinle
JJ: TNFα and SOCS3 regulate IRS-1 to increase retinal endothelial
cell apoptosis. Cell Signal. 24:1086–1092. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Montero JA, Ruiz-Moreno JM and Correa ME:
Intravitreal anti-VEGF drugs as adjuvant therapy in diabetic
retinopathy surgery. Curr Diabetes Rev. 7:176–184. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hammes HP, Feng Y, Pfister F and Brownlee
M: Diabetic retinopathy: targeting vasoregression. Diabetes.
60:9–16. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bressler NM, Beck RW and Ferris FL III:
Panretinal photocoagulation for proliferative diabetic retinopathy.
N Engl J Med. 365:1520–1526. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang CY, Parton LE, Ye CP, Krauss S, Shen
R, Lin CT, Porco JA Jr and Lowell BB: Genipin inhibits
UCP2-mediated proton leak and acutely reverses obesity- and high
glucose-induced beta cell dysfunction in isolated pancreatic
islets. Cell Metab. 3:417–427. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brand MD and Esteves TC: Physiological
functions of the mitochondrial uncoupling proteins UCP2 and UCP3.
Cell Metab. 2:85–93. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bouillaud F: UCP2, not a physiologically
relevant uncoupler but a glucose sparing switch impacting ROS
production and glucose sensing. Biochim Biophys Acta. 1787:377–383.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wilkinson-Berka JL, Rana I, Armani R and
Agrotis A: Reactive oxygen species, Nox and angiotensin II in
angiogenesis: implications for retinopathy. Clin Sci (Lond).
124:597–615. 2013. View Article : Google Scholar
|
|
12
|
Zheng Z, Chen H, Ke G, Fan Y, Zou H, Sun
X, Gu Q, Xu X and Ho PC: Protective effect of perindopril on
diabetic retinopathy is associated with decreased vascular
endothelial growth factor-to-pigment epithelium-derived factor
ratio: involvement of a mitochondria-reactive oxygen species
pathway. Diabetes. 58:954–964. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
He Y, Wang N, Shen Y, Zheng Z and Xu X:
Inhibition of high glucose-induced apoptosis by UCP2 in human
umbilical vein endothelial cells. Int J Mol Med. 33:1275–1281.
2014.PubMed/NCBI
|
|
14
|
Luan Z, He Y, Alattar M, Chen Z and He F:
Targeting the prohibitin scaffold-CRAF kinase interaction in
RAS-ERK-driven pancreatic ductal adenocarcinoma. Mol Cancer.
13:382014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zorov DB, Filburn CR, Klotz LO, Zweier JL
and Sollott SJ: Reactive oxygen species (ROS)-induced ROS release:
a new phenomenon accompanying induction of the mitochondrial
permeability transition in cardiac myocytes. J Exp Med.
192:1001–1014. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hausenloy DJ and Yellon DM: The
mitochondrial permeability transition pore: its fundamental role in
mediating cell death during ischaemia and reperfusion. J Mol Cell
Cardiol. 35:339–341. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Davidson SM, Hausenloy D, Duchen MR and
Yellon DM: Signalling via the reperfusion injury signalling kinase
(RISK) pathway links closure of the mitochondrial permeability
transition pore to cardioprotection. Int J Biochem Cell Biol.
38:414–419. 2006. View Article : Google Scholar
|
|
18
|
Lim SY, Davidson SM, Paramanathan AJ,
Smith CC, Yellon DM and Hausenloy DJ: The novel adipocytokine
visfatin exerts direct cardioprotective effects. J Cell Mol Med.
12:1395–1403. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bass DA, Parce JW, Dechatelet LR, Szejda
P, Seeds MC and Thomas M: Flow cytometric studies of oxidative
product formation by neutrophils: a graded response to membrane
stimulation. J Immunol. 130:1910–1917. 1983.PubMed/NCBI
|
|
20
|
van Reyk DM, King NJ, Dinauer MC and Hunt
NH: The intracellular oxidation of 2′,7′-dichlorofluorescin in
murine T lymphocytes. Free Radic Biol Med. 30:82–88. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shao C, Stewart V, Folkard M, Michael BD
and Prise KM: Nitric oxide-mediated signaling in the bystander
response of individually targeted glioma cells. Cancer Res.
63:8437–8442. 2003.PubMed/NCBI
|
|
22
|
Jezek P and Garlid KD: Mammalian
mitochondrial uncoupling proteins. Int J Biochem Cell Biol.
