Introduction
Atherosclerosis (AS) is a disease which affect the
arteries and causes high mortality and morbidity in industrialized
countries (1). AS was
traditionally considered to be triggered by hyperlipemia, which
causes multiple metabolic disturbances (2). However, other research has suggested
that the initiation and development of AS involves chronic
lipid-related inflammation of the vessel wall, mainly caused by
activated mononuclear cells such as dendritic cells (DCs) which are
present in the vascular subendothelium. The activation of DCs
promoted the progression of atherosclerotic plaques by affecting
cellular recruitment, interaction and secretory products (3). Thus, DCs play an important role in
triggering development of AS.
MicroRNAs (miRNAs) are single-stranded RNAs and are
approximately 22 nucleotides in length. In a sequence-specific
manner, miRNAs induce the formation of silencing complexes that
lead to translational repression and/or mRNA decay in target genes
(4). miRNAs have been shown to be
involved in a variety of physiological and pathological activities
and the development of various conditions and diseases, including
hypertrophic remodeling, heart failure and arrhythmia (5–10).
Previous research has shown that a number of miRNAs are involved in
mediating the functions of DCs and thus play important roles in the
pathogenesis of AS (11). miR-155
is encoded within an exon of a non-coding RNA transcribed from the
B-cell integration cluster (BIC), which is located on chromosome 21
(12). Previous research has
indicated that miR-155 is involved in regulating various biological
processes, including hematopoietic differentiation, viral
infection, AS and tumorigenesis (11,13–16). Several previous studies have also
demonstrated that miR-155 is involved in various immune-mediated
inflammatory diseases although the exact role of miR-155 in the
vasculogenesis, development and progression of AS remains a matter
of debate. For instance, Nazari-Jahantigh et al (17) and other researchers have reported
that miR-155 promotes the development of AS (18). On the other hand, research from
our laboratory suggests that miR-155 plays a protective role in the
development and progression of AS (19). In a murine model of
hyperlipidemia, miR-155 was found to act as an anti-inflammatory
and protective factor against AS (20).
Despite those controversial reports regarding the
role of miR-155 in AS, it has been reported in a number of studies
that treatment with oxidized low-density lipoprotein (oxLDL), which
plays a well-known pathogenic role in the development and
progression of AS, induces the expression of miR-155 in various
cell types under different (patho)physiological conditions
(21–23). However, exactly how miRNA-155 was
regulated in DCs treated with oxLDL was not fully understood. The
aim of the present study was thus to investigate the molecular
mechanisms by which miR-155 expression is mediated by oxLDL in
DCs.
Materials and methods
Cell culture
In the present study, experiments using human
peripheral blood were undertaken according to the principles of the
Declaration of Helsinki and were approved by the Ethics Committee
of Zhejiang University (Hangzhou, China). We also obtained
appropriate consent from the healthy volunteer donors. Human
peripheral blood was obtained from healthy volunteer adults for
subsequent isolation of monocytes using a lymphocyte separation
liquid system (Sigma-Aldrich, St. Louis, MO, USA). Cells were
cultured in complete medium [RPMI-1640, 10% fetal calf serum and
1,000 U/ml granulocyte-macrophage colony-stimulating factor
(GM-CSF)] and 1,000 U/ml interleukin-4 (IL-4) (all from Peprotech,
Inc., Rocky Hill, NJ, USA) for 5 days. Complete medium was replaced
every other day. oxLDL (40 µg/ml) (Yiyuan Biotechnology,
Guangzhou, China) was added at day 5, and cells were harvested at
day 7. All inhibitors, namely SB203580, SP600125, UO126, PDTC and
AG490 (all purchased from Sigma-Aldrich), were added 30 min prior
to oxLDL (40 µg/ml; Yiyuan Biotechnology) treatment for 24
h. Dimethyl sulphoxide (DMSO; Sigma-Aldrich) was used as the
control of inhibitors since these chemical inhibitors were
dissolved in DMSO. Finally, the cells were harvested for later
experiments.
siRNA transfection
DCs were transfected with siRNA against scavenger
receptor A (SRA), lectin-like oxidized low-density lipoprotein
(LDL) receptor-1 (LOX-1), cluster determinant 36 (CD36), signal
transducer and activator of transcription 3 (STAT3), miR-155
inhibitor, Yin Yang 1 (YY1) or V-Myb avian myeloblastosis viral
oncogene homolog (MYB) (50 nM) (Baiao Biotech, Inc., Changchun,
China) using Lipofectamine RNAiMAX reagent (Life Technologies,
Grand Island, NY, USA) according to the manufacturer's
instructions. Control siRNA against no significant targets (Baiao
Biotech, Inc.) was also used as the negative control (NC). Briefly,
2 µl siRNA, at a final concentration of 50 nM, and 5
µl transfection reagent were mixed and added to DCs in
serum-free medium. Cells were harvested 24 h post-transfection.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated using an miRcute miRNA
isolation kit (Tiangen Biotech Co., Ltd., Beijing, China). cDNA was
generated using a PrimeScript miRNA RT reagent kit, and qPCR was
carried out using SYBR-Green Premix Ex Taq (both from Takara Bio,
Inc., Otsu, Japan). PCR was performed using an ABI PRISM 7500
Sequence Detection system (Applied Biosystems Life Technologies,
Foster City, CA, USA). U6 was used as an endogenous control. The
sequences of primers used for qPCR are: U6 forward,
5-ACTTGCTCATCAAGGTGTCAG-3′ and reverse, 5-TGACCAGCGTTTGTTCAATGT-3′;
miR-155 forward, 5-UUAAUGCUAAUCGUGAGAGGGGU-3′ and reverse uni-miR
qPCR primer, 5-TTTTTTTTTTTTTTTTTTTT-3′ (Takara Bio, Inc.).
Western blot analysis
DCs were lysed and a total of 60 µg protein
lysates were subjected to electrophoresis on 10% polyacrylamide SDS
gel, followed by transfer onto PVDF membranes (Millipore Corp.,
Billerica, MA, USA). The membranes were subsequently probed using
primary antibodies: rabbit anti-YY1 (1:1,000; #2185), rabbit
anti-MYB (1:1,000; #12319) and rabbit anti-Janus kinase 1/2
(JAK1/2) (1:1,000; H-103) (all from Cell Signaling Technology,
Beverly, MA, USA) or rabbit anti-GAPDH (1:1,000; CW101M) (Changwei,
China). An ECL kit (Pierce Biotechnology, Inc., Rockford, IL, USA)
was then used to detect chemiluminescence.
