Introduction
Doxorubicin (DOX) is one of the most widely used
anticancer drugs because of its potent therapeutic effects on
various types of cancer, including leukemia, lymphoma and breast
cancer. However, the clinical use of DOX is limited by severe
cardiotoxicity, which may lead to dilated cardiomyopathy and
congestive heart failure (1). The
production of reactive oxygen species (ROS) is involved in the
toxic effect elicited by DOX on cardiomyocytes and endothelial
cells, and in the promotion of endothelial dysfunction (2) and apoptosis (3). A number of pharmacological
interventions have been suggested as therapies to protect against
DOX-induced cardiotoxicity.
Hydrogen sulfide (H2S) is considered a
toxic gas, and has been classified as the third gasotransmitter,
together with nitric oxide and carbon monoxide, and exerts various
effects on the cardiovascular system (4). Previous findings have shown that
H2S protects the heart from myocardial
ischemia-reperfusion (IR) injury (5). In a previous study, it was
demonstrated that the increased generation of endogenous
H2S in the early reperfusion phase has an important
function in ischemic pre-conditioning-elicited protection in
isolated hearts (6).
The forkhead box class O (FoxO) subfamily of
forkhead transcription factors comprises the members FoxO1, FoxO3a
and FoxO4, which are downstream targets of Akt (7). FoxO3a is involved in the regulation
of various cell processes, including proliferation and apoptosis,
as well as protection against oxidative stress and metabolism
(8). A model of β-amyloid-induced
neuron death was used to demonstrate that FoxO3a is activated,
translocates to the nucleus, and subsequently mediates neuron death
through Bim (9). In neonatal rat
ventricular myocytes (NRVMs), hyperglycemia was demonstrated to
markedly enhance the apoptosis of NRVMs through the trans-location
of FoxO3a to the nucleus, and the resultant enhanced
transcriptional activity of FoxO3a (10). Li et al found that the
PI3K/Akt/FoxO3a pathway is involved in neuronal apoptosis in the
brain of a developing rat (11).
In a previous study, it was demonstrated that resveratrol protects
PC12 cells against high glucose-induced oxidative stress and
apoptosis through the activation of the PI3K/Akt/FoxO3a signaling
pathway (12). In addition,
sodium tanshinone IIA sulfonate (13) and bromelain (14) have been found to protect rat
hearts against IR injury through the activation of the
PI3K/Akt/FoxO3a pathway. Therefore, we hypothesized that the
PI3K/Akt/FoxO3a pathway may be involved in the protective effect of
exogenous H2S against DOX-induced cardiotoxicity in H9c2
cardiac cells.
To examine this hypothesis, H9c2 cells were treated
with 5 µM DOX in the present study to establish a model of
chemotherapy-induced cardiotoxicity as previously described
(15). Subsequently, we examined:
i) the effect of DOX on the phosphorylation of Akt and FoxO3a; ii)
the effect of exogenous H2S on the DOX-induced
translocation of FoxO3a to the nucleus where it subsequently
mediates H9c2 cardiac cell death through Bim; and iii) whether
exogenous H2S protects H9c2 cells against DOX-induced
cardiotoxicity through the PI3K/Akt/FoxO3a pathway.
Materials and methods
Materials
Methyl thiazolyl tetrazolium (MTT), Hoechst 33258,
2′,7′-dichlorofluorescein diacetate (DCFH-DA), DOX, sodium
hydrosulfide (NaHS), and N-acetyl-L-cysteine (NAC) were purchased
from Sigma-Aldrich (St. Louis, MO, USA). LY294002 was purchased
from Calbiochem (Billerica, MA, USA). Cell culture medium
components were purchased from Thermo Fisher Scientific (Waltham,
MA, USA) unless otherwise noted. The H9c2 cardiac myocytes were
obtained from the Shanghai Cell Library of China [originally from
the American Type Culture Collection (ATCC); Manassas, VA,
USA].
Cell culture
The H9c2 cardiac cells were cultured in Dulbecco's
modified Eagle's medium (DMEM) supplemented with 10% fetal bovine
serum (FBS), 100 µg/ml streptomycin (Life Technologies,
Carlsbad, CA, USA) and 100 U/ml penicillin-streptomycin (Life
Technologies) in a humidified 5% CO2 atmosphere at 37°C.
The H9c2 cardiac myocytes were passaged every 2 days. Subsequently,
they were seeded at a density of 2×106 cells/dish in
100-mm dishes with 10% calf serum, incubated for 24 h and then, the
medium was replaced with 0.5% FBS DMEM for 24-h serum starvation.
To determine the degree of apoptosis, the H9c2 cardiac myocytes
were treated with NaHS (100 µM) for 30 min or NAC (1,000
µM) for 60 min, followed by exposure to DOX for 24 h. In
some experiments, the H9c2 cells were treated with LY294002 (50
µM) prior to NaHS stimulation.
MTT assay
The MTT assay is a standard method used to assess
cell viability. Prior to each experiment, the H9c2 cardiac myocytes
(5×102 cells/well) were seeded in 96-well microtiter
plates. After incubation with the phosphatidylinositol-3-kinase
(PI3K) inhibitor LY294002 (50 µM) and/or NaHS for 30 min,
the cells were exposed to 5 µM DOX for a further 24 h.
Subsequently, 10 µl MTT solution was added to each well, and
the plates were incubated for 4 h at 37°C. The absorbance was
measured at 470 nm using a SpectraMax 190 Absorbance Microplate
Reader (Molecular Devices LLC, Sunnyvale, CA, USA) and used to
calculate the relative ratio of cell viability. Three independent
experiments were performed for each experimental condition.
Assessment of H9c2 cell apoptosis
The analysis of apoptosis was performed by
fluorescence microscopy with the chromatin dye Hoechst 33258. After
various treatments, the cells were fixed in ice-cold 4%
paraformaldehyde dissolved in phosphate-buffered saline (PBS) at
room temperature for 20 min. Non-specific binding was blocked using
5% normal goat serum in 0.01 M PBS containing 0.3% Triton X-100
(PBS+T). The cells were washed twice with PBS and incubated with 10
µg/ml Hoechst 33258 for 15 min at room temperature in the
dark. The cells were visualized under a fluorescence microscope
(BX50-FLA; Olympus, Tokyo, Japan). Apoptotic cells exhibited
condensed, fractured or distorted nuclei, whereas viable cells
exhibited normal nuclear size and uniform fluorescence.
Measurement of intracellular ROS
levels
The determination of intracellular ROS levels was
performed by measuring the level of a fluorescent product formed by
the oxidation of DCFH-DA. Briefly, the culture medium was plated
into 96-well microtiter plates. After various treatments, the cells
were washed with PBS 3 times. Following the addition of fresh
culture medium, the cells were incubated with DCFH-DA at a final
concentration of 10 µmol/l for 30 min at 37°C. The cells
were washed again with PBS 3 times and then lysed with 500
µl 90% DMSO and 10% PBS for 10 min at room temperature in
the dark. The supernatant (200 µl) was transferred to
another 96-well microtiter plate. The fluorescence intensity of the
oxidized product, 2′,7′-dichlorofluorescein (DCF), was measured
using a Model F-4500 fluorescence spectrophotometer (Hitachi,
Tokyo, Japan) at 495 nm (excitation) and 520 nm (emission). The
values were expressed as the percentage of fluorescence intensity
relative to the control wells.