30:1163–1168. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Derdak Z, Mark NM, Beldi G, Robson SC,
Wands JR and Baffy G: The mitochondrial uncoupling protein-2
promotes chemoresistance in cancer cells. Cancer Res. 68:2813–2819.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Baffy G: Uncoupling protein-2 and cancer.
Mitochondrion. 10:243–252. 2010. View Article : Google Scholar
|
|
25
|
Williams IL, Wheatcroft SB, Shah AM and
Kearney MT: Obesity, atherosclerosis and the vascular endothelium:
mechanisms of reduced nitric oxide bioavailability in obese humans.
Int J Obes Relat Metab Disord. 26:754–764. 2002.PubMed/NCBI
|
|
26
|
Bhattacharjee PS, Huq TS, Potter V, Young
A, Davenport IR, Graves R, Mandal TK, Clement C, McFerrin HE,
Muniru-zzaman S, et al: High-glucose-induced endothelial cell
injury is inhibited by a peptide derived from human apolipoprotein
E. PLoS One. 7:e521522012. View Article : Google Scholar
|
|
27
|
Titchenell PM and Antonetti DA: Using the
past to inform the future: anti-VEGF therapy as a road map to
develop novel therapies for diabetic retinopathy. Diabetes.
62:1808–1815. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ababneh OH, Yousef YA, Gharaibeh AM, Abu
Ameerh MA, Abu-Yaghi NE and Al Bdour MD: Intravitreal bevacizumab
in the treatment of diabetic ocular neovascularization. Retina.
33:748–755. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Verma A, Shan Z, Lei B, Yuan L, Liu X,
Nakagawa T, Grant MB, Lewin AS, Hauswirth WW, Raizada MK and Li Q:
ACE2 and Ang-(1-7) confer protection against development of
diabetic retinopathy. Mol Ther. 20:28–36. 2012. View Article : Google Scholar :
|
|
30
|
Zhang Z, Meng X, Wu Z, Zou W, Zhang J, Zhu
D, Chen T and Zhang Q: Changes in choroidal thickness after
panretinal photocoagulation for diabetic retinopathy: a 12-week
longitudinal study. Invest Ophthalmol Sci. 56:2631–2638. 2015.
View Article : Google Scholar
|
|
31
|
Kumar B, Gupta SK, Saxena R and Srivastava
S: Current trends in the pharmacotherapy of diabetic retinopathy. J
Postgrad Med. 58:132–139. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu BC, Chen Q, Luo DD, Sun J, Phillips
AO, Ruan XZ and Liu NF: Mechanisms of irbesartan in prevention of
renal lesion in streptozotocin-induced diabetic rats. Acta
Pharmacol Sin. 24:67–73. 2003.PubMed/NCBI
|
|
33
|
Devi TS, Hosoya K, Terasaki T and Singh
LP: Critical role of TXNIP in oxidative stress, DNA damage and
retinal pericyte apoptosis under high glucose: implications for
diabetic retinopathy. Exp Cell Res. 319:1001–1012. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rask-Madsen C and King GL: Vascular
complications of diabetes: mechanisms of injury and protective
factors. Cell Metab. 17:20–33. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Fu D, Wu M, Zhang J, Du M, Yang S, Hammad
SM, Wilson K, Chen J and Lyons TJ: Mechanisms of modified
LDL-induced pericyte loss and retinal injury in diabetic
retinopathy. Diabetologia. 55:3128–3140. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kowluru RA, Mohammad G, dos Santos JM and
Zhong Q: Abrogation of MMP-9 gene protects against the development
of retinopathy in diabetic mice by preventing mitochondrial damage.
Diabetes. 60:3023–3033. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tyas L, Brophy VA, Pope A, Rivett AJ and
Tavaré JM: Rapid caspase-3 activation during apoptosis revealed
using fluorescence-resonance energy transfer. EMBO Rep. 1:266–270.
2000. View Article : Google Scholar
|
|
38
|
Mattiasson G, Shamloo M, Gido G, Mathi K,
Tomasevic G, Yi S, Warden CH, Castilho RF, Melcher T,
Gonzalez-Zulueta M, et al: Uncoupling protein-2 prevents neuronal
death and diminishes brain dysfunction after stroke and brain
trauma. Nat Med. 9:1062–1068. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tian XY, Wong WT, Xu A, Lu Y, Zhang Y,
Wang L, Cheang WS, Wang Y, Yao X and Huang Y: Uncoupling protein-2
protects endothelial function in diet-induced obese mice. Circ Res.
110:1211–1216. 2012. View Article : Google Scholar : PubMed/NCBI
|