Introduction
Vascular smooth muscle cells (VSMCs) play a major
role in the morphogenesis of blood vessels (1). Following vascular injury, the rate
of VSMC proliferation and migration is greatly increased, as well
as the synthetic capacity (1).
VSMCs play a role in a number of vascular diseases, including
atherosclerosis and restenosis. Abnormal proliferation and
migration of VSMCs is pivotal in the development of atherosclerosis
(1). Platelet-derived growth
factor (PDGF) is known to stimulate the migration and proliferation
of VSMCs following injury, which form an intimal vascular lesion
(2,3). In addition, PDGF influences the
proliferation of VSMCs via the phosphorylation of extracellular
signal-regulated kinase 1/2 (ERK1/2), p38 mitogen-activated protein
kinase (p38 MAPK), c-Jun N-terminal kinase (JNK), and AKT signaling
pathways (4,5).
Cell cycle progression is regulated by
cyclin-dependent kinases (CDKs) that are activated by cyclins,
which are regulatory subunits, to form cyclin/CDK complexes
(6,7). During proliferation, VSMCs initially
undergo a G1- to S-phase transition followed by further rounds of
proliferation (1,8). The cyclin-dependent kinase
inhibitors (CDKIs) p21WAF1 and p27KIP1 negatively control the G1-
to S-phase cell cycle progression by inhibiting the kinase
activities of cyclin/CDK complexes (9,10).
Previous studies have noted the positive regulatory role which
p21WAF1 plays in the activation of cyclin/CDK as an effector of
proliferation (11,12).
Matrix metalloproteinase-9 (MMP-9) is a gelatinase
that degrades type IV collagen, leading to the migration and
invasion of VSMCs, which results in the vascular plaque instability
characteristic of vascular diseases, such as atherosclerosis
(13,14). Previous research using in
vitro and in vivo experiments has suggested that the
elevated expression of MMP-9 is closely associated with the
formation of arterial lesions (14–17). The expression of MMP-9 is markedly
induced by growth factors and cytokines although the basal levels
of MMP-9 expression are very low (14–17). Previous studies have demonstrated
that the induction of MMP-9 expression is strictly regulated at the
transcriptional level through the binding activity of nuclear
factor-κB (NF-κB), specificity protein 1 (Sp1) and activator
protein-1 (AP-1) motifs (18,19).
Rosa hybrida is used in the cosmetics
industry as a source of aromatic oils (20). Previously, researchers have
identified gallic acid and volatiles (1-butanol, dodecyl acrylate
and cyclododecane) as the main components of R. hybrida
(21). Previous studies have
established various pharmacological effects for the extract of
R. hybrida: it has been noted for its antioxidant,
antimicrobial, anti-inflammatory and neuroprotective effects
(20–22). However, the inhibitory effect of
water extract of Rosa hybrida (WERH) on vascular diseases
has not previously been studied to the best of our knowledge. In
the present study, we investigated the anti-atherogenic role of
WERH in regulating proliferation, migration, and invasion in
PDGF-stimulated VSMCs.
Materials and methods
Antibodies
Polyclonal antibodies against cyclin E (sc-481),
CDK2 (sc-163) and CDK4 (sc-260), p21WAF1 (sc-756), p53 (sc-126),
p27 (sc-528) and GAPDH (sc-20357) were all obtained from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA). Monoclonal antibody
against cyclin D1 (04-221) was obtained from Millipore (Temecula,
CA, USA). Polyclonal antibodies against ERK (9102), phosphorylated
(p)-ERK (9101), p38 MAPK (9212), p-p38 MAPK (9211), JNK (9258),
p-JNK (9251), AKT (9272) and p-AKT (9271) were all purchased from
Cell Signaling Technology (Lakewood, NJ, USA). Polyclonal MMP-9
antibody (AB19016) was purchased from Millipore. HRP-conjugated
secondary antibodies against goat anti-rabbit IgG-HRP (sc-2004),
goat anti-mouse IgG-HRP (sc-2005) and donkey anti-goat IgG-HRP
(sc-2020) were purchased from Santa Cruz Biotechnology, Inc. All
procedures and protocols used in the present study were approved by
the Ethics Committee of Chung-Ang University (Anseong, Korea).
Preparation of extract
We used a total of 100 g of air-dried Rosa
Hybrida flowers (purchased from Jincheon Agricultural
Technology Center, Jincheon-gun, Korea), which were finely crushed
and soaked in water, followed by heating at 100°C. The extract was
concentrated via a rotary evaporator (Rotavapor-R; Brinkmann
Instruments, Westbury, NY, USA), lyophilized, and freeze-dried. The
yield of the final extract was approximately 10% w/w, and the
extract was diluted in saline buffer.
Cell cultures
VSMCs were obtained from the aortas of 2 young male
Sprague-Dawley rats (8 weeks old, weighing 200–250 g; DHbiolink,
Eumseong, Korea) using enzymatic digestion, as described previ
ously (23). Briefly, the rats
were sacrificed by CO2 inhalation, the vessels were
obtained under sterile conditions and were washed several times in
Hanks' balanced salt solution (HBSS). Following removal of the
adventitia from the aortas, digestion of the aortas in 5 ml of
digestion solution (0.125 mg/ml elastase, 0.25 mg/ml soybean
trypsin inhibitor, 10 mg/ml collagenase I, 2.0 mg/ml crystallized
bovine albumin and 15 mM HEPES; all from Sigma-Aldrich; St. Louis,
MO, USA) was performed at 37°C for 45 min. The digested cells were
filtered with a sterile 100-µm nylon mesh (BD Biosciences,
Franklin Lakes, NJ, USA), centrifuged at 1,000 rpm for 10 min and
rinsed twice in Dulbecco's modified Eagle's medium (DMEM; Corning,
Inc., Corning, NY, USA) containing 10% fetal calf serum. They were
then cultured in DMEM. To characterize the cells as VSMCs,
immunofluorescence staining was performed with a monoclonal
antibody against smooth muscle-α-actin (Sigma-Aldrich). The
explants were incubated in DMEM containing 10% FBS (Corning, Inc.),
2 mM glutamine (Sigma-Aldrich), 50 µg/ml gentamicin
(Sigma-Aldrich), and 50 µl/ml amphotericin B (Sigma-Aldrich)
at 37°C in an atmosphere with 5% CO2. The cells were
used at passages five to eight. For use in the experiments, the
cells were grown to 80–90% confluence and rendered quiescent by
serum-starvation for at least 24 h in DMEM without FBS.