Dual-luciferase reporter assay
Genomic DNA was extracted from DCs using a Genomic
DNA Extraction kit (Kangwei, Beijing, China). Promoter regions of
miRNA-155, 2,000, 1,500 or 1,000 bp upstream of the transcription
initiation site were amplified using PCR. The PCR products were
gel-purified and then subcloned into a pGL3 luciferase reporter
vector (Promega Corp., Madison, WI, USA) at HindIII and
SacI sites. The sequences of the primers used to subclone
these cis-regulatory elements are shown below: 2k-promoter forward,
5′-CAAGCACTGCCGACTACAATA-3′ and reverse,
5′-CCAGGCTGATTCATCCCACTG-3′; 1.5k-promoter forward,
5′-GCCGACTCAGGCACTACAATA-3′ and reverse,
5′-CCAGGCTGATTCATCCCACTG-3′; and 1k-promoter forward,
5′-GACTACAATCACGAACTGCCA-3′ and reverse,
5′-CCAGGCTGATTCATCCCACTG-3′. The reporter was transfected into DCs
together with transcription factors (TFs) of interest and activity
was determined 48 h post-transfection. Firefly luciferase activity
was also measured using a dual-luciferase reporter assay system
(Promega Corp.). Relative reporter activity was subsequently
obtained by normalization to Renilla control luciferase
activity.
TF filter plate assay
After treatment, DCs were harvested to extract
nuclear proteins. A TF filter plate assay (Signosis Inc.,
Sunnyvale, CA, USA) was performed according to the manufacturer's
instructions. In brief, a TF DNA complex was mixed and subsequently
separated from free probes, prior to elution of bound probes.
Hybridization of the eluted probe with a hybridization plate was
then performed and finally detected using a luminometer (BioTek,
Winooski, VT, USA).
Co-immunoprecipitation (Co-IP) assay
Protein lysates were purified from DCs, as mentioned
above. One milligram of protein lysate was incubated with 5
µg of the appropriate antibody [anti-MYB (#12319) or rabbit
IgG (#14708); Cell Signaling Technology] on a rotator (JinDun,
Ningbo, China), at 4°C, overnight, followed by further incubation
for a 2–4-h period with 10 µl protein A-agarose beads (Cell
Signaling Technology). After being washed three times, the
immuno-complexes were precipitated by centrifugation at 3,000 rpm
for 3 min at 4°C. The bound proteins were released with 2X SDS
buffer, boiled for 5 min, and subjected to western blot
analysis.
Bioinformatics analysis
For the purposes of the present study, the promoter
sequence of miR-155 was obtained from the website http://genome.ucsc.edu/cgi-bin/hgNear.
Transcription factor binding sites were predicted using the website
http://www.cbrc.jp/research/db/TFSEARCH.html by typing
the promoter sequence of miR-155.
Chromatin immunoprecipitation (ChIP)
assay
DCs were cross-linked with 1% formaldehyde in medium
for 10 min at 25°C and washed with ice-cold PBS before resuspension
in 1 ml SDS lysis buffer. The cell suspension was sonicated to
average 300–900-nt length DNA fragments before being pre-cleared
with 20 µl protein A-agarose beads for 30 min at 4°C. After
the beads were removed, the chromatin solution was
immunoprecipitated with an antibody (anti-YY1 or anti-MYB) at 4°C
overnight, followed by incubation with protein A-agarose beads for
an additional 1 h at 4°C. The immune complex was then eluted using
150 µl elution buffer (1% SDS and 0.1 M NaHCO3).
Formaldehyde cross-linking was reversed by heating at 65°C for 4 h
with proteinase K and NaCl (5 M). Genomic DNA was purified from the
immunoprecipitate and analyzed by PCR using specifically designed
primers to detect target regions of interest. Primers to amplify
the region of YY1-a were: forward, 5′-GTTCCAACACAAACTCTT-3′ and
reverse, 5′-CTCTAATCAGGCAATTCG-3′; primers to amplify the region of
'YY1-b' were: forward, 5′-CTTATTATATAAAGGCGC-3′ and reverse,
5′-GGTGACCCATCACGAAAGG-3′; primers to amplify the region of 'MYB'
were: forward, 5′-GACACAGTCTTACGTTCGA-3′ and reverse,
5′-CGACCTCACTGTCGTATA-3′.
Statistical analysis
Statistically significant differences between groups
were calculated using unpaired t-tests. Values are expressed as the
means ± SD, and a p-value <0.05 was considered to indicate a
statistically significant difference. Every experiment was
performed at least 5 times (n≥5).
Results
miR-155 is upregulated by oxLDL through
modulation of SRA and CD36
miR-155 has been established as an
inflammatory-related miRNA, and its role in AS has been examined;
consistent with our previous research (19), we found that miR-155 was increased
in DCs in response to oxLDL treatment in a dose-dependent manner
(Fig. 1A). Subsequently, to
explore the molecular basis underlying this upregulation, we used
siRNA to knock down three scavenger receptors, SRA, CD36 and LOX-1,
all of which are known to mediate endocytosis of oxLDL in
macrophages (24), and then
tested whether any of these scavenger receptors was involved in the
upregulation of miR-155 by oxLDL in DCs. As shown in Fig. 1B, siRNA knockdown of either CD36
or SRA, but not LOX-1, significantly decreased the levels of
miR-155 induced by oxLDL. Thus, we conclude that CD36 and SRA are
involved in the oxLDL-mediated miR-155 upregulation in DCs.