Western blot analysis
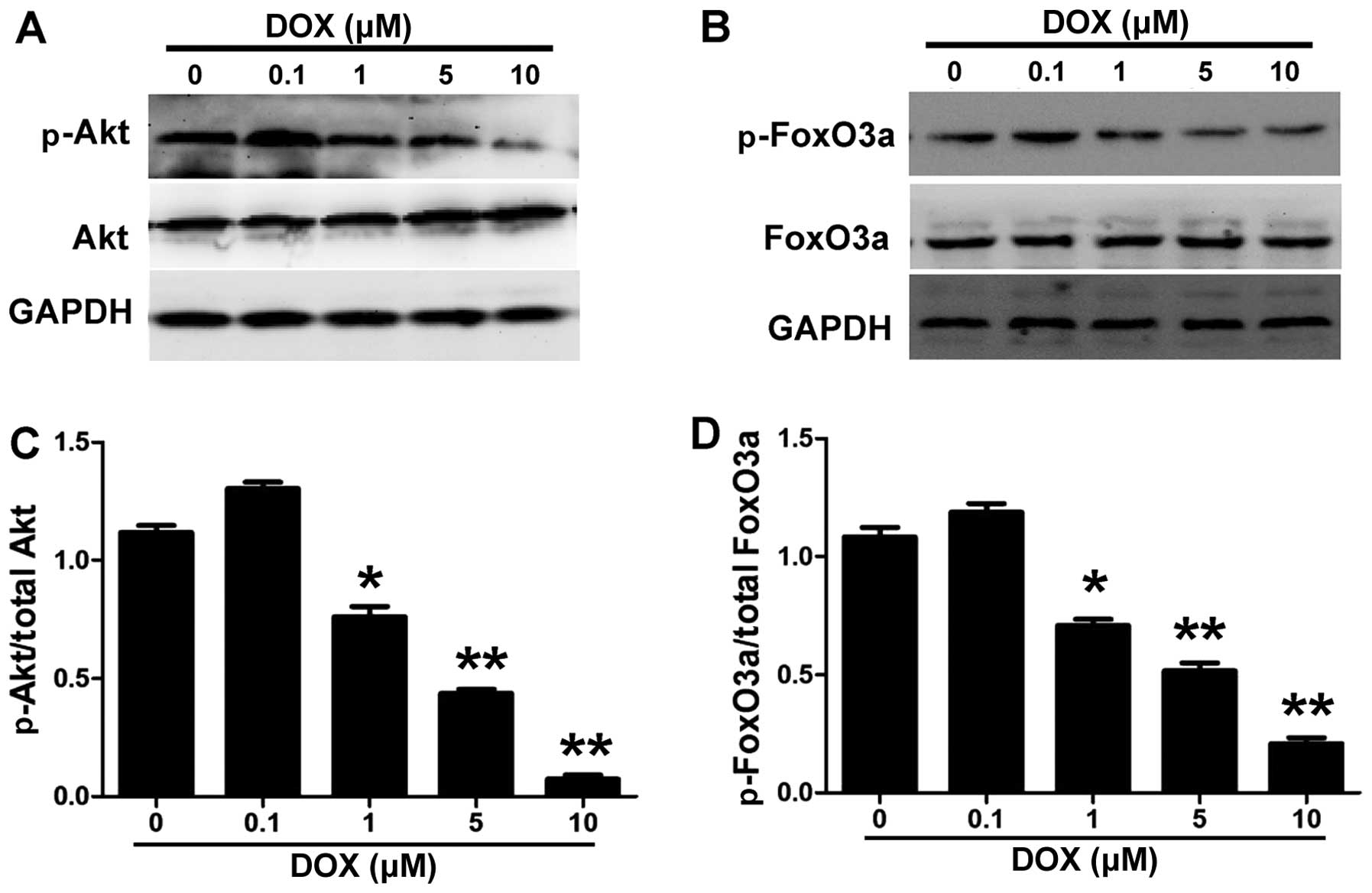
H9c2 cells were treated with 0.1, 1, 5 and 10
µM DOX for 24 h (Fig. 1)
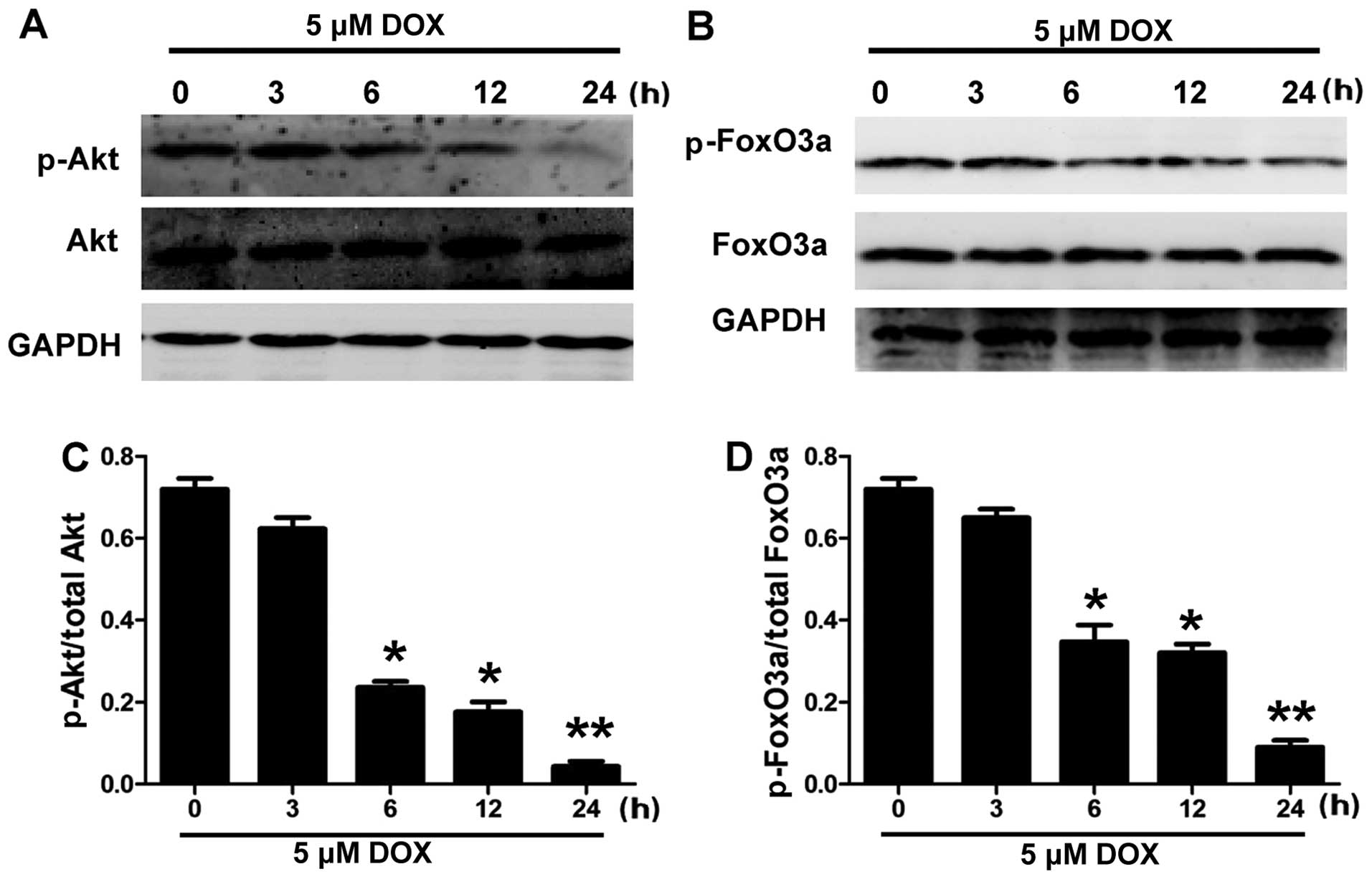
and exposed to 5 µM DOX for 3, 6, 12 and 24 h (Fig. 2). The cells were homogenized
directly in cell lysis buffer (Cell Signaling Technology, Danvers,
MA, USA) and phosphatase inhibitor cocktail (Sigma-Aldrich). The
lysates were centrifuged at 12000 × g for 10 min at 4°C. The
protein concentration was determined with the use of a BCA protein
assay kit according to the manufacturer's instructions. For
nuclear/cytoplasmic fractionation, the cultured H9c2 cells were
fractionated into nuclear and cytoplasmic lysates using a Nuclear
and Cytoplasmic Protein Extraction kit (Beyotime Institute of
Biotechnology, Shanghai, China) according to the manufacturer's
instructions. The extracted proteins were mixed with 5% sodium
dodecyl sulfate (SDS)-PAGE sample buffer, boiled at 100°C for 7 min
and then separated by electrophoresis on a 10% SDS-polyacrylamide
gel. Following electrophoresis, the proteins were transferred to
polyvinylidene difluoride membranes (Beyotime Institute of
Biotechnology). The membranes were blocked in Tris-buffered saline
(TBS) containing 0.1% Tween-20 (TBS-T) with 5% non-fat dry milk,
for 2 h at room temperature, with rotation. After blocking, the
membranes were incubated with the following antibodies: rabbit
anti-Akt polyclonal antibody (cat. no. 9272, 1:2,000), rabbit
anti-phosphorylated (p-) Akt (Ser 473) monoclonal antibody (cat.
no. 4060, 1:2,000), rabbit anti-FoxO3a polyclonal antibody (cat.
no. 12829, 1:2,000), rabbit anti-p-FoxO3a (ser 253) polyclonal
antibody (cat. no. 9466, 1:1,000) (all from Cell Signaling
Technology) and rabbit anti-Bim poly-clonal antibody (ab32158,
1:200; Abcam, Cambridge, MA, USA). The membranes were then
incubated in 5% milk or bovine serum albumin overnight at 4°C. The
primary antibody was removed by washing the membranes 3 times in
TBS-T, and subsequently incubating the membranes for 2 h with the
appropriate horseradish peroxidase-conjugated secondary antibodies.