Cell viability assay
The cells were cultured to 80–90% confluence, and
then starved while being cultured in serum-free DMEM for 24 h. The
cells were incubated with various concentrations of WERH (100, 200,
400 and 600 µg/ml) for 40 min and then cultured with or
without PDGF (20 ng/ml; Millipore) for a further 24 h. A modified
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sigma-Aldrich) assay was used to determine cell viability, as
described previously (12,23).
Thymidine incorporation assay
After 24 h starvation, the cells were treated with
the indicated concentrations of WERH and then cultured with or
without PDGF (20 ng/ml) for a further 24 h. This was followed by
the addition of 1 µCi/ml [methyl-3H]thymidine
(PerkinElmer NEN., Boston, MA, USA) for 4 h. Incorporated thymidine
was precipitated with cold 10% trichloroacetic acid, solubilized in
0.2 M NaOH, and counted using a scintillation counter (PerkinElmer
NEN, Inc., Waltham, MA, USA).
Cell cycle analysis using
fluorescence-activated cell sorting (FACS)
The cells were collected and fixed in 70% ethanol
(Millipore) for 2 h at 4°C. After washing with ice-cold
phosphate-buffered saline (PBS; Sigma-Aldrich), the cells were
incubated with RNase (0.1 mg/ml; Sigma-Aldrich) and propidium
iodide (50 µg/ml; Sigma-Aldrich) for 30 min. Cell cycle
analysis was performed using a Becton-Dickinson FACStar flow
cytometer equipped with Becton-Dickinson Cell Fit software (both
from BD Biosciences), as previously described (12,23).
Immunoblot analysis
The cells were lysed using 250 µl lysis
buffer [containing, in mmol/l, HEPES (pH 7.5) 50, NaCl 150, EDTA 1,
EGTA 2.5, DTT 1, β-glycerophosphate 10, NaF 1,
Na3VO4 0.1, and phenylmethylsulfonylfluoride
0.1 and 10% glycerol, 0.1% Tween-20, 10 µg/ml of leupeptin,
and 2 µg/ml aprotinin; all from Sigma-Andrich]. After
centrifugation at 12,000 rpm for 20 min at 4°C, the protein
concentrations of the cell lysates were determined by Bradford
assay. Equal amounts of the samples were resolved via
electrophoresis on SDS-polyacrylamide (10%) gels, and then
electroblotted onto nitrocellulose membranes (Hybond; Amersham
Corp., Arlington Heights, IL, USA). The membranes were incubated
overnight with specific primary antibodies at 4°C followed by
peroxidase-conjugated secondary antibodies for 1 h. Immunocomplexes
were visualized using an ECLPlus™ Western Blot Detection system
(Amersham Biosciences, Piscataway NJ, USA). Experiments were
performed at least 3 times.
Immunoprecipitation and immune complex
kinase assays
For the immunoprecipitation assay, equal amounts of
cell lysates were incubated with specific antibodies overnight at
4°C. Immunocomplexes were added using protein A-sepharose beads
(Santa Cruz Biotechnology, Inc.), followed by incubation at 4°C for
2 h. The immunoprecipitates were washed with lysis buffer 3 times,
and then resuspended in SDS-PAGE sample buffer containing
β-mercaptoethanol (Bio-Rad, Richmond, CA, USA). The samples were
analyzed by immunoblot analysis. For immune complex kinase assays,
the immunoprecipitated samples were washed 3 times with lysis
buffer and twice with kinase buffer (containing, in mM/l, HEPES 50,
MgCl2 10, DTT 1, β-glycerophosphate 10, NaF 1, and
sodium orthovanadate 0.1). The pellet was resuspended in 25
µl kinase buffer containing either 1 µg glutathione
S-transferase (GST)-pRb C-terminal (pRb amino acids 769–921 fusion
protein; Santa Cruz Biotechnology, Inc.) or 5 µg histone
H1 (Life Technologies, Grand Island, NY, USA), 20
µM/l ATP, and 5 µCi [γ32P]ATP (4,500
µCi/mmol; ICN, Costa Mesa, CA, USA), and incubated for 20
min at 30°C. The reaction was then stopped by the addition of 25
µl of 2X concentrated Laemmli sample buffer (Santa Cruz
Biotechnology, Inc.). Electrophoresis was performed with the
samples using an SDS-polyacrylamide (10%) gel. The phosphorylated
forms of pRb and histone H1 were visualized with a BAS
2000 bioimaging analyzer (Fujifilm, Tokyo, Japan).
Wound healing migration assay
Serum-starved cells were grown to 90% confluence,
and then damaged with a pipette tip. The medium was replaced with
fresh medium containing different concentrations of WERH in the
presence or absence of PDGF (20 ng/ml). After 24 h incubation, a
phase-contrast microscope (Optika, Ponteranica, Italy) was use to
capture images and to evaluate the wound closure effected by cell
migration.
Invasion assay
An invasion assay was then performed using modified
Boyden chambers with a polycarbonate nucleopore membrane (Corning,
Inc.). The serum-starved cells were cultured in DMEM containing
WERH with or without PDGF and they were then poured into each of
the upper chamber plates for 24 h to allow for cell invasion
through the membrane. Non-invading cells on the upper surface were
removed using a cotton swab, and the cells that had invaded the
membrane were stained with 0.1% crystal violet solution (containing
2.5 mM crystal violet, 1% MeOH, 4% paraformaldehyde) for 20 min.
Invasive cells were then quantified by counting the cells, using a
microscope, in 6 independent areas at ×20 magnification/well.
Gelatin zymography
Conditioned medium was resolved via electrophoresis
of polyacrylamide gels supplemented with 1 mg/ml gelatin. After
washing the gels twice with 2.5% Triton X-100 at room temperature
for 2 h, they were reacted with a buffer (10 mM CaCl2,
150 mM NaCl, and 50 mM Tris-HCl, pH 7.5) at 37°C overnight. The
gels were then stained with 0.2% Coomassie blue, followed by
destaining with 50% methanol and 5% acetic acid. Areas of
gelatinase activity were visualized as clear bands against a dark
blue field.
Nuclear extracts and electrophoretic
mobility shift assay (EMSA)
Nuclear extracts and EMSA were performed as
described previously (12,18).
The cultured cells were harvested by centrifugation, washed and
suspended in a buffer [10 mM HEPES (pH 7.9), 10 mM KCl, 0.1 mM
EDTA, 0.1 mM EGTA, 1 mM DTT and 0.5 mM PMSF] for 15 min on ice.