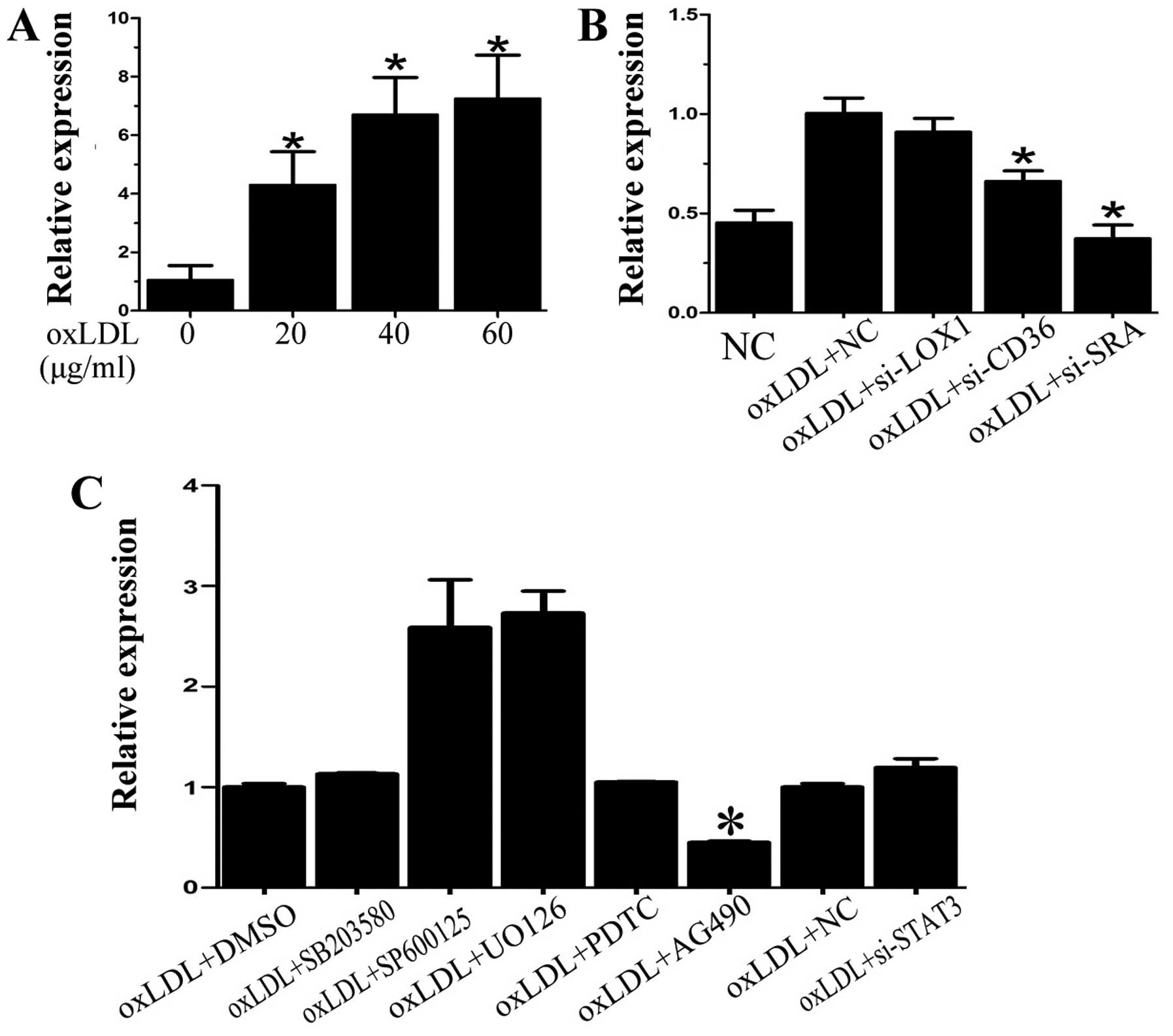 | Figure 1miR-155 expression is upregulated by
oxidized low-density lipoprotein (oxLDL) through scavenger receptor
A (SRA) and Janus kinase 1/2 (JAK1/2) signaling. (A) miR-155 was
increased in dendritic cells (DCs) following oxLDL treatment. (B)
Knockdown of cluster determinant 36 (CD36) or SRA both decreased
miR-155 expression induced by oxLDL. (C) p38, c-Jun N-terminal
kinase (JNK), extracellular signal-regulated kinase (ERK)1/2 and
nuclear factor-κB (NF-κB) signaling did not mediate miR-155
activation caused by oxLDL. SB203580, SP600125, UO126 and PDTC,
inhibitors of p38, JNK, ERK1/2 and NF-κB, respectively, were
applied before oxLDL treatment. Signal transducer and activator of
transcription 3 (STAT3) siRNA also exerted almost no effect on
miR-155 expression. AG490, the inhibitor of JAK2, significantly
decreased oxLDL-induced miR-155 expression. Data are presented as
the means ± SD. n=5 and *p<0.05, compared with
control group [negative control (NC) or oxLDL + DMSO]. |
miR-155 is upregulated by oXLDL and the
JAK1/2 signaling pathway is involved
Subsequently, we sought to probe the potential
pathways by which oxLDL triggers miR-155 transcription. Since
mitogen-activated protein kinase (MAPK) and nuclear factor-κB
(NF-κB) pathways have been established as key signaling pathways
driving inflammatory responses, we pre-treated DCs with specific
inhibitors for p38, c-Jun N-terminal kinase (JNK), extracellular
signal-regulated kinase (ERK)1/2 or NF-κB prior to oxLDL treatment.
Neither MAPK nor NF-κB signaling was directly involved in mediating
miR-155 activation caused by oxLDL (Fig. 1C). Since previous research has
postulated a potential role of JAK and STAT3 in miR-155
transcription (25), we then
examined whether they were involved in oxLDL-induced miR-155
upregulation using specific inhibitors for JAK and STAT3. The
inhibition of JAK2 by AG490 significantly reduced the induction of
miR-155 in response to oxLDL, but inhibition of STAT3 using STAT3
siRNA did not exert any significant effect on miR-155 expression
induced by oxLDL (Fig. 1C).