After washing the membranes 3 times in TBS-T, the antigen-antibody
bands were detected using an enhanced chemiluminescence reagent kit
and quantified using a densitometry program. The data from the
western blot analysis of p-Akt and p-FoxO3a were presented as a
ratio of the p-forms to their total forms, respectively. The
immunoblot of Bim was corrected to the bands of GAPDH.
Statistical analysis
The results are presented as the means ± SEM.
Statistical analysis was performed using the Student's t-test or
analysis of variance (ANOVA) with SPSS 13.0 software (SPSS, Inc.,
Chicago, IL, USA). In all cases, P<0.05 was considered to
indicate a statistically significant difference.
Results
DOX decreases the phosphorylation of Akt
and FoxO3a in H9c2 cells
To investigate the role of the PI3K/Akt/FoxO3a
pathway in DOX-induced cytotoxicity, we investigated the
phosphorylation of Akt and FoxO3a in the H9c2 cells following
exposure to DOX. The H9c2 cells were treated with DOX at different
concentrations for different time periods, and the effect on the
phosphorylation of Akt and FoxO3a was determined using western blot
analysis. Figs. 1 and 2 show that DOX decreased the
phosphorylation of Akt and FoxO3a in the H9c2 cells in a
concentration- and time-dependent manner. DOX inhibited the
phosphorylation of Akt and FoxO3a in the H9c2 cells at a
concentration of 1 µM and the maximal effect was reached at
a concentration of 10 µM (Fig.
1). Fig. 2 shows that 5
µM DOX induced a significant decrease in the levels of p-Akt
and p-FoxO3a at 6 and 12 h, and it almost completely abolished the
phosphorylation of Akt and FoxO3a at 24 h in the H9c2 cells.
Exogenous H2S ameliorates the
DOX-induced decrease in the levels of p-Akt and p-FoxO3a in H9c2
cells
To determine whether the cytoprotective effect of
H2S against DOX-induced toxicity was associated with the
PI3K/Akt/FoxO3a pathway in H9c2 cells, we examined the effect of
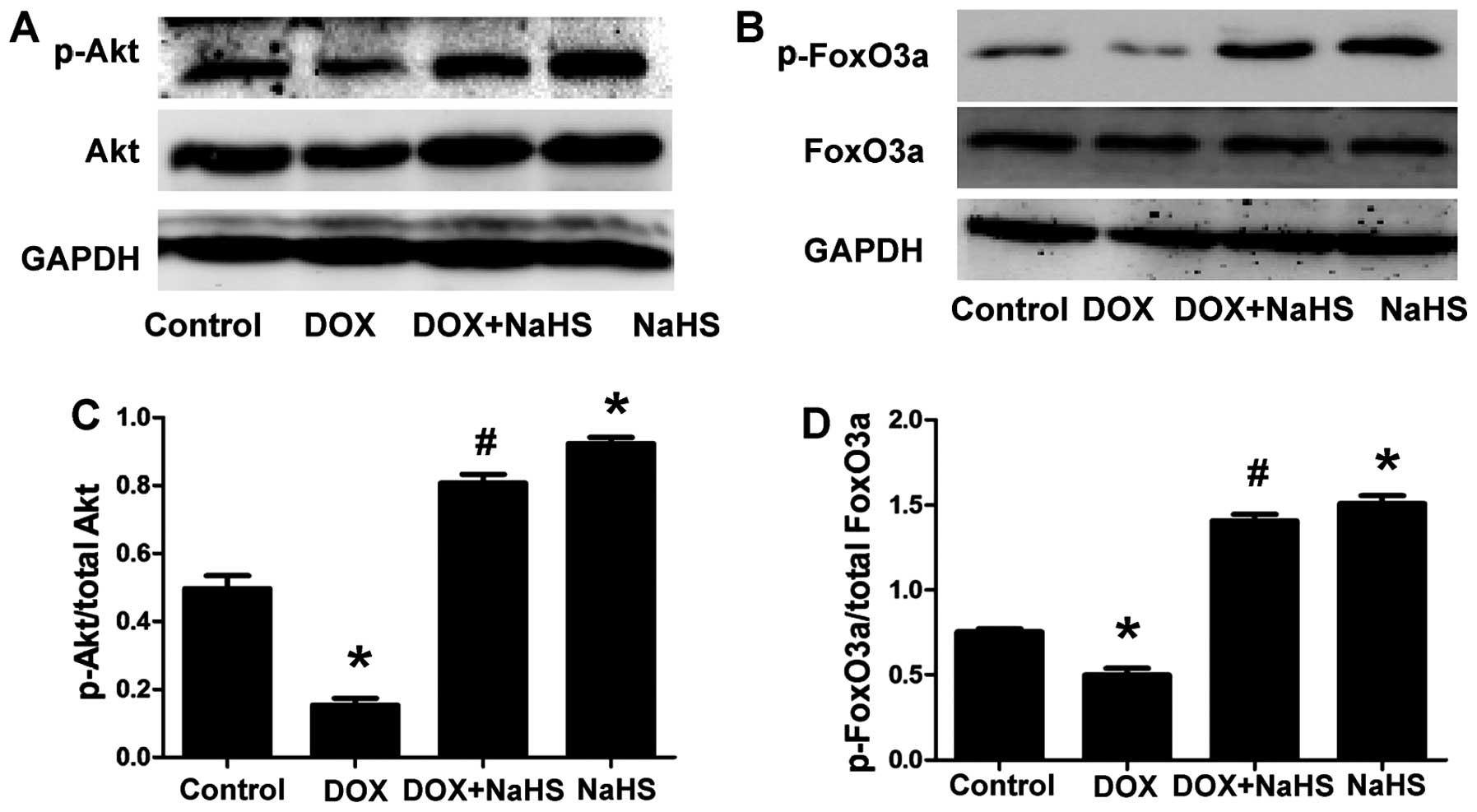
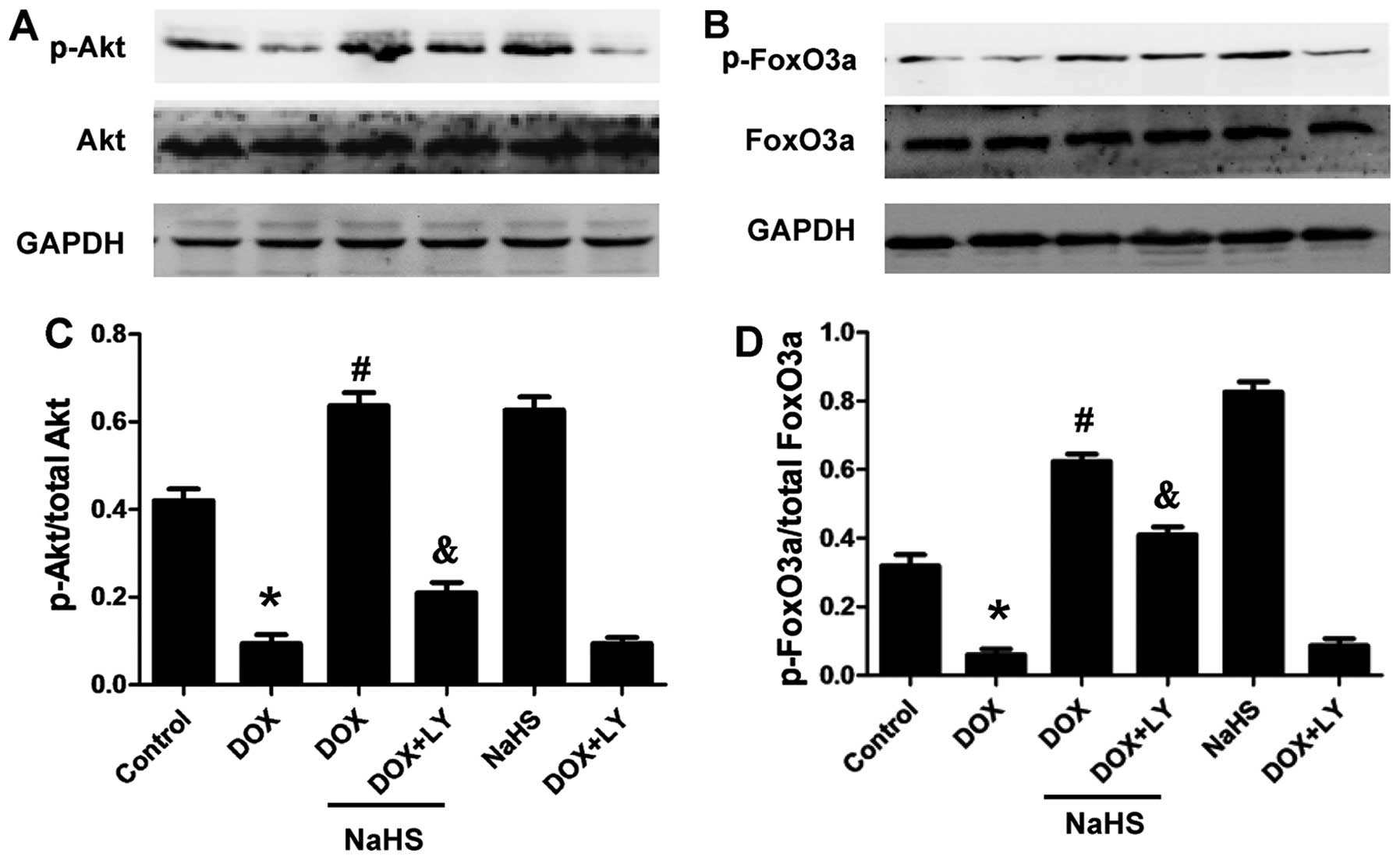
NaHS on the expression of p-Akt and p-FoxO3a. The results showed
that treating the H9c2 cells with 100 µM NaHS (a donor of
H2S) for 30 min prior to exposure to 5 µM DOX for
24 h significantly increased the phosphorylation of Akt and FoxO3a
(Fig. 3). Furthermore, NaHS
treatment alone also significantly increased the levels of p-Akt
and p-FoxO3a compared with the DOX-treated groups. The total Akt
and FoxO3a levels remained unchanged among the four groups. These
results suggested that the PI3K/Akt/FoxO3a pathway was involved in
the protective effect of H2S.