After vortexing the cells with 0.5% Nonidet P-40, a nuclear pellet
was then assembled by centrifugation at 12,000 rpm for 1–2 min at
4°C and extracted in buffer (20 mM HEPES pH 7.9, 0.4 M NaCl, 1 mM
EDTA, 1 mM EGTA, 1 mM DTT and 1 mM PMSF) for 15 min at 4°C. The
nuclear extract (10–20 µg) was reacted at 4°C for 30 min
using the 100-fold excess of an unlabeled oligonucleotide
containing the -79 MMP-9 cis element of interest (Bioneer,
Daejeon, Korea). The sequences used were as follows: AP-1,
CTGACCCCTGAGTCAGCACTT; NF-κB, CAGTGGAATTCCCCAGCC; and Sp1,
GCCCATTCCTTCCGCCCCCAGATGAAGCAG. The binding reaction mixture was
then incubated at 4°C for 20 min in a buffer (25 mM HEPES buffer pH
7.9, 0.5 mM EDTA, 0.5 mM DTT, 0.05 M NaCl, and 2.5% glycerol) with
2 µg poly(dI/dC) (Bioneer) and 5 fmol (2×104 cpm)
of a Klenow end-labeled (32P-ATP) 30-mer oligonucleotide
(Bioneer), which spanned the DNA binding site in the MMP-9
promoter. The reaction mixture was electrophoresed at 4°C in a 6%
polyacrylamide gel with TBE (89 mM Tris, 89 mM boric acid and 1 mM
EDTA) running buffer. The gel was washed with water, dried and
exposed to X-ray film overnight. The shifted bands were detected
and evaluated using ImageQuant software (GE Healthcare Life
Sciences, Pittsburgh, PA, USA).
Statistical analysis
In the present study, the results are represented as
the means ± SE. Statistical comparisons between groups were
analyzed using factorial ANOVA analysis and Fisher's least
significant difference test. A P-value <0.05 was considered to
indicate a statistically significant difference.
Results
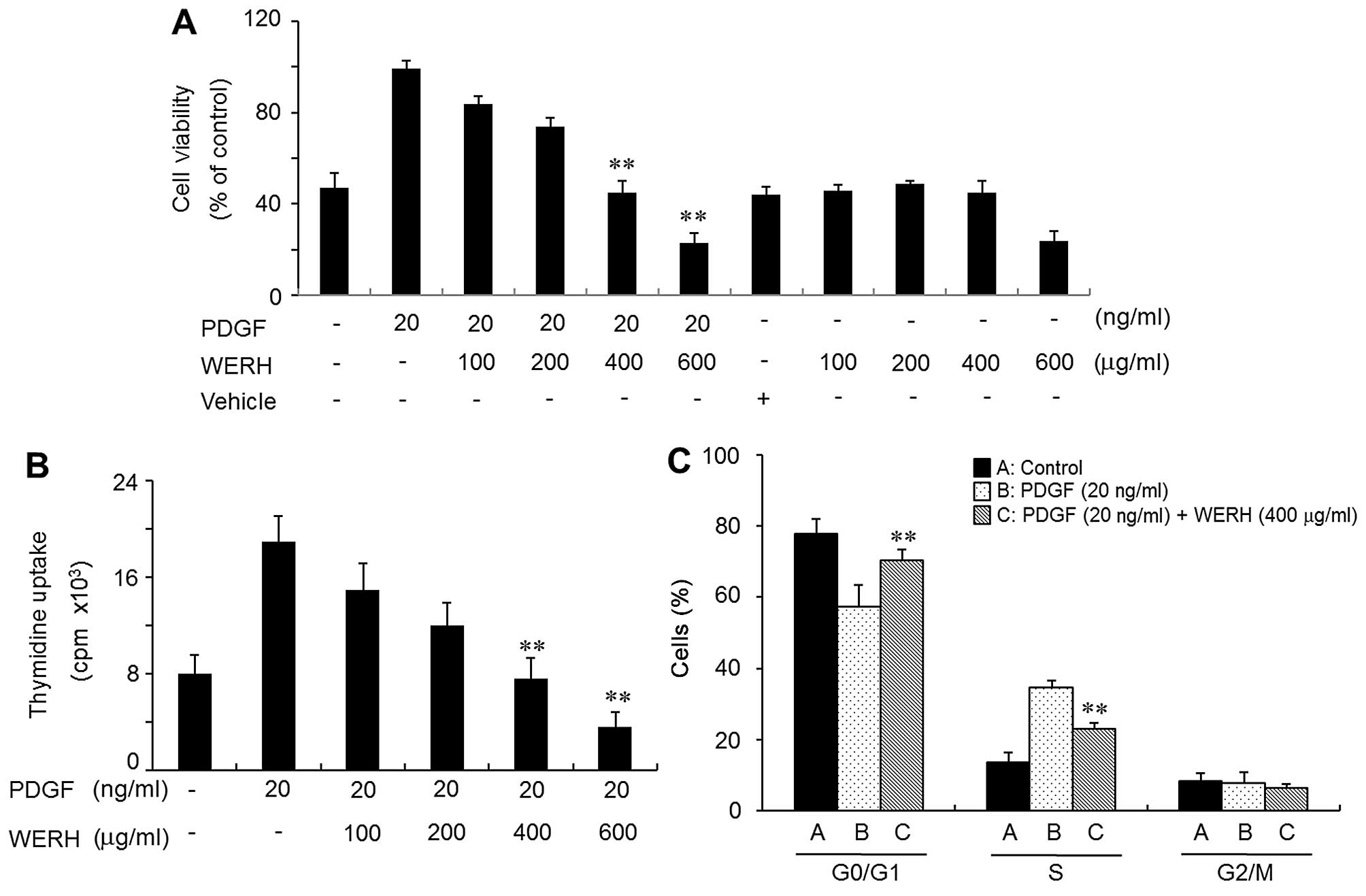
WERH inhibits the PDGF-stimulated
proliferation of VSMCs
To evaluate the effect of WERH on cell
proliferation, VSMCs were treated with WERH, and this was followed
by the addition of PDGF. VMSC proliferation was induced by PDGF,
and this was verified by MTT assay and a thymidine incorporation
assay. The PDGF-induced proliferative effects were reversed to a
control level (non-treatment) at a dose of 400 µg/ml WERH
and cell toxicity was not noted (Fig.
1A and B). Cell death was not observed in the vehicle-treated
group (Fig. 1A). The survival of
VSMCs was significantly reduced at a 600 µg/ml concentration
of WERH (Fig. 1A). Similar
results were obtained in a thymidine incorporation experiment
(Fig. 1B). Subsequent experiments
were performed using a non-toxic cellular concentration of WERH
(below 400 µg/ml). These results demonstrated that WERH
inhibited VSMC proliferation induced by PDGF, without causing cell
death.