TFs YY1 and MYB participate in miR-155
transcriptional regulation
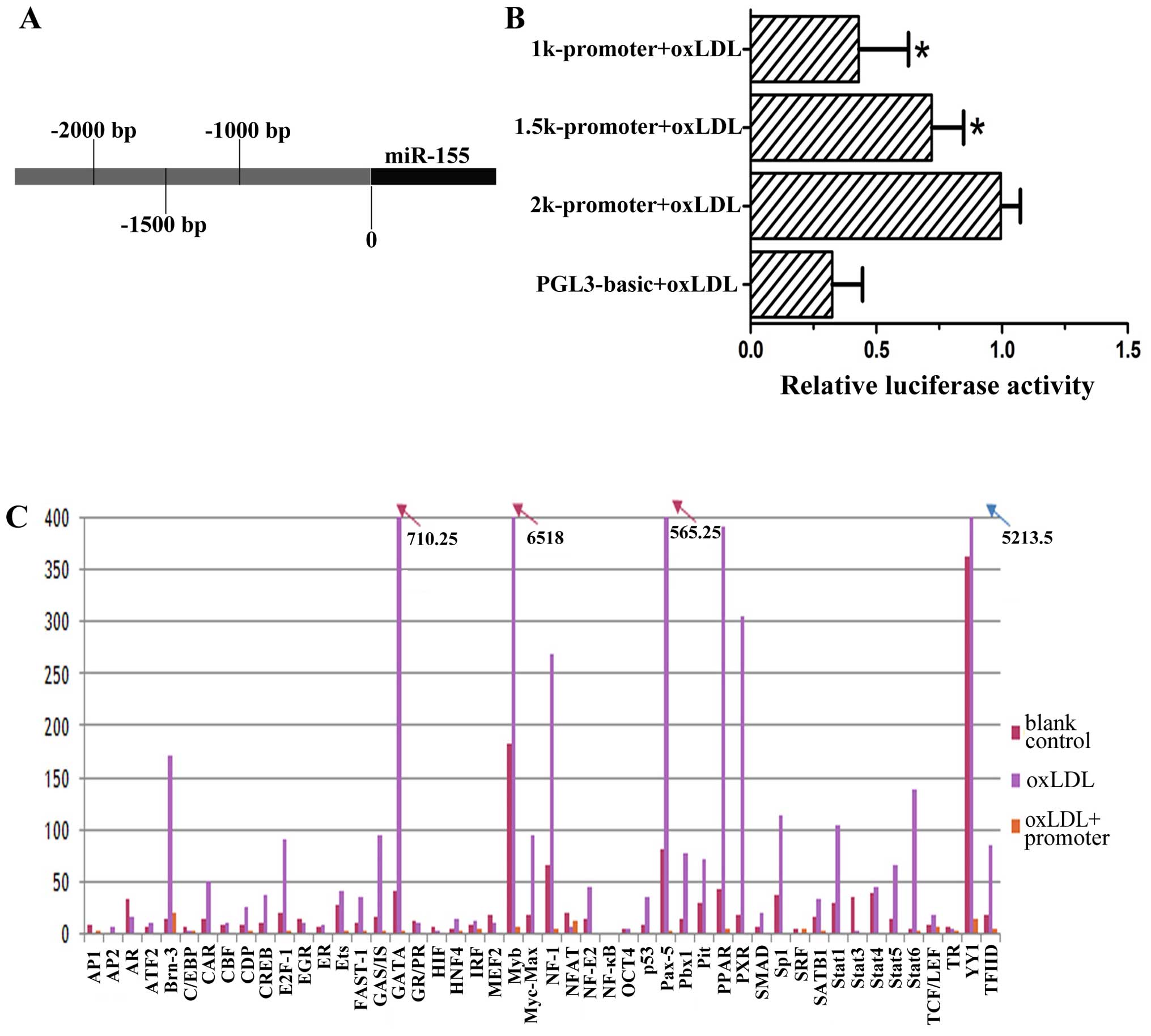
To study the regulatory elements in the promoter of
miR-155, we constructed a series of luciferase reporter plasmids
including 1,000, 1,500 and 2,000 bp upstream of the transcription
initiation site of miR-155 (Fig.
2A), as previously described (26). Data from the dual luciferase
reporter assays showed that the 2,000 bp region exhibited the
highest activity in response to oxLDL treatment, whereas the 1,000
bp sequence showed the lowest, but still significant, activation
(Fig 2B). Subsequently, we
performed TF filter plate assays using the 2,000 bp sequence of
miR-155 promoter. As shown in Fig.
2C, YY1 and MYB had more significant bindings to this promoter
region than other 46 TFs examined.
YY1 and MYB promote miR-155 transcription
by directly binding to the miR-155 promoter
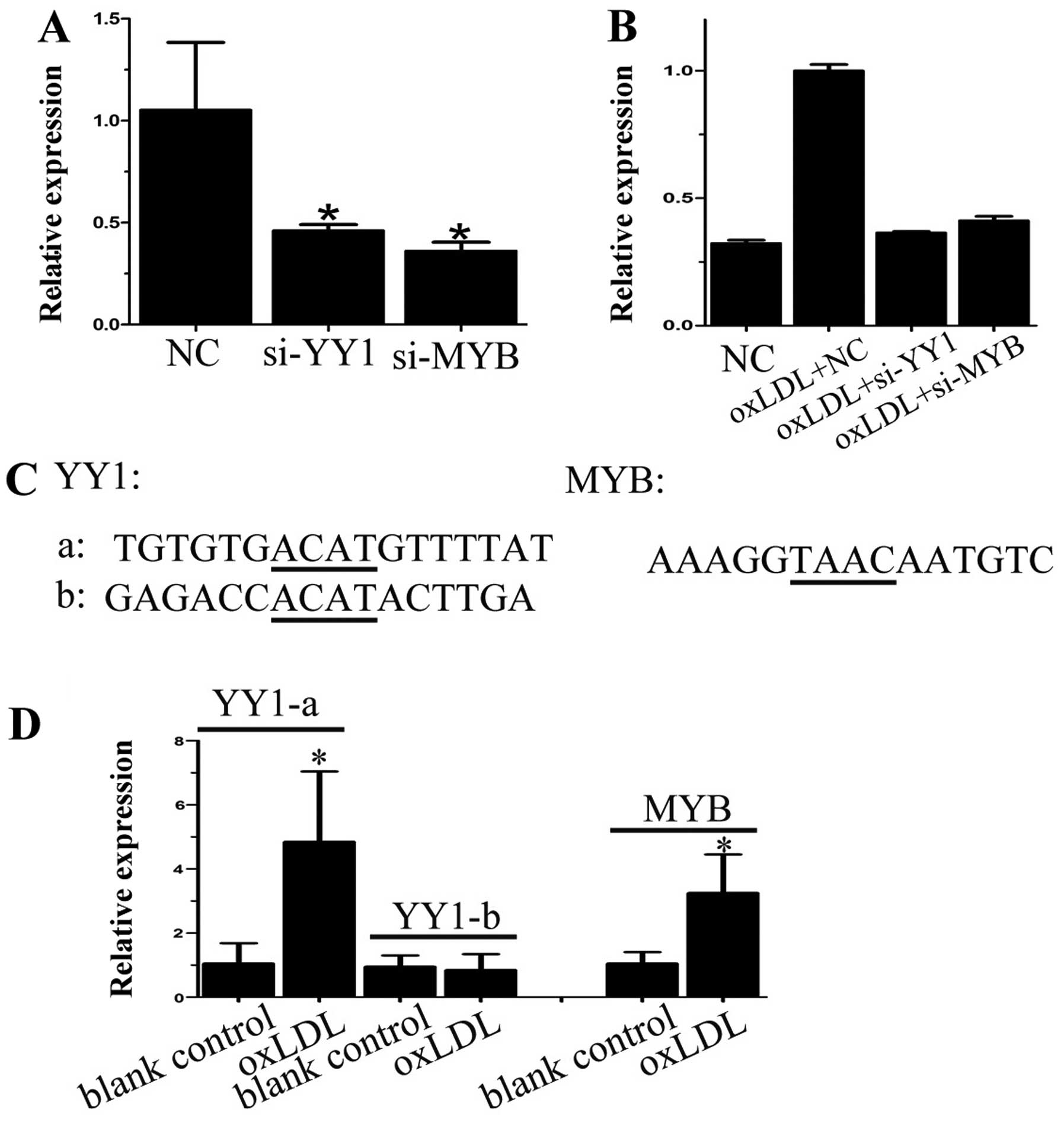
We next attempted to elucidate whether YY1 and/or
MYB was involved in oxLDL-induced miR-155 transcription in DCs. By