NAC ameliorates the DOX-induced decrease
in the levels of p-Akt and p-FoxO3a in H9c2 cells
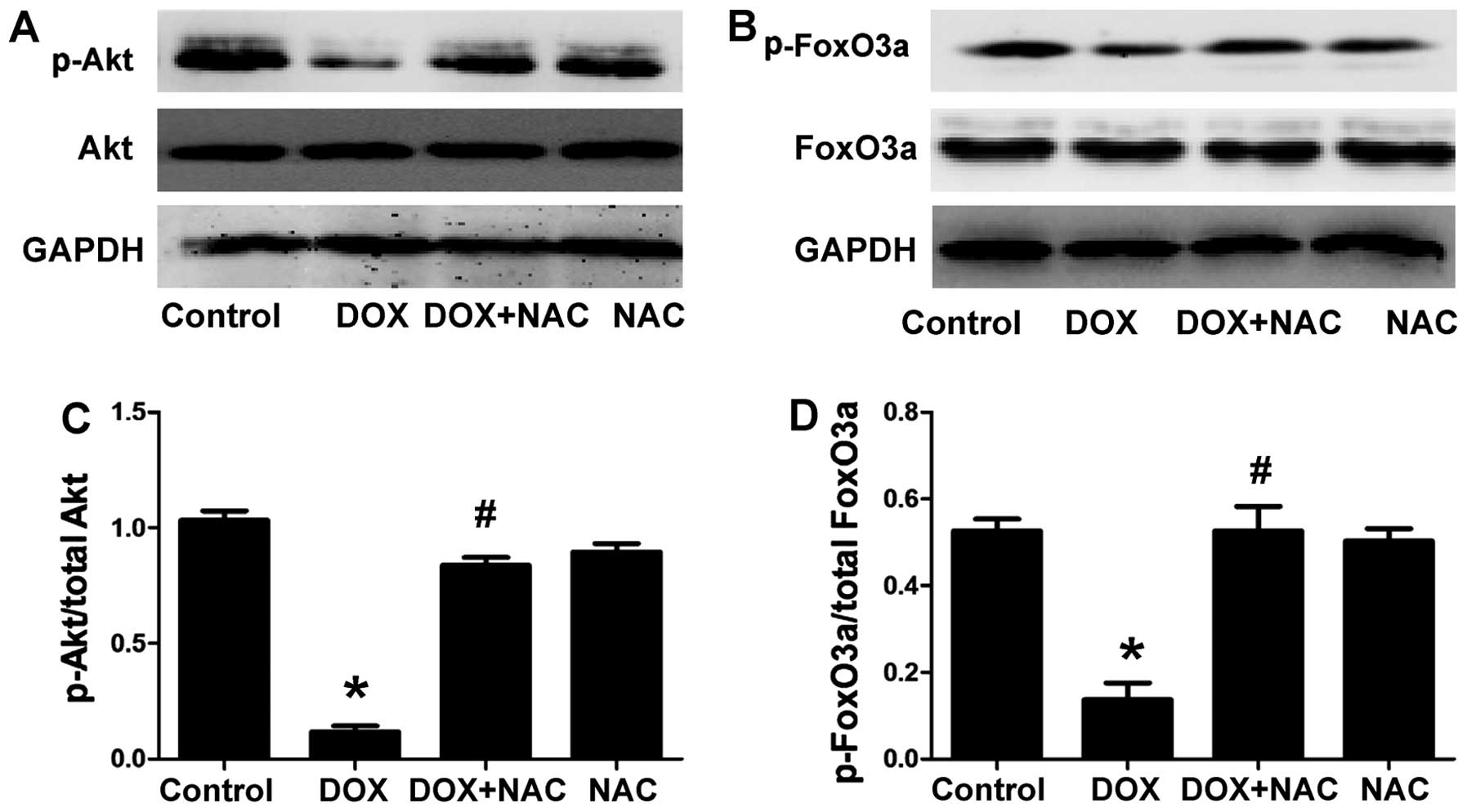
The H9c2 cells were treated with 1,000 µM NAC
(ROS scavenger) for 60 min prior to exposure to 5 µM DOX for
24 h, to confirm whether the protective effect of H2S on
the DOX-induced decrease in p-Akt and p-FoxO3a is associated with
antioxidation. As shown in Fig.
4, the pretreatment of the H9c2 cells with NAC markedly
increased the expression of p-Akt and p-FoxO3a and this was similar
to the protective effect observed with NaHS pre-treatment. The
total Akt and FoxO3a levels remained unchanged in the four groups.
The results revealed that an antioxidant effect contributed to the
protective effect of H2S against DOX-induced
injuries.
Exogenous H2S induces Akt and
FoxO3a phosphorylation through the PI3K/Akt pathway in H9c2
cells
To investigate the role of the PI3K/Akt pathway in
the protective effects of H2S, the H9c2 cells were
treated with the PI3K inhibitor LY294002 prior to exposure to NaHS
plus DOX. The activation of Akt and FoxO3a was determined as
described above. LY294002 abolished the stimulation of p-Akt and
p-FoxO3a in the presence of NaHS (Fig. 5), but elicited no effect on the
expression levels of total Akt and FoxO3a. These results suggested
that the PI3K/Akt/FoxO3a pathway was involved in the protective
effect of H2S.