WERH induces G1-phase cell cycle arrest
by reducing the expression of cyclins/CDKs in PDGF-stimulated
VSMCs
We then investigated whether WERH induced changes in
cell cycle distribution in PDGF-stimulated VSMCs. As shown in
Fig. 1C, flow cytometric analysis
indicated that in PDGF-stimulated VSMCs WERH treatment induced an
accumulation of cells in the G1-phase cell cycle and reduced the
number of cells in the S-phase cell cycle, compared with
PDGF-stimulated VSMCs which were not treated with WERH. These
results suggest that growth inhibition promoted by WERH in VSMCs
was caused by G1-phase cell cycle arrest. Subsequent experiments
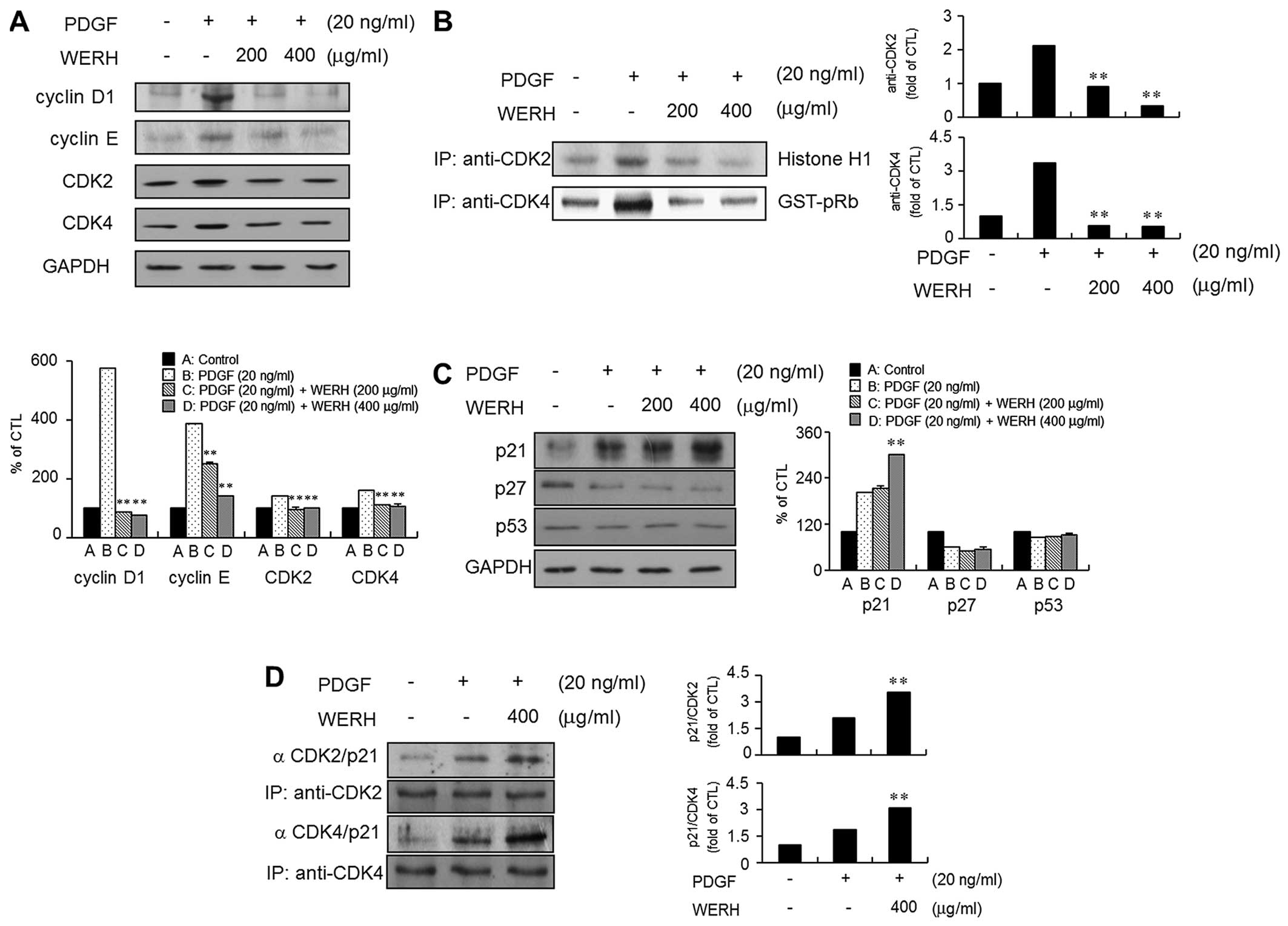
examined the cell cycle-regulatory proteins that are closely
associated with the G1-phase cell cycle. The expression levels of
cyclin D1, cyclin E, CDK2 and CDK4 were upregulated in
PDGF-stimulated VMSCs (Fig. 2A).
The treatment of VSMCs with WERH resulted in a significant
reduction in the PDGF-stimulated expression levels of cyclin D1,
cyclin E, CDK2 and CDK4 (Fig.
2A). The kinase activity of CDKs, as cyclin-CDK complexes, is
known to be essential to G1- to S-phase cell cycle progression
(6,7). Thus, the following experiment
investigated the effect of WERH on the kinase activity of CDKs in
PDGF-stimulated VSMCs. PDGF stimulation increased the kinase
activity of CDK2 and CDK4 in VSMCs. The increased kinase activity
of CDK2 and CDK4 induced by PDGF was suppressed in the presence of
WERH (Fig. 2B).
Upregulation of p21WAF1 is associated
with WERH-mediated G1-phase cell cycle arrest in PDGF-stimulated
VSMCs
As CDKIs are known to be negative regulators that
control the transition from the G1- to S-phases of the cell cycle
(9,10), the expression levels of the CDKIs
p21WAF1 and p27KIP1 were examined. PDGF treatment induced the
expression of p21WAF1 and decreased the expression of p27KIP1 in
VSMCs (Fig. 2C). Stimulation of
VSMCs to PDGF had almost no effect on the expression level of tumor
suppressor protein p53. In addition, WERH treatment increased
p21WAF1 expression in PDGF-stimulated VSMCs. However, WERH had no
effect on the PDGF-induced inhibition of p27KIP1 expression in
VSMCs. The level of p53 expression was not markedly affected by the
addition of WERH in the presence of PDGF (Fig. 2C). To further investigate whether
the observed inhibitory effect on the CDKs was due to interactions
with p21WAF1, immunoprecipitation analysis was performed using cell
lysates in the presence of WERH. An increase in the association of
p21WAF1 with CDK2 and CDK4 was detected in the PDGF-stimulated
VSMCs (Fig. 2D). WERH treatment
increased the levels of p21WAF1/CDK2 and p21WAF1/CDK4 in VSMCs
following stimulation with PDGF (Fig.