transfecting cells with siRNA against YY1 and MYB, we demonstrated
that knockdown of both YY1 and MYB reduced baseline miR-155
expression both on their own and also with oxLDL treatment
(Fig. 3A and B), suggesting that
YY1 and MYB regulated miR-155 expression both at basal levels and
in response to oxLDL. Bioinformatics analysis revealed two
potential binding sites for YY1 and one for MYB on the miR-155
promoter (Fig. 3C), and we
confirmed that oxLDL treatment strengthened the binding of YY1 to
the first binding site (Fig. 3D),
and that oxLDL also induced binding between MYB and the miR-155
promoter (Fig. 3D). Taken
together, our results suggest that both YY1 and MYB mediate miR-155
expression via directly binding to the cis-regulatory sequence of
miR-155.
YY1 and MYB cooperatively induce miR-155
expression through JAK1/2 kinase activation
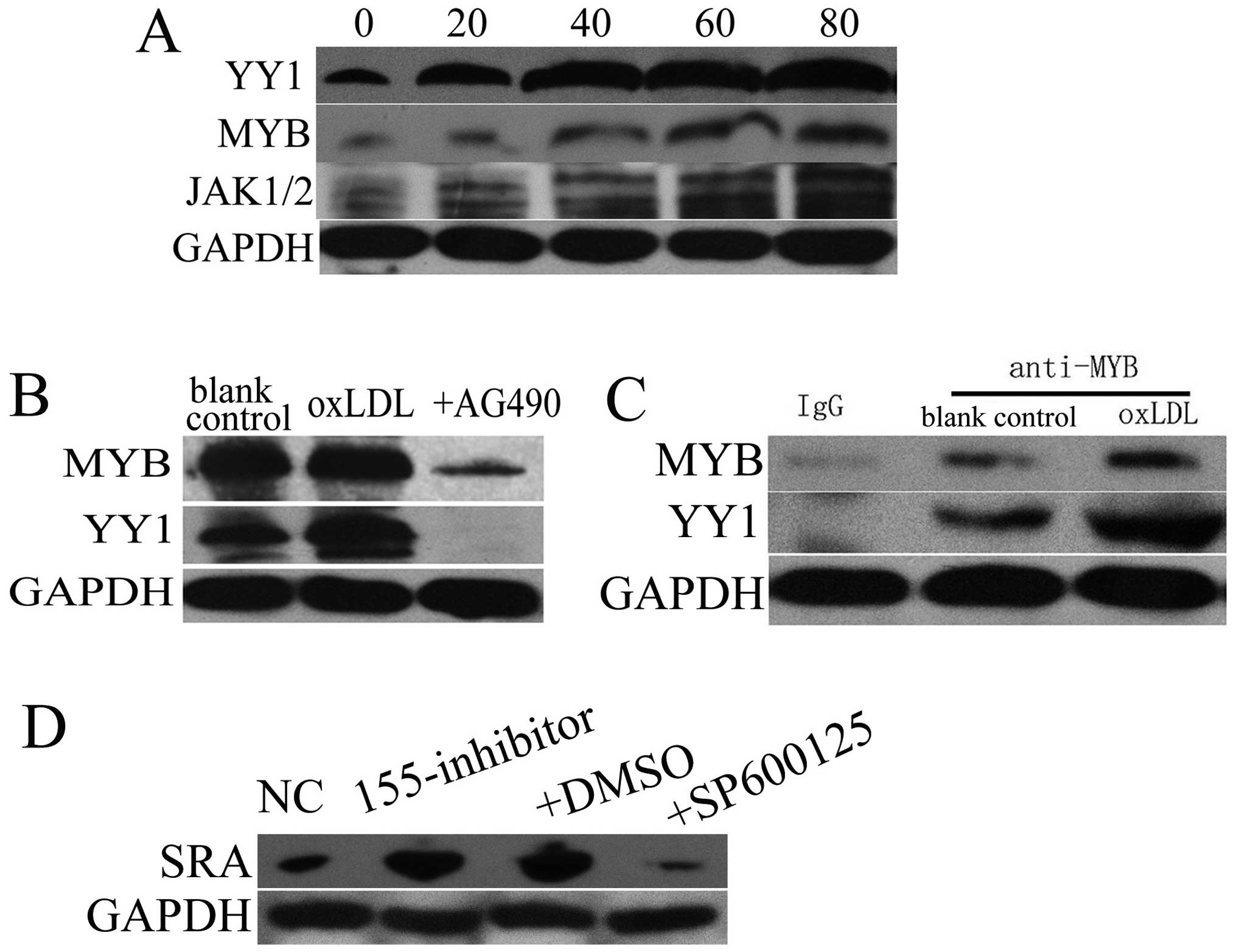
To further examine whether YY1 and MYB were also
activated by JAK1/2 or other kinases, we firstly tested whether the
expression of YY1, MYB and JAK1/2 was altered by oxLDL treatment.