DOX enhances the nuclear localization of
FoxO3a in H9c2 cells whereas H2S blocks the effect of
DOX
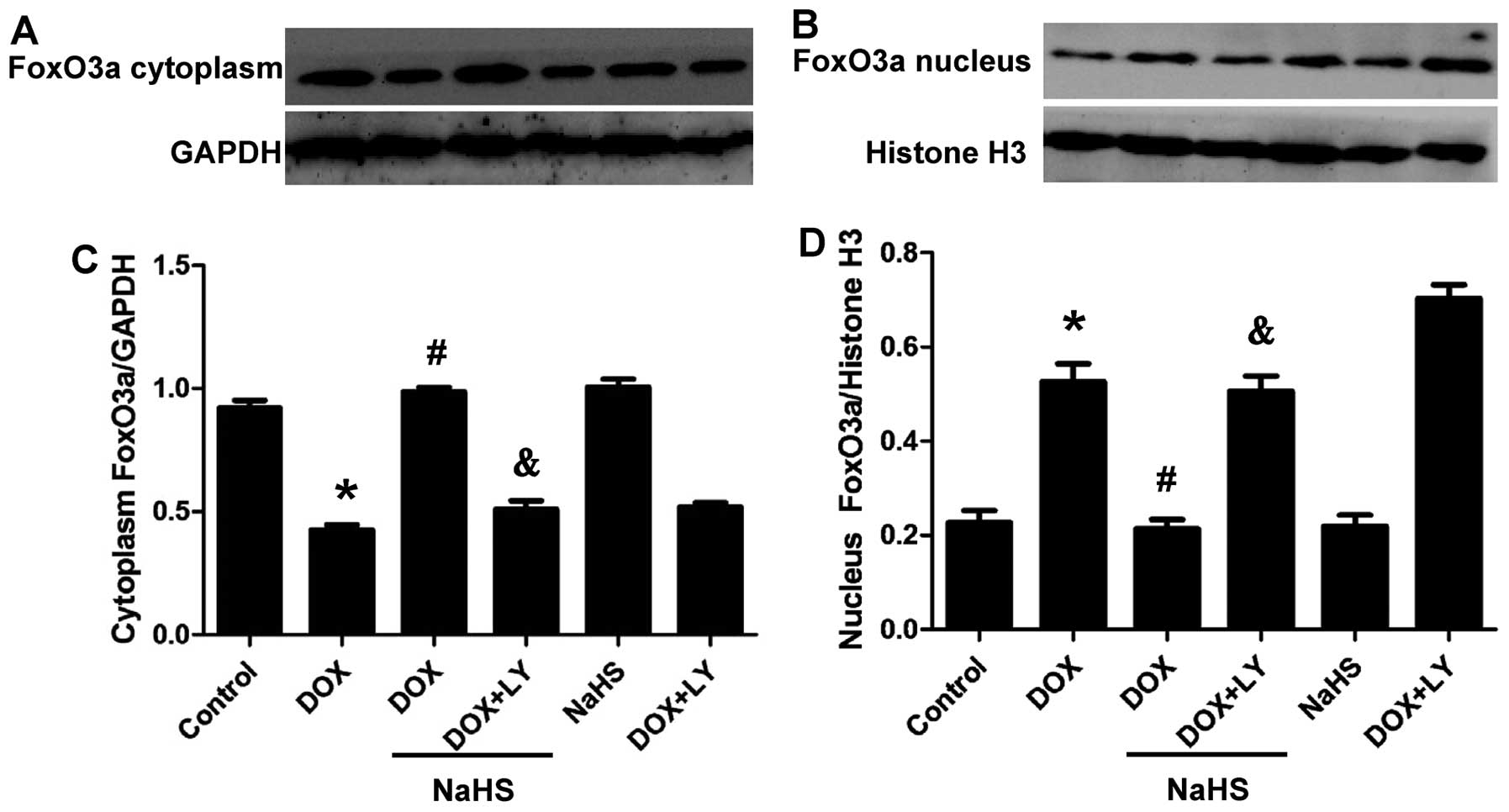
The transcription factor FoxO3a functions through
its phosphorylation and subcellular localization. The
phosphorylation of FoxO3a by Akt causes it to localize in the
cytoplasm and inhibit the functions of FoxO3a, including
pro-apoptotic effects (25). By
contrast, dephosphorylation of this protein promotes the
translocation of FoxO3a to the nucleus and triggers apoptosis. To
investigate the effects of H2S and DOX on FoxO3a, we
studied the subcellular localization of FoxO3a following exposure
to these reagents. Nuclear and cytosolic proteins from H9c2 cells
were extracted, and the subcellular localization of FoxO3a was
determined. Fig. 6 shows that DOX
enhanced the nuclear localization of FoxO3a in the H9c2 cells,
whereas NaHS blocked the effect of DOX. Co-treatment with the PI3K
inhibitor, LY294002, abolished the protective effect of NaHS.
Exogenous H2S downregulates
Bim expression through the PI3K/Akt-dependent signaling
pathway
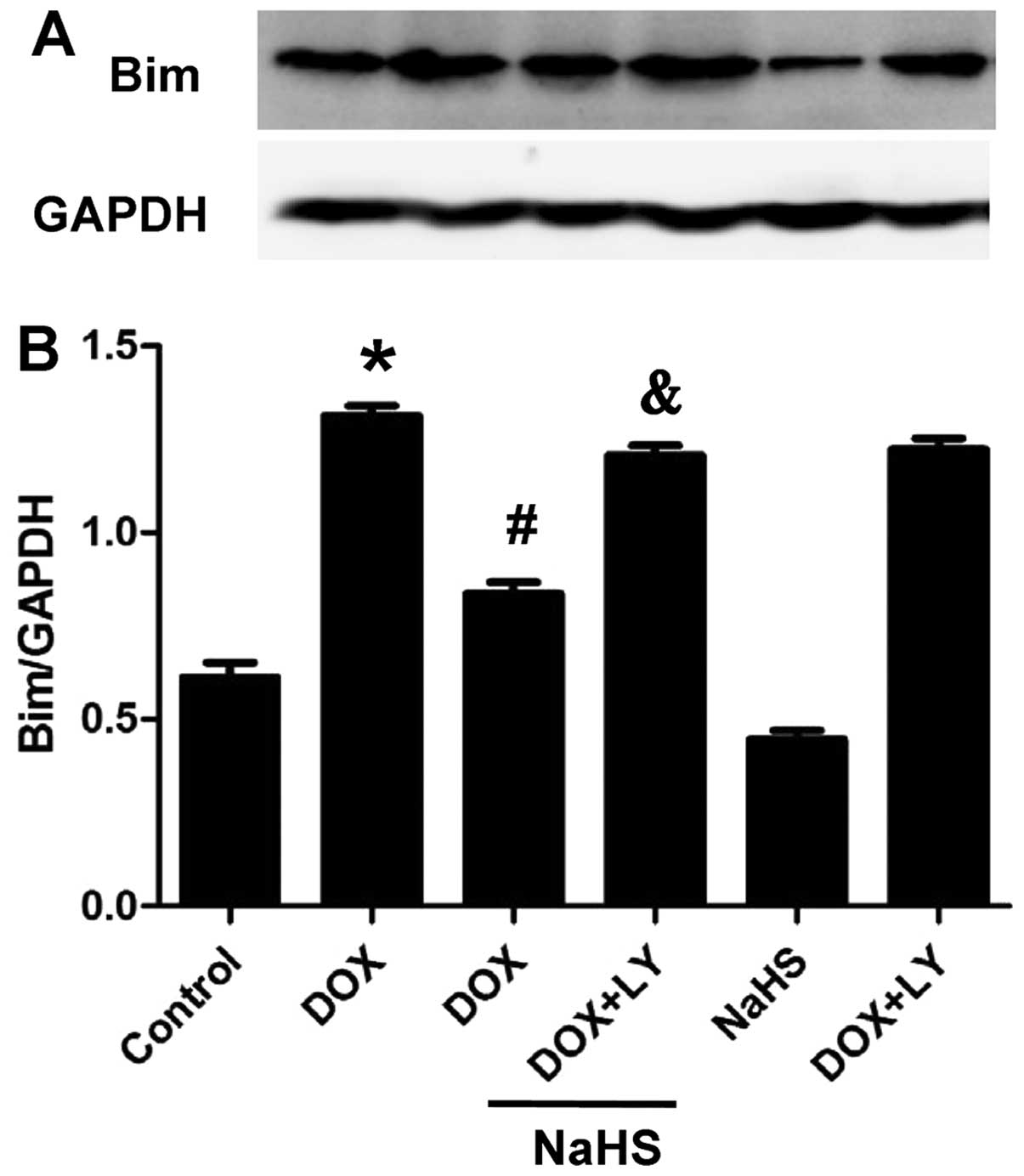
A significant down-regulation of Bim protein was
observed in the NaHS + DOX group compared with the DOX-treated
group. Furthermore, co-treatment with LY294002 increased Bim
protein expression compared with the control group (Fig. 7). These results indicated that
pre-treatment with NaHS downregulated the expression of Bim through
a PI3K/Akt-dependent signaling pathway.