2D). These results demonstrated that WERH suppressed the
PDGF-stimulated proliferation of VSMCs, through p21WAF1-mediated
G1-phase cell cycle arrest, which involved inhibition of the kinase
activity of CDKs.
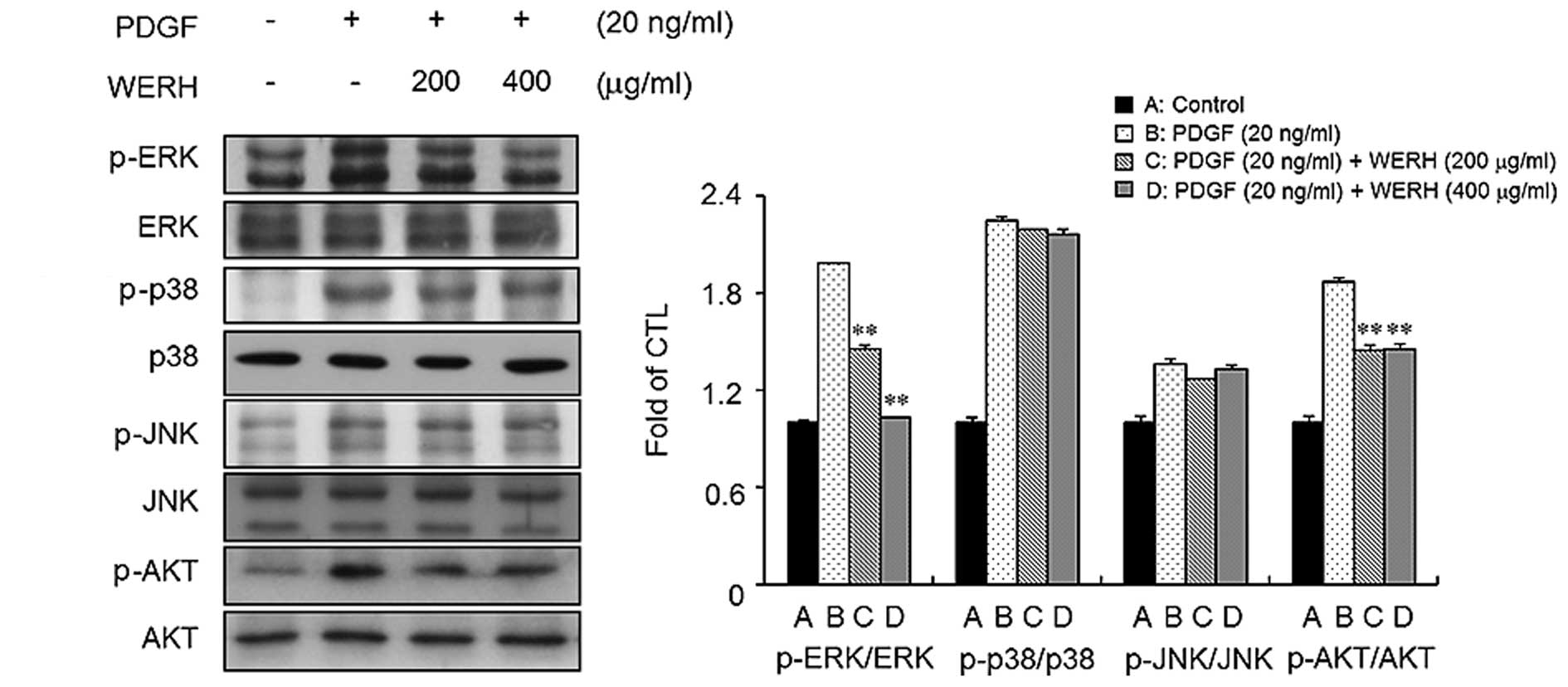
WERH inhibits the phosphorylation of
ERK1/2 and AKT in PDGF-treated VSMCs
To identify the signaling pathways involved in the
PDGF responses in VSMCs, we examined the phosphorylation of MAPKs
and AKT in VSMCs. Stimulation of VSMCs with PDGF led to an increase
in the phosphorylation of ERK1/2, JNK, p38 MAPK, and AKT (Fig. 3). We next investigated whether
WERH is capable of inhibiting the phosphorylation of ERK1/2, JNK,
p38 MAPK and AKT in PDGF-stimulated VSMCs. As shown in Fig. 3, treatment with WERH significantly
suppressed the PDGF-induced phosphorylation of ERK1/2 and AKT in
VSMCs. However, WERH treatment did not markedly affect the
phosphorylation of JNK and p38 MAPK induced by PDGF in VSMCs. These
results indicated that WERH blocks the proliferation of
PDGF-stimulated VSMCs by suppressing ERK1/2 and AKT signaling.