Western blot analysis showed that YY1, MYB and JAK1/2 were
increased following treatment with oxLDL, in a dose-dependent
fashion (Fig. 4A). Inhibition of
JAK1/2 using AG490 abolished oxLDL-induced promotion of YY1 and MYB
expression (Fig. 4B). Moreover,
we observed the interaction between YY1 and MYB, which was enhanced
by oxLDL treatment (Fig. 4C).
Thus, we conclude that YY1 and MYB cooperatively induce miR-155
expression through JAK1/2 kinase.
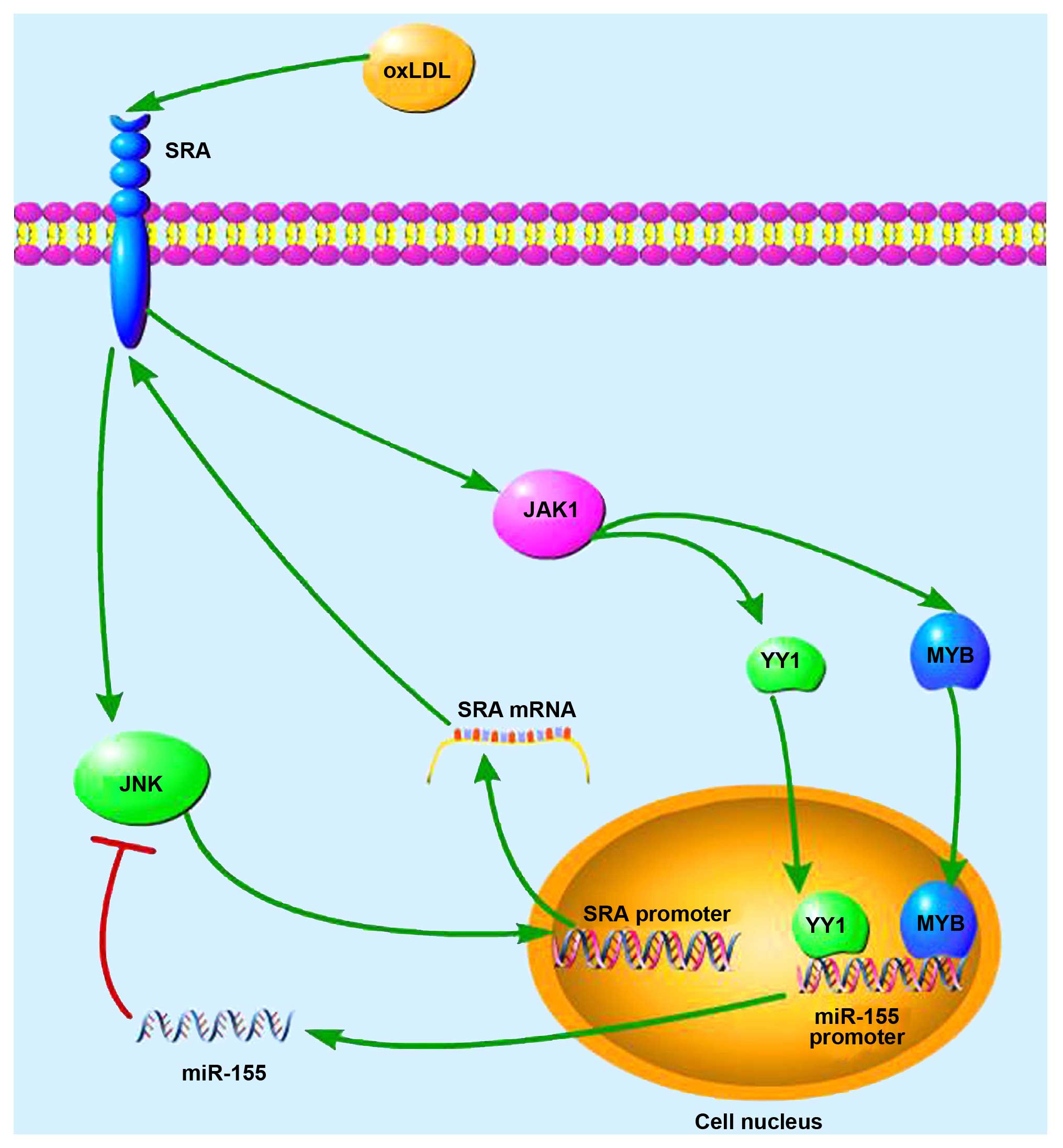
Negative feedback and the induction of
miR-155 by oxLDL
We observed that oxLDL promoted miR-155 expression
partially through binding to SRA. Our previous research had shown
that miR-155 inhibited SRA expression and the JNK pathway (24), and JNK signaling has been
demonstrated to control SRA expression in macrophages. Thus, we
questioned whether miR-155 inhibited SRA expression through
suppressing the JNK pathway. Western blot analysis showed that
miR-155 inhibition upregulated SRA whereas SP600125, a specific JNK
inhibitor, abrogated the promoting effect caused by miR-155
inhibitor on SRA expression (Fig.
4D). Thus, a negative feedback loop exists between
miR-155-SRA-JNK-miR-155 in response to oxLDL, as is illustrated in
Fig. 5.
Discussion
Previous research has demonstrated that miR-155 is
involved in numerous biological processes, including haematopoietic
lineage commitment and tumor formation (13,27,28). BIC/miR-155 expression is greatly
increased in activated B and T cells and DCs (29). Previous research has revealed that
miR-155-deficient mice exhibited impaired antigen-presenting
capacities (27). Furthermore,
increased expression of BIC/miR-155 was observed in patients with
Hodgkin's lymphoma (30) and
miR-155 was found to be overexpressed in the bone marrow of
patients with certain subtypes of acute myeloid leukemia (AML)
(13), suggesting that miR-155 is
linked to human diseases. Indeed, miRNA-155 has been shown to play
specific roles in regulating various diseases and may thus be a
good candidate for predicting clinical prognosis and generating
potential therapeutic treatments for a number of diseases,
including renal cell carcinoma (15), and breast (16) and colon cancer (28). Moreover, miR-155 appears to be
involved in biochemical mechanisms associated with viral
infections. For example, miR-155 contributes to Epstein-Barr virus
(EBV) immortalization by regulating NF-κB signaling and suppressing
the host innate immunity to latent viral infection (14). Therefore, miR-155 exhibits a wide
spectrum of physiological and pathological function.