Exogenous H2S inhibits
DOX-induced cytotoxicity
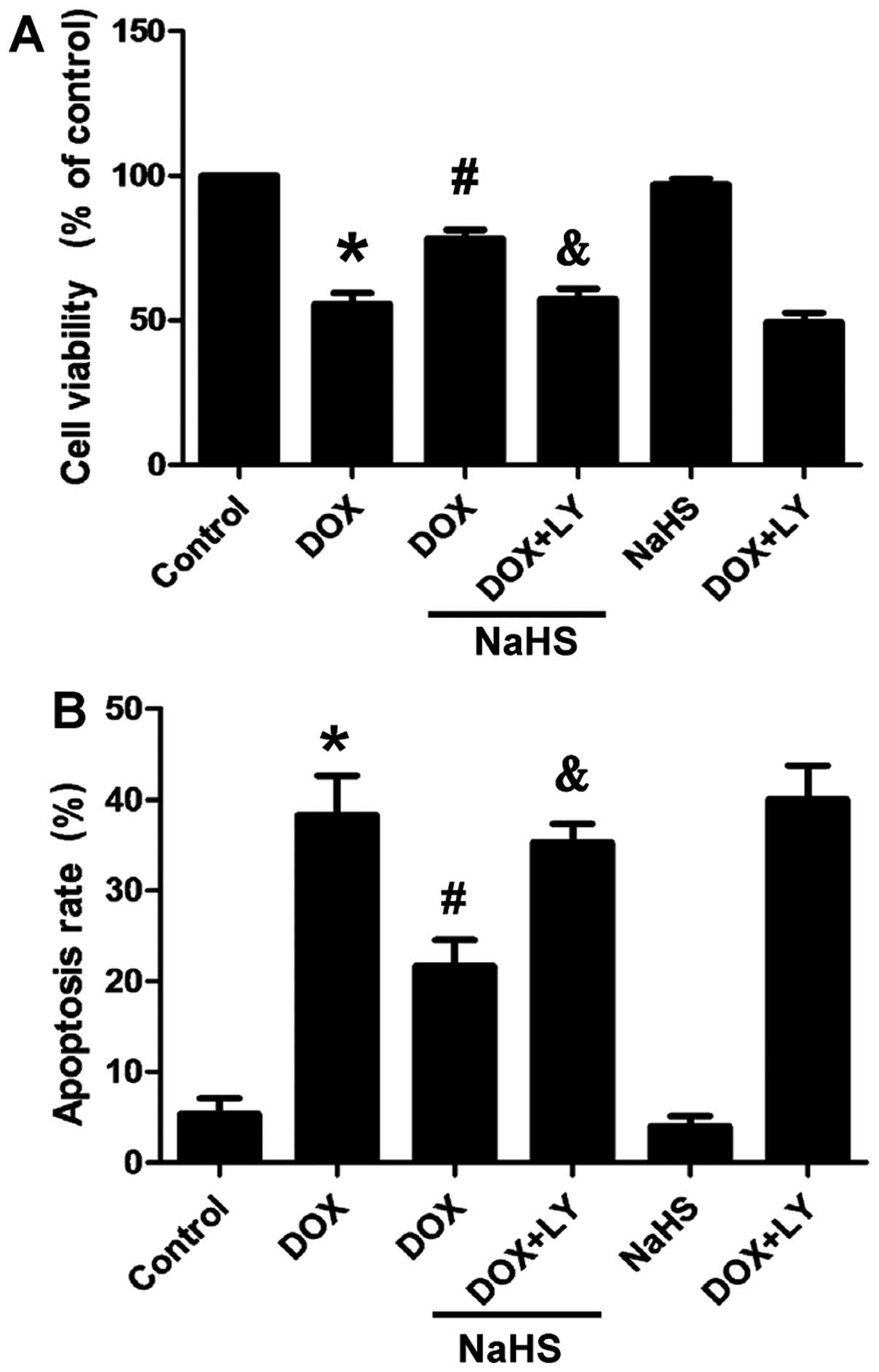
Fig. 8A shows that
the exposure of the H9c2 cells to DOX for 24 h induced marked
cytotoxicity, leading to a decrease in cell viability. However,
cell pre-treatment with 100 µM NaHS for 30 min prior to DOX
exposure significantly attenuated the degree of DOX-induced
cytotoxicity, as demonstrated by an increase in cell viability. The
preceding results (Figs. 3 and
5) showed that H2S
attenuated the DOX-induced decrease in p-Akt in the H9c2 cells.
Thus, we aimed to confirm whether the PI3K/Akt signaling pathway is
involved in the protective effect of H2S. The treatment
of the H9c2 cells with LY294002 and NaHS for 30 min prior to DOX
exposure for 24 h abolished the protective effect of
H2S, leading to an decrease in cell viability (Fig. 8A). NaHS alone did not alter the
viability of the H9c2 cells. These findings suggested that
H2S exerts a protective effect against DOX-induced
cytotoxicity, which may occur through the PI3K/Akt signaling
pathway.
Exogenous H2S reduces
DOX-induced apoptosis in H9c2 cells
The effects of H2S on DOX-induced
apoptosis were observed. Fig. 8B
shows that the apoptotic rate of the H9c2 cells exposed to 5
µM of DOX for 24 h increased significantly. However, cell
pre-treatment with 100 µM NaHS for 30 min prior to DOX
exposure markedly decreased the DOX-induced increase in the
apoptotic rate. To ascertain whether the PI3K/Akt signaling pathway
is involved in apoptosis induced by DOX, the H9c2 cells were
treated with LY294002 prior to exposure to NaHS plus DOX. The
results revealed that pretreatment with LY294002 abolished the
protective effect of H2S. NaHS alone did not markedly
alter the percentage of apoptotic H9c2 cells. These data strongly
suggest that H2S protects DOX-exposed H9c2 cells against
apoptosis and this may occur through the PI3K/Akt signaling
pathway.
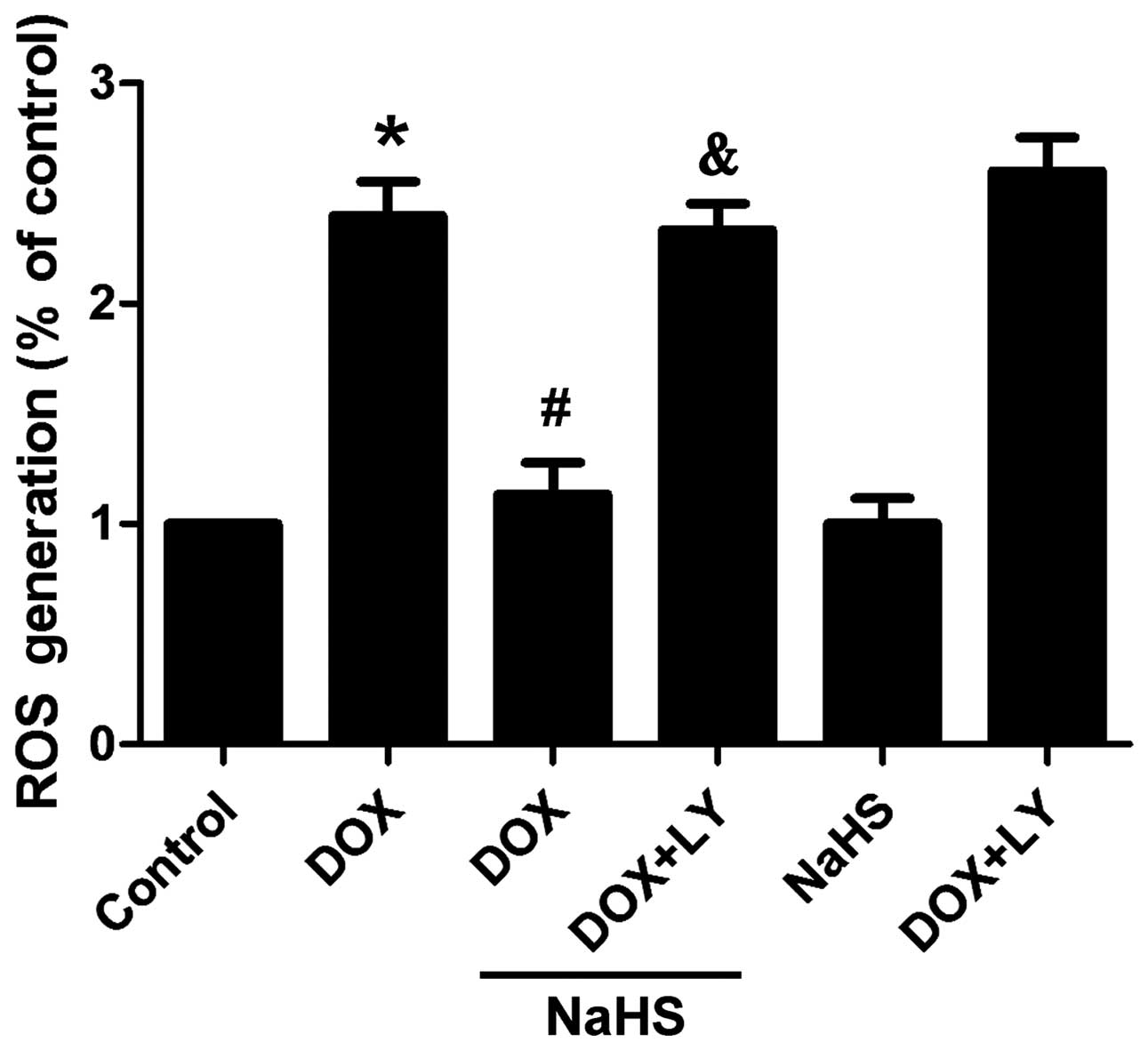
Exogenous H2S reduces
DOX-induced oxidative stress in H9c2 cells
The effect of H2S on the DOX-induced
production of ROS was investigated to elucidate whether the
antioxidant activity of H2S affords a cytoprotective
effect against DOX-induced cardiotoxicity. Fig. 9 shows that the exposure of H9c2
cells to 5 µM DOX evidently enhanced the generation of
intracellular ROS. However, NaHS pre-conditioning for 30 min
markedly attenuated the DOX-elicited generation of ROS. Notably,
pre-treatment with LY294002 abolished the protective effect of
H2S. However, NaHS alone did not alter the basal levels
of intracellular ROS.
Discussion
DOX is one of the most widely used and successful
antitumor drugs, although the clinical use of DOX is limited by
cumulative and dose-dependent cardiotoxic effects. With an
increasing population of cancer survivors, there is a growing need
to develop preventive strategies and effective therapies against
DOX-induced cardiotoxicity, and particularly, late-onset
cardio-myopathy. Although the cardiotoxic effects of DOX have been
previously examined, the underlying mechanisms responsible for
these effects remain to be elucidated. Mounting evidence supports
the hypothesis that free radical-induced oxidative stress, which
leads to cardiomyocyte death by apoptosis and necrosis, is a key
contributor to DOX-induced cardiotoxicity (16).
Previous findings have demonstrated that exogenous
H2S offers protection against DOX-induced cardiotoxicity
through antoixidant effects and the downregulation of inflammatory
responses (15,17,18). Guo et al have demonstrated
that exogenous H2S attenuates DOX-induced inflammation
and cytotoxicity through the inhibition of the p38 MAPK/NF-κB
pathway in H9c2 cells (15).
H2S also attenuates DOX-induced cardiotoxicity through
the inhibition of endoplasmic reticulum stress in H9c2 cells
(17). Su et al have
demonstrated that the downregulation of endogenously generated
H2S is probably involved in the pathogenesis of
DOX-induced cardiomyopathy, as H2S reduces lipid
peroxidation, increases the activity of antioxidant enzyme systems
and inhibits oxidative stress-induced injury (18).
Erythropoietin has been found to protect the
myocardium against DOX-induced impairment of heart function and
inhibits the apoptosis of cardiomyocytes by activating the PI3K/Akt
cell-survival pathway (19). In
addition, neureg-ulin-1 (20),
tanshinone IIA (21), and
urotensin II (22) have been
demonstrated to prevent the apoptosis of cardiomyocytes exposed to
DOX, partly through the Akt signaling pathways. Thus, these agents
may promote cell survival and exert cardioprotective effects.
FoxO3a is regulated by the PI3K/Akt pathway and plays an important
role in mediating the cytotoxic effects of DOX (23). It has also been demonstrated that
in the presence of serum and growth factors, the survival kinase
Akt is phosphorylated, which in turn phosphorylates FoxO
transcription factors, thereby leading to nuclear exclusion,
cytoplasmic retention and the inactivation of FoxO transcription
factors (24). Conversely,
oxidative stress has been shown to induce the re-localization of
FoxO transcription factors from the cytoplasm to the nucleus and
activate the target genes of FoxO transcription factors, including
the pro-apoptotic gene Bim, with subsequent cell apoptosis
(25).
The roles of FoxO3a in oxidative stress-induced
cardio-toxicity have received attention. Cardiac microvascular
endothelial cells (CMECs) are some of the predominant cells that
are immediately damaged after myocardial I/R injury. High glucose
(26) and hypoxia (27) have been observed to reduce the
phosphorylation of Akt and FoxO3a, induce FoxO3a activation, and
lead to ROS production and apoptosis in CMECs. Wang et al
reported that venlafaxine protects PC12 cells against
corticosterone-induced cell death by modulating the activity of the
PI3K/Akt/FoxO3a pathway (7).
Simvastatin inhibits rapamycin-induced dysfunction and apoptosis of
CMECs, probably through the activation of the PI3K/Akt/FoxO3a
signaling pathway (28).
Furthermore, erythropoietin activates the PI3K/Akt/FoxO3a signaling
pathway and protects neurons from 6-hydroxydopamine
(6-OHDA)-induced apoptosis (29).
In addition, sodium tanshinone IIA sulfonate (13) and bromelain (14) have been demonstrated to inhibit
the FoxO3a pathway and apoptosis of cardiomyocytes. In the present
study, we have demonstrated that a statistically significant
reduction in the phosphorylation of Akt and FoxO3a protein was
observed in the DOX-treated H9c2 cells. The data suggest that the
PI3K/Akt/FoxO3a signaling pathway is important in DOX-induced
cytoxicity in cardiomyocytes.
To elucidate the potential protective effects of
H2S against DOX-induced cardiotoxicity as well as the
mechanisms responsible for these effects, we observed the effect of
NaHS on the phosphorylation of Akt and FoxO3a protein induced by
DOX exposure. The findings of the present study show that the
treatment of H9c2 cells with NaHS significantly prevented the
DOX-induced reduction in the levels of p-Akt and p-FoxO3a, and this
was accompanied by an increase in cell viability, indicating that
the PI3K/Akt/FoxO3a pathway may be involved in the protective
effects of exogenous H2S against DOX-induced
cardiotoxicity.
Notably, the results of the present study provide
novel evidence that an interaction between ROS and FoxO3a exists in
DOX-exposed H9c2 cells, because H2S attenuated the
DOX-induced reduction in p-Akt and p-FoxO3a levels. Similar to
exogenous H2S, the treatment of H9c2 cells with NAC (ROS
scavenger) prior to DOX exposure attenuated the phosphorylation of
Akt and FoxO3a. These results indicated that targeting the
interaction that occurs between ROS and FoxO3a in DOX-induced
cardiotoxicity may aid in the treatment and prevention of cardiac
injury. Another important novel finding of this study was that
exogenous H2S protects against DOX-induced
cardiotoxicity by activating the PI3K/Akt/FoxO3a pathway in H9c2
cells. The findings of the present study support this hypothesis.
The treatment of H9c2 cells with NaHS (a donor of H2S)
prior to DOX exposure significantly ameliorated the reduction in
p-Akt and p-FoxO3a, attenuated the nuclear localization of FoxO3a
and the DOX-induced apoptosis of H9c2 cells, and exerted an
inhibitory effect on Bim expression. In addition, pre-treatment
with LY294002, a selective inhibitor of PI3K/Akt, reversed the
protective effect of H2S on DOX-induced cardiotoxicity,
as demonstrated by an increase in the number of apoptotic cells, a
reduction in cell viability and the phosphorylation of Akt and
FoxO3a, as well as a simultaneous increase in Bim expression.
Therefore, our results suggest that the protective effects of
H2S on FoxO3a nuclear translocation and Bim expression
are mediated by the PI3K/Akt pathway.
In conclusion, to the best of our knowledge, the
present study has demonstrated for the first time, that FoxO3a
plays a central role in the DOX-induced apoptosis of H9c2 cells.
Furthermore, data from the present study have revealed that
H2S protects H9c2 cardiomyocytes against DOX-induced
cytotoxicity through the activation of the PI3K/Akt/FoxO3a pathway.
To the best of our knowledge, this is the first study to show that
H2S is capable of acting on the PI3K/Akt/FoxO3a pathway
to enhance the survival of cardiomyocytes, thereby suggesting that
the FoxO3a pathway may be a novel therapeutic target in
cardiovascular disease.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (81470435, Z.-S.J.)
and the Graduate student research innovation project of Hunan
province (CX2013B397, M.-H.L.).
References
|
1
|
Magnano LC, Martínez Cibrian N, Andrade
González X and Bosch X: Cardiac complications of chemotherapy: role
of prevention. Curr Treat Options Cardiovasc Med. 16:3122014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Truong J, Yan AT, Cramarossa G and Chan
KK: Chemotherapy-induced cardiotoxicity: detection, prevention, and
management. Can J Cardiol. 30:869–878. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Spagnuolo RD, Recalcati S, Tacchini L and
Cairo G: Role of hypoxia-inducible factors in the
dexrazoxane-mediated protection of cardiomyocytes from
doxorubicin-induced toxicity. Br J Pharmacol. 163:299–312. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang Y, Tang ZH, Ren Z, Qu SL, Liu MH,
Liu LS and Jiang ZS: Hydrogen sulfide, the next potent preventive
and therapeutic agent in aging and age-associated diseases. Mol
Cell Biol. 33:1104–1113. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Osipov RM, Robich MP, Feng J, Liu Y,
Clements RT, Glazer HP, Sodha NR, Szabo C, Bianchi C and Sellke FW:
Effect of hydrogen sulfide in a porcine model of myocardial
ischemia-reperfusion: comparison of different administration
regimens and characterization of the cellular mechanisms of
protection. J Cardiovasc Pharmacol. 54:287–297. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huang YE, Tang ZH, Xie W, Shen XT, Liu MH,
Peng XP, Zhao ZZ, Nie DB, Liu LS and Jiang ZS: Endogenous hydrogen
sulfide mediates the cardioprotection induced by ischemic
post-conditioning in the early reperfusion phase. Exp Ther Med.
4:1117–1123. 2012.PubMed/NCBI
|
|
7
|
Wang H, Zhou X, Huang J, Mu N, Guo Z, Wen
Q, Wang R, Chen S, Feng ZP and Zheng W: The role of Akt/FoxO3a in
the protective effect of venlafaxine against corticosterone-induced
cell death in PC12 cells. Psychopharmacology (Berl). 228:129–141.
2013. View Article : Google Scholar
|
|
8
|
van der Vos KE and Coffer PJ: The
extending network of FOXO transcriptional target genes. Antioxid
Redox Signal. 14:579–592. 2011. View Article : Google Scholar
|
|
9
|
Sanphui P and Biswas SC: FoxO3a is
activated and executes neuron death via Bim in response to
β-amyloid. Cell Death Dis. 4:e6252013. View Article : Google Scholar
|
|
10
|
Bao W, Pan F, Chen L, Su G, Gao X, Li Y,
Sun Q, Sun J, He K and Song H: The PI3K/AKT pathway and FOXO3a
transcription factor mediate high glucose-induced apoptosis in
neonatal rat ventricular myocytes. Iran Red Crescent Med J.
16:e149142014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li D, Qu Y, Mao M, Zhang X, Li J, Ferriero
D and Mu D: Involvement of the PTEN-AKT-FOXO3a pathway in neuronal
apoptosis in developing rat brain after hypoxia-ischemia. J Cereb
Blood Flow Metab. 29:1903–1913. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu MH, Yuan C, He J, Tan TP, Wu SJ, Fu
HY, Liu J, Yu S, Chen YD, Le QF, et al: Resveratrol protects PC12
cells from high glucose-induced neurotoxicity via PI3K/Akt/FoxO3a
pathway. Cell Mol Neurobiol. 35:513–522. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang MQ, Zheng YL, Chen H, Tu JF, Shen Y,
Guo JP, Yang XH, Yuan SR, Chen LZ, Chai JJ, et al: Sodium
tanshinone IIA sulfonate protects rat myocardium against
ischemia-reperfusion injury via activation of PI3K/Akt/FOXO3A/Bim
pathway. Acta Pharmacol Sin. 34:1386–1396. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Juhasz B, Thirunavukkarasu M, Pant R, Zhan
L, Penumathsa SV, Secor ER Jr, Srivastava S, Raychaudhuri U, Menon
VP, Otani H, et al: Bromelain induces cardioprotection against
ischemia-reperfusion injury through Akt/FOXO pathway in rat
myocardium. Am J Physiol Heart Circ Physiol. 294:H1365–H1370. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Guo R, Wu K, Chen J, Mo L, Hua X, Zheng D,
Chen P, Chen G, Xu W and Feng J: Exogenous hydrogen sulfide
protects against doxorubicin-induced inflammation and cytotoxicity
by inhibiting p38MAPK/NFκB pathway in H9c2 cardiac cells. Cell
Physiol Biochem. 32:1668–1680. 2013.
|
|
16
|
Tocchetti CG, Carpi A, Coppola C,
Quintavalle C, Rea D, Campesan M, Arcari A, Piscopo G, Cipresso C,
Monti MG, et al: Ranolazine protects from doxorubicin-induced
oxidative stress and cardiac dysfunction. Eur J Heart Fail.
16:358–366. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang XY, Yang CT, Zheng DD, Mo LQ, Lan AP,
Yang ZL, Hu F, Chen PX, Liao XX and Feng JQ: Hydrogen sulfide
protects H9c2 cells against doxorubicin-induced cardiotoxicity
through inhibition of endoplasmic reticulum stress. Mol Cell
Biochem. 363:419–426. 2012. View Article : Google Scholar
|
|
18
|
Su YW, Liang C, Jin HF, Tang XY, Han W,
Chai LJ, Zhang CY, Geng B, Tang CS and Du JB: Hydrogen sulfide
regulates cardiac function and structure in adriamycin-induced
cardiomyopathy. Circ J. 73:741–749. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim KH, Oudit GY and Backx PH:
Erythropoietin protects against doxorubicin-induced cardiomyopathy
via a phosphatidylinositol 3-kinase-dependent pathway. J Pharmacol
Exp Ther. 324:160–169. 2008. View Article : Google Scholar
|
|
20
|
An T, Zhang Y, Huang Y, Zhang R, Yin S,
Guo X, Wang Y, Zou C, Wei B, Lv R, et al: Neuregulin-1 protects
against doxorubicin-induced apoptosis in cardiomyocytes through an
Akt-dependent pathway. Physiol Res. 62:379–385. 2013.PubMed/NCBI
|
|
21
|
Hong HJ, Liu JC, Chen PY, Chen JJ, Chan P
and Cheng TH: Tanshinone IIA prevents doxorubicin-induced
cardiomyocyte apoptosis through Akt-dependent pathway. Int J
Cardiol. 157:174–179. 2012. View Article : Google Scholar
|
|
22
|
Chen YL, Loh SH, Chen JJ and Tsai CS:
Urotensin II prevents cardiomyocyte apoptosis induced by
doxorubicin via Akt and ERK. Eur J Pharmacol. 680:88–94. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ho KK, McGuire VA, Koo CY, Muir KW, de
Olano N, Maifoshie E, Kelly DJ, McGovern UB, Monteiro LJ, Gomes AR,
et al: Phosphorylation of FOXO3a on Ser-7 by p38 promotes its
nuclear localization in response to doxorubicin. J Biol Chem.
287:1545–1555. 2012. View Article : Google Scholar :
|
|
24
|
Wang Y, Zhou Y and Graves DT: FOXO
transcription factors: their clinical significance and regulation.
Biomed Res Int. 2014:9253502014.PubMed/NCBI
|
|
25
|
Storz P: Forkhead homeobox type O
transcription factors in the responses to oxidative stress.
Antioxid Redox Signal. 14:593–605. 2011. View Article : Google Scholar :
|
|
26
|
Peng C, Ma J, Gao X, Tian P, Li W and
Zhang L: High glucose induced oxidative stress and apoptosis in
cardiac microvascular endothelial cells are regulated by FoxO3a.
PLoS One. 8:e797392013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang S, Zhao Y, Xu M, Yu L, Zhao Y, Chen
J, Yuan Y, Zheng Q and Niu X: FoxO3a modulates hypoxia stress
induced oxidative stress and apoptosis in cardiac microvascular
endothelial cells. PLoS One. 8:e803422013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pan Q, Xie X, Guo Y and Wang H:
Simvastatin promotes cardiac microvascular endothelial cells
proliferation, migration and survival by phosphorylation of p70 S6K
and FoxO3a. Cell Biol Int. 38:599–609. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jia Y, Mo SJ, Feng QQ, Zhan ML, OuYang LS,
Chen JC, Ma YX, Wu JJ and Lei WL: EPO-dependent activation of
PI3K/Akt/FoxO3a signalling mediates neuroprotection in in vitro and
in vivo models of Parkinson's disease. J Mol Neurosci. 53:117–124.
2014. View Article : Google Scholar : PubMed/NCBI
|