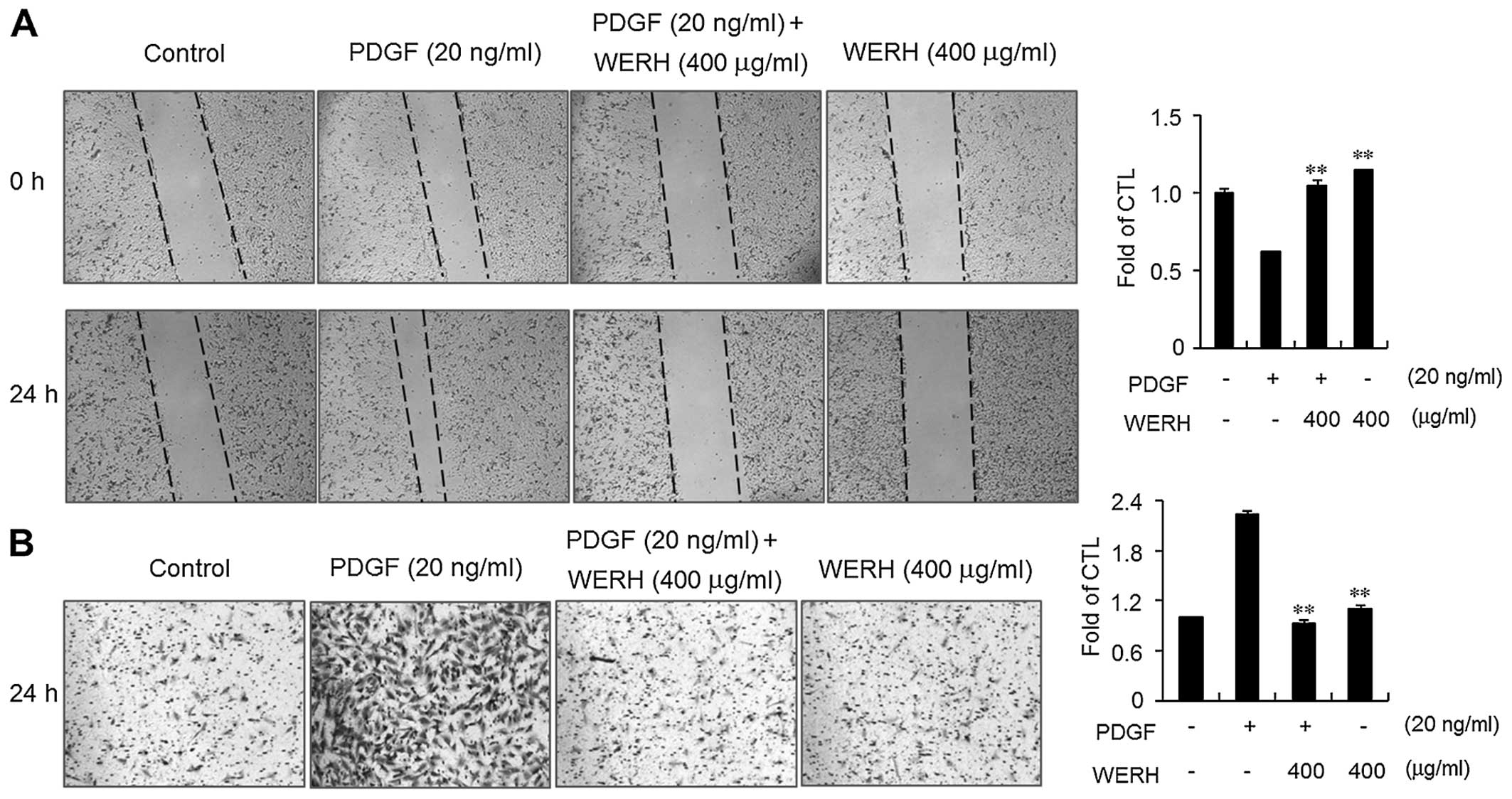
WERH suppresses PDGF-induced migration
and invasion
The migration and invasion of VSMCs are critical
steps in the formation of vascular lesions that lead to the
development of atherosclerosis (13,14). An in vitro wound healing
assay and an invasion assay were performed to determine the effects
of WERH on the PDGF-induced migration and invasion of VSMCs. PDGF
treatment significantly increased the motility and invasiveness of
VSMCs within 24 h (Fig. 4).
Treatment of VSMCs with WERH resulted in decreased migration of
VSMCs (Fig. 4A). In addition, the
invasiveness of PDGF-stimulated VSMCs was significantly obstructed
in the presence of WERH (Fig.
4B). These results clearly suggest that WERH plays an
inhibitory role in the migration and invasion of VSMCs induced by
PDGF.
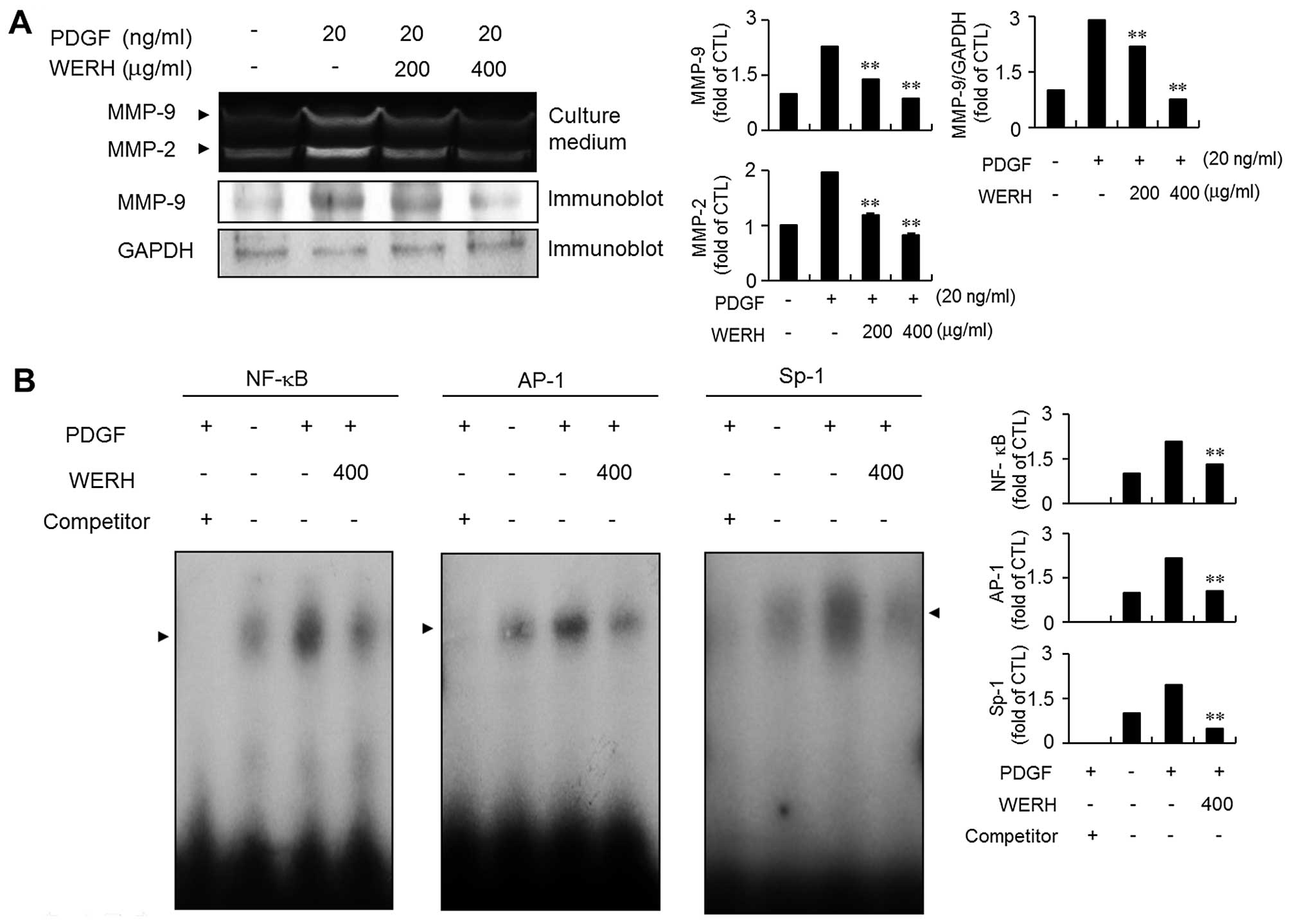
WERH impedes PDGF-induced MMP-9
expression by suppressing NF-κB, AP-1 and Sp-1 binding
activities
MMP-9 expression is involved in the migration and
invasion of VSMCs during the development of vascular lesions
(13,14). Thus, the next experiment examined
whether WERH inhibited the expression of MMP-9 in PDGF-stimulated
VSMCs using a gelatin zymographic assay and immunoblot analysis.