Interestingly, miR-155 has also been found to be
involved in cardiovascular disease: overexpression of this
miRNA-155 was accompanied by impaired angiotensin II type 1
receptor (AT1R) activity and low blood pressure (31). Our previous research has shown
that miR-155 contributed to the prevention of AS development and
progression (19). Although
previous research has demonstrated the role of miR-155 in multiple
diseases, relatively little is known about how its expression is
mediated in a disease setting, particularly in an
immuno-inflammatory context.
Several studies have shown that the increase in
miR-155 expression is related to the ERK and JNK signaling pathways
(32). For example, B-cell
receptor activation induced BIC/miR-155 expression via a conserved
AP-1 element (32). miR-155
expression was also induced in response to LPS, resulting in a
decrease in Src homology domain 2 (SH2)-containing inositol
phosphatase (SHIP1) expression and allowing PI3K activation of
NF-κB and MAPK to proceed and promote the pro-inflammatory response
(33). However, in the presence
of interleukin (IL)-10, miR-155 expression was inhibited through
the STAT3 signaling pathway, which led to the recovery of SHIP1
expression and the conversion of phosphatidylinositol (3,4,5)-trisphosphate (PIP3) back to its
inactive phosphatidylinositol (4,5)-bisphosphate (PIP2) state (25). As a result, pro-inflammatory
responses were switched off (25). Indeed, previous data from our
laboratory showed that miR-155 contributed to the prevention of
inflammation and its progression by regulating the MAPK pathway via
specifically targeting mitogen-activated protein kinase kinase
kinase 10 (19).
In the present study, neither MAPK, NF-κB nor STAT3
pathways were found to be involved in mediating the expression of
miR-155, which was upregulated in response to oxLDL. On the
contrary, we demonstrated that oxLDL promoted miR-155 expression
through the SRA receptor and JAK2 signaling pathway, as the
inhibition of SRA or JAK1/2 signaling significantly decreased the
miR-155 levels which had risen in response to oxLDL. Moreover,
miR-155 negatively regulated SRA expression by suppressing the JNK
pathway. Furthermore, we identified response elements present in
the miR-155 promoter region that mediated the activity of the
cis-regulatory sequence of miR-155. These response elements were
recognized by the transcription factors YY1 and MYB, both of which
exhibited physical interaction which was potentiated by oxLDL
stimulation. Interestingly, YY1 and MYB participated in mediating
miR-155 transcription by binding to the miR-155 promoter through
JAK1/2 kinase. Taken together, the results of our present study
demonstrated the molecular mechanisms by which miR-155 was
regulated upon oxLDL treatment. Given that oxLDL is an important
promoter of the development of AS, and DCs are also critically
involved in the inflammatory reaction that occurs during AS
initiation and progression, our findings provide novel insights
into how miR-155 expression and activity is mediated in DCs during
the course of AS, in which YY1 and MYB may also play an important
regulatory role. Consistent with these findings, YY1 was previously
shown to be induced by vascular cell injury (34), whereas MYB was also involved in AS
(35). However, we do not rule
out the possibility that other TFs also play a role in mediating
miR-155 transcription. It will also be interesting to further
explore more roles of miR-155 in DC functions, and clarify the
exact role of miR-155 in the development of AS.
In conclusion, our study revealed that oxLDL
promotes miR-155 expression partially through the SRA receptor and
the JAK2 signaling pathway in human peripheral blood-derived DCs.
oxLDL induces the expression of TFs MYB and YY1, which subsequently
bind to the regulatory sequence of miRNA-155 to activate its
transcription. Furthermore, in the present study we uncovered a
negative feedback loop miR-155-SRA-JNK-miR-155 involved in the
activation of miR-155 by oxLDL. Therefore, our findings provide
novel insights into the mechanisms underlying the increased
expression of miR-155 by oxLDL in DCs.
Acknowledgments
The present study was funded by the Natural Science
Foundation of China (grant no. 81200214/H0215).
References
|
1
|
Hansson GK and Hermansson A: The immune
system in atherosclerosis. Nat Immunol. 12:204–212. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Libby P, Ridker PM and Hansson GK:
Progress and challenges in translating the biology of
atherosclerosis. Nat Immunol. 473:317–325. 2011.
|
|
3
|
Niessner A and Weyand CM: Dendritic cells
in atherosclerotic disease. Clin Immunol. 134:25–32. 2010.
View Article : Google Scholar :
|
|
4
|
Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee
JK, Sohn SY, Cho Y, Zhang BT and Kim VN: Molecular basis for the
recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell.
125:887–901. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sayed D, Hong C, Chen IY, Lypowy J and
Abdellatif M: MicroRNAs play an essential role in the development
of cardiac hypertrophy. Circ Res. 100:416–424. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tatsuguchi M, Seok HY, Callis TE, Thomson
JM, Chen JF, Newman M, Rojas M, Hammond SM and Wang DZ: Expression
of microRNAs is dynamically regulated during cardiomyocyte
hypertrophy. J Mol Cell Cardiol. 42:1137–1141. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thum T, Galuppo P, Wolf C, Fiedler J,
Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J,
Haverich A, et al: MicroRNAs in the human heart: a clue to fetal
gene reprogramming in heart failure. Circulation. 116:258–267.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
van Rooij E and Olson EN: microRNAs put
their signatures on the heart. Physiol Genomics. 31:365–366. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
van Rooij E and Olson EN: MicroRNAs:
powerful new regulators of heart disease and provocative
therapeutic targets. J Clin Invest. 117:2369–2376. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
van Rooij E, Sutherland LB, Liu N,
Williams AH, McAnally J, Gerard RD, Richardson JA and Olson EN: A
signature pattern of stress-responsive microRNAs that can evoke
cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA.