The expression of MMP-9 was stimulated by exposure to PDGF as
demonstrated by zymographic assay (Fig. 5A). Treatment with WERH
significantly abolished PDGF-stimulated MMP-9 expression in VSMCs.
The immunoblot analysis results were similar (Fig. 5A). To further investigate the
exact molecular mechanism of action of the inhibitory effect of
WERH, an EMSA experiment was employed using three motifs, NF-κB,
AP-1 and Sp-1 cis-elements, that are responsible for MMP-9
expression (18,19). PDGF treatment significantly
stimulated the binding activity of the NF-κB, AP-1 and Sp-1 motifs
in VSMCs. The addition of WERH markedly reduced the increased
binding ability of NF-κB, AP-1 and Sp-1 motifs in PDGF-stimulated
VSMCs (Fig. 5B). These results
demonstrated that the transcription factors NF-κB, AP-1 and Sp-1
are involved in the WERH-induced inhibition of MMP-9 expression in
PDGF-stimulated VSMCs.
Discussion
The proliferation and migration of mitogen- and
growth factor-induced VSMCs is involved in the formation of
vascular lesions, which leads to the development of vascular
diseases, including atherosclerosis and restenosis (1–3).
The role and importance of PDGF in regulating the proliferation and
migration of VSMCs has attracted considerable attention (2,3).
It is now well established that PDGF, produced by several types of
cells, plays a pivotal role as a specific mitogen in VSMCs
(2,3). A natural product such as WERH that
targets PDGF responses in VSMCs is a potential therapeutic strategy
in the prevention or inhibition of vascular diseases. The extract
of Rosa hybrida is known to be used in the cosmetics
industry (20), and it is also
known to exert various pharmacological effects, including
antioxidant, anti-inflammatory, antimicrobial and neuroprotective
effects (20–22). However, the effect of Rosa
hybrida extract on cardiovascular diseases has not yet been
examined to the best of our knowledge. In the present study, we
investigated the novel suppressive effects of WERH that are
involved in the molecular events underlying the proliferation,
migration, and invasion of VSMCs in response to PDGF.
One of the key processes that occurs in
atherosclerosis is the proliferation of VSMCs stimulated by PDGF
(1–3). In the present study, WERH
concentrations at and below 400 µg/ml were found to inhibit
the proliferation of VSMCs in response to PDGF, in a non-toxic
manner. It has previously been pointed out that after vascular
injury involving PDGF stimulation, VSMCs proliferate abnormally
upon re-entry into the cell cycle (1,8).
The findings of the present study showed that WERH induced G1-phase
cell cycle arrest in PDGF-stimulated VSMCs. WERH-mediated G1-phase
cell cycle arrest was associated with the inhibited expression of
cyclins and CDKs. The data also indicated that WERH treatment
induced a significant upregulation of p21WAF1 without altering
p27KIP1 expression, during G1-phase cell arrest in PDGF-stimulated
VSMCs. Taking into consideration the results from the immunoblot
analysis demonstrating the increased expression of p21WAF1 in
PDGF-stimulated VSMCs, we suggest that p21WAF1 played an important
role, as also evidenced by cell proliferation assay. These results
clearly demonstrated that the preventive effect of WERH in
PDGF-stimulated VSMCs is attributable to p21WAF1-mediated G1-phase
cell cycle arrest and inhibition of the kinase activity of
cyclin/CDK complexes.
PDGF signaling is involved in the MAPKs and AKT
pathways, which are involved in the proliferation of VSMCs
(4,5). In the present study, WERH inhibited
the PDGF-induced phosphorylation of ERK1/2 in VSMCs. Our results
were consistent with those from previous studies demonstrating that
phosphorylation of ERK1/2 was inhibited by the hexane fraction of
Rosa hybrida in RAW264.7 cells stimulated with
lipopolysaccharide (LPS) (24).
In addition, we noted that the phosphorylation of AKT was
attenuated by the addition of WERH when cells were stimulated with
PDGF. This observation is the first evidence of WERH-mediated
inhibition of AKT phosphorylation in PDGF-stimulated VSMCs, to the
best of our knowledge. However, unexpectedly, treatment with WERH
barely affected the phosphorylation of p38 MAPK and JNK signaling
in PDGF-stimulated VSMCs. These data demonstrated that a reduction
in ERK1/2 phosphorylation and a decrease in AKT phosphorylation
occurred when WERH exerted an inhibitory effect on the
proliferation of VSMCs stimulated by PDGF.
Migration and invasion of VSMCs are central to
vascular lesion formation during atherosclerosis and restenosis
(1–3). MMPs have been noted to act as
important regulators of the migration and invasion of VSMCs
(14–17). Notably, enhanced levels of MMP-9
expression have been observed during the formation of arterial
lesions in vitro and in vivo (14–17). Previous studies have reported that
MMP-9 expression is regulated by transcription factors such as
NF-κB, Sp1 and AP-1 motifs (18,19). Thus, we hypothesized that WERH
affects the sequential machinery of
migration/invasion/MMP-9/transcription factors in PDGF-stimulated
VSMCs. Consistent with this hypothesis, our results demonstrated a
significant decrease in the wound-healing and invasive ability of
PGDF-stimulated VSMCs that had been treated with WERH. Following
analysis of the migration and invasion, our data also showed that
WERH suppressed the expression of MMP-9 in PDGF-treated VSMCs.
Finally, treating VSMCs with WERH significantly inhibited the
binding activity of NF-κB, Sp1 and AP-1, which were increased by
PDGF in VSMCs. These results demonstrated that WERH treatment
abolished PDGF-induced MMP-9 expression in VSMCs by suppressing the
binding activity of NF-κB, AP-1 and Sp-1; we hypothesize that this
led to an inhibition of the breakdown of ECM and its influence on
the migratory and invasive abilities of VSMCs.