103:18255–18260. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Busch M and Zernecke A: microRNAs in the
regulation of dendritic cell functions in inflammation and
atherosclerosis. J Mol Med (Berl). 90:877–885. 2012. View Article : Google Scholar
|
|
12
|
Lagos-Quintana M, Rauhut R, Yalcin A,
Meyer J, Lendeckel W and Tuschl T: Identification of
tissue-specific microRNAs from mouse. Curr Biol. 12:735–739. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
O'Connell RM, Rao DS, Chaudhuri AA, Boldin
MP, Taganov KD, Nicoll J, Paquette RL and Baltimore D: Sustained
expression of microRNA-155 in hematopoietic stem cells causes a
myeloproliferative disorder. J Exp Med. 205:585–594. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lu F, Weidmer A, Liu CG, Volinia S, Croce
CM and Lieberman PM: Epstein-Barr virus-induced miR-155 attenuates
NF-kappaB signaling and stabilizes latent virus persistence. J
Virol. 82:10436–10443. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shinmei S, Sakamoto N, Goto K, Sentani K,
Anami K, Hayashi T, Teishima J, Matsubara A, Oue N, Kitadai Y and
Yasui W: MicroRNA-155 is a predictive marker for survival in
patients with clear cell renal cell carcinoma. Int J Urol.
20:468–477. 2013. View Article : Google Scholar
|
|
16
|
Sun Y, Wang M, Lin G, Sun S, Li X, Qi J
and Li J: Serum microRNA-155 as a potential biomarker to track
disease in breast cancer. PLoS One. 7:e470032012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nazari-Jahantigh M, Wei Y, Noels H, Akhtar
S, Zhou Z, Koenen RR, Heyll K, Gremse F, Kiessling F, Grommes J, et
al: MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in
macrophages. J Clin Invest. 122:4190–4202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wei Y, Zhu M, Corbalán-Campos J, Heyll K,
Weber C and Schober A: Regulation of Csf1r and Bcl6 in macrophages
mediates the stage-specific effects of microRNA-155 on
atherosclerosis. Arterioscler Thromb Vasc Biol. 35:796–803. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhu J, Chen T, Yang L, Li Z, Wong MM,
Zheng X, Pan X, Zhang L and Yan H: Regulation of microRNA-155 in
atherosclerotic inflammatory responses by targeting MAP3K10. PLoS
One. 7:e465512012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Donners MM, Wolfs IM, Stöger LJ, van der
Vorst EP, Pöttgens CC, Heymans S, Schroen B, Gijbels MJ and de
Winther MP: Hematopoietic miR155 deficiency enhances
atherosclerosis and decreases plaque stability in hyperlipidemic
mice. PLoS One. 7:e358772012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tian FJ, An LN, Wang GK, Zhu JQ, Li Q,
Zhang YY, Zeng A, Zou J, Zhu RF, Han XS, et al: Elevated
microRNA-155 promotes foam cell formation by targeting HBP1 in
atherogenesis. Cardiovasc Res. 103:100–110. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu GF, Yang LX, Guo RW, Liu H, Shi YK,
Wang H, Ye JS, Yang ZH and Liang X: miR-155 inhibits oxidized
low-density lipoprotein-induced apoptosis of RAW264.7 cells. Mol
Cell Biochem. 382:253–261. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang YH, Xia LH, Jin JM, Zong M, Chen M
and Zhang B: Expression level of miR-155 in peripheral blood. Asian
Pac J Trop Med. 8:214–219. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen T, Yan H, Li Z, Jing T, Zhu W, Ge J,
Zheng X, Pan X, Yan H and Zhu J: MicroRNA-155 regulates lipid
uptake, adhesion/chemokine marker secretion and SCG2 expression in
oxLDL-stimulated dendritic cells/macrophages. Int J Cardiol.
147:446–447. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McCoy CE, Sheedy FJ, Qualls JE, Doyle SL,
Quinn SR, Murray PJ and O'Neill LA: IL-10 inhibits miR-155
induction by toll-like receptors. J Biol Chem. 285:20492–20498.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li P, Grgurevic S, Liu Z, Harris D,
Rozovski U, Calin GA, Keating MJ and Estrov Z: Signal transducer
and activator of transcription-3 induces microRNA-155 expression in
chronic lymphocytic leukemia. PLoS One. 8:e646782013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rodriguez A, Vigorito E, Clare S, Warren
MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska
EA, et al: Requirement of bic/microRNA-155 for normal immune
function. Science. 316:608–611. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Volinia S, Calin GA, Liu CG, Ambs S,
Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et
al: A microRNA expression signature of human solid tumors defines
cancer gene targets. Proc Natl Acad Sci USA. 103:2257–2261. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Faraoni I, Antonetti FR, Cardone J and
Bonmassar E: miR-155 gene: a typical multifunctional microRNA.
Biochim Biophys Acta. 1792:497–505. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
van den Berg A, Kroesen BJ, Kooistra K, de
Jong D, Briggs J, Blokzijl T, Jacobs S, Kluiver J, Diepstra A,
Maggio E and Poppema S: High expression of B-cell receptor
inducible gene BIC in all subtypes of Hodgkin lymphoma. Genes
Chromosomes Cancer. 37:20–28. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Martin MM, Buckenberger JA, Jiang J,
Malana GE, Nuovo GJ, Chotani M, Feldman DS, Schmittgen TD and Elton
TS: The human angiotensin II type 1 receptor +1166 A/C polymorphism
attenuates microRNA-155 binding. J Biol Chem. 282:24262–24269.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yin Q, Wang X, McBride J, Fewell C and
Flemington E: B-cell receptor activation induces BIC/miR-155
expression through a conserved AP-1 element. J Biol Chem.
283:2654–2662. 2008. View Article : Google Scholar
|
|
33
|
Lind EF, Millar DG, Dissanayake D, Savage
JC, Grimshaw NK, Kerr WG and Ohashi PS: miR-155 upregulation in
dendritic cells is sufficient to break tolerance in vivo by
negatively regulating SHIP1. J Immunol. 195:4632–4640. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Santiago FS, Lowe HC, Bobryshev YV and
Khachigian LM: Induction of the transcriptional repressor Yin
Yang-1 by vascular cell injury. Autocrine/paracrine role of
endogenous fibroblast growth factor-2. J Biol Chem.
276:41143–41149. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Martin-McNulty B, Tham DM, da Cunha V, Ho
JJ, Wilson DW, Rutledge JC, Deng GG, Vergona R, Sullivan ME and
Wang YX: 17 Beta-estradiol attenuates development of angiotensin
II-induced aortic abdominal aneurysm in apolipoprotein E-deficient
mice. Arterioscler Thromb Vasc Biol. 23:1627–1632. 2003. View Article : Google Scholar : PubMed/NCBI
|