The present study elucidates important, novel
molecular mechanisms for the effects of WERH in PDGF-stimulated
VSMC responses. First, our results suggested that WERH inhibited
the proliferation of VSMCs stimulated by PDGF through
p21WAF1-mediated G1-phase cell cycle arrest, which was attributed
to the reduced kinase activity of cyclin D1/CDK4 and cyclin E/CDK2
complexes. In addition, the treatment of VSMCs with WERH blocked
the phosphorylation of ERK1/2 and AKT signaling in response to
PDGF. Furthermore, WERH treatment impeded the PDGF-induced
migration and invasion of VSMCs, which was regulated through
diminished MMP-9 expression, by a decrease in the binding activity
of NF-κB, AP-1 and Sp1 motifs. The findings of the present study
suggest that WERH has the potential to be used in the prevention
and treatment of atherosclerosis and re-stenosis. Further study is
required to identify the active components of WERH and to elucidate
its efficacy in animal models.
Acknowledgments
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF), funded by the Ministry of Education, Science and Technology
(no. 2012R1A6A3A01040720). This study was also supported by the
Chung-Ang University Excellent Student Scholarship in 2014.
References
|
1
|
Ross R: Cell biology of atherosclerosis.
Annu Rev Physiol. 57:791–804. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jawien A, Bowen-Pope DF, Lindner V,
Schwartz SM and Clowes AW: Platelet-derived growth factor promotes
smooth muscle migration and intimal thickening in a rat model of
balloon angioplasty. J Clin Invest. 89:507–511. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ferns GA, Raines EW, Sprugel KH, Motani
AS, Reidy MA and Ross R: Inhibition of neointimal smooth muscle
accumulation after angioplasty by an antibody to PDGF. Science.
253:1129–1132. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Owens GK, Kumar MS and Wamhoff BR:
Molecular regulation of vascular smooth muscle cell differentiation
in development and disease. Physiol Rev. 84:767–801. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhan Y, Kim S, Izumi Y, Izumiya Y, Nakao
T, Miyazaki H and Iwao H: Role of JNK, p38, and ERK in
platelet-derived growth factor-induced vascular proliferation,
migration, and gene expression. Arterioscler Thromb Vasc Biol.
23:795–801. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sherr CJ: G1 phase progression: cycling on
cue. Cell. 79:551–555. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sherr CJ: Cancer cell cycles. Science.
274:1672–1677. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gordon D, Reidy MA, Benditt EP and
Schwartz SM: Cell proliferation in human coronary arteries. Proc
Natl Acad Sci USA. 87:4600–4604. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xiong Y, Hannon GJ, Zhang H, Casso D,
Kobayashi R and Beach D: p21 is a universal inhibitor of cyclin
kinases. Nature. 366:701–704. 1993. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Toyoshima H and Hunter T: p27, a novel
inhibitor of G1 cyclin-Cdk protein kinase activity, is related to
p21. Cell. 78:67–74. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Weiss RH, Joo A and Randour C:
p21(Waf1/Cip1) is an assembly factor required for platelet-derived
growth factor-induced vascular smooth muscle cell proliferation. J
Biol Chem. 275:10285–10290. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Moon SK, Kim HM, Lee YC and Kim CH:
Disialoganglioside (GD3) synthase gene expression suppresses
vascular smooth muscle cell responses via the inhibition of ERK1/2
phosphorylation, cell cycle progression, and matrix
metalloproteinase-9 expression. J Biol Chem. 279:33063–33070. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Senior RM, Griffin GL, Fliszar CJ, Shapiro
SD, Goldberg GI and Welgus HG: Human 92- and 72-kilodalton type IV
collagenases are elastases. J Biol Chem. 266:7870–7875.
1991.PubMed/NCBI
|
|
14
|
Newby AC and Zaltsman AB: Molecular
mechanisms in intimal hyperplasia. J Pathol. 190:300–309. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cho A and Reidy MA: Matrix
metalloproteinase-9 is necessary for the regulation of smooth
muscle cell replication and migration after arterial injury. Circ
Res. 91:845–851. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Galis ZS, Johnson C, Godin D, Magid R,
Shipley JM, Senior RM and Ivan E: Targeted disruption of the matrix
metalloproteinase-9 gene impairs smooth muscle cell migration and
geometrical arterial remodeling. Circ Res. 91:852–859. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cho A, Graves J and Reidy MA:
Mitogen-activated protein kinases mediate matrix
metalloproteinase-9 expression in vascular smooth muscle cells.
Arterioscler Thromb Vasc Biol. 20:2527–2532. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Moon SK, Cha BY and Kim CH: ERK1/2
mediates TNF-alpha-induced matrix metalloproteinase-9 expression in
human vascular smooth muscle cells via the regulation of NF-kappaB
and AP-1: involvement of the ras dependent pathway. J Cell Physiol.
198:417–427. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bond M, Chase AJ, Baker AH and Newby AC:
Inhibition of transcription factor NF-kappaB reduces matrix
metalloproteinase-1, -3 and -9 production by vascular smooth muscle
cells. Cardiovasc Res. 50:556–565. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Choi EM and Hwang JK: Investigations of
anti-inflammatory and antinociceptive activities of Piper cubeba,
Physalis angulata and Rosa hybrida. J Ethnopharmacol. 89:171–175.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang G, Park D, Lee SH, Bae DK, Yang YH,
Kyung J, Kim D, Choi EK, Hong JT, Jeong HS, et al: Neuroprotective
effects of a butanol fraction of Rosa hybrida petals in a middle
cerebral artery occlusion model. Biomol Ther (Seoul). 21:454–461.
2013. View Article : Google Scholar
|
|
22
|
Lee HR and Lee JM, Choi NS and Lee JM: The
antioxidative and antimicrobial ability of ethanol extracts from
Rosa hybrida. Korean J Food Sci Technol. 35:373–378. 2003.
|
|
23
|
Lee SJ, Park SS, Kim WJ and Moon SK:
Gleditsia sinensis thorn extract inhibits proliferation and
TNF-α-induced MMP-9 expression in vascular smooth muscle cells. Am
J Chin Med. 40:373–386. 2012. View Article : Google Scholar
|
|
24
|
Lee HJ, Kim HS, Kim ST, Park D, Hong JT,
Kim YB and Joo SS: Anti-inflammatory effects of hexane fraction
from white rose flower extracts via inhibition of inflammatory
repertoires. Biomol Ther (Seoul). 19:331–335. 2011. View Article : Google Scholar